
Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. MCAO Model Mice
2.3. Measurement of Evans Blue (EB) Extravasation
2.4. Isolation of Brain Microvascular Endothelial Cells
2.5. Western Blot Analysis
2.6. Immunofluorescence (IF)
2.7. Cell Culture and Deferoxamine (DFO) Treatment
2.8. Oxygen and Glucose Deprivation and Reperfusion
2.9. Measurement of Cell Viability
2.10. Assessment of Apoptosis by Flow Cytometry
2.11. Measurement of Lipid ROS and Mitochondrial ROS
2.12. Measurement of Malondialdehyde (MDA) and Superoxide Dismutases (SODs)
2.13. Statistical Analysis
3. Results
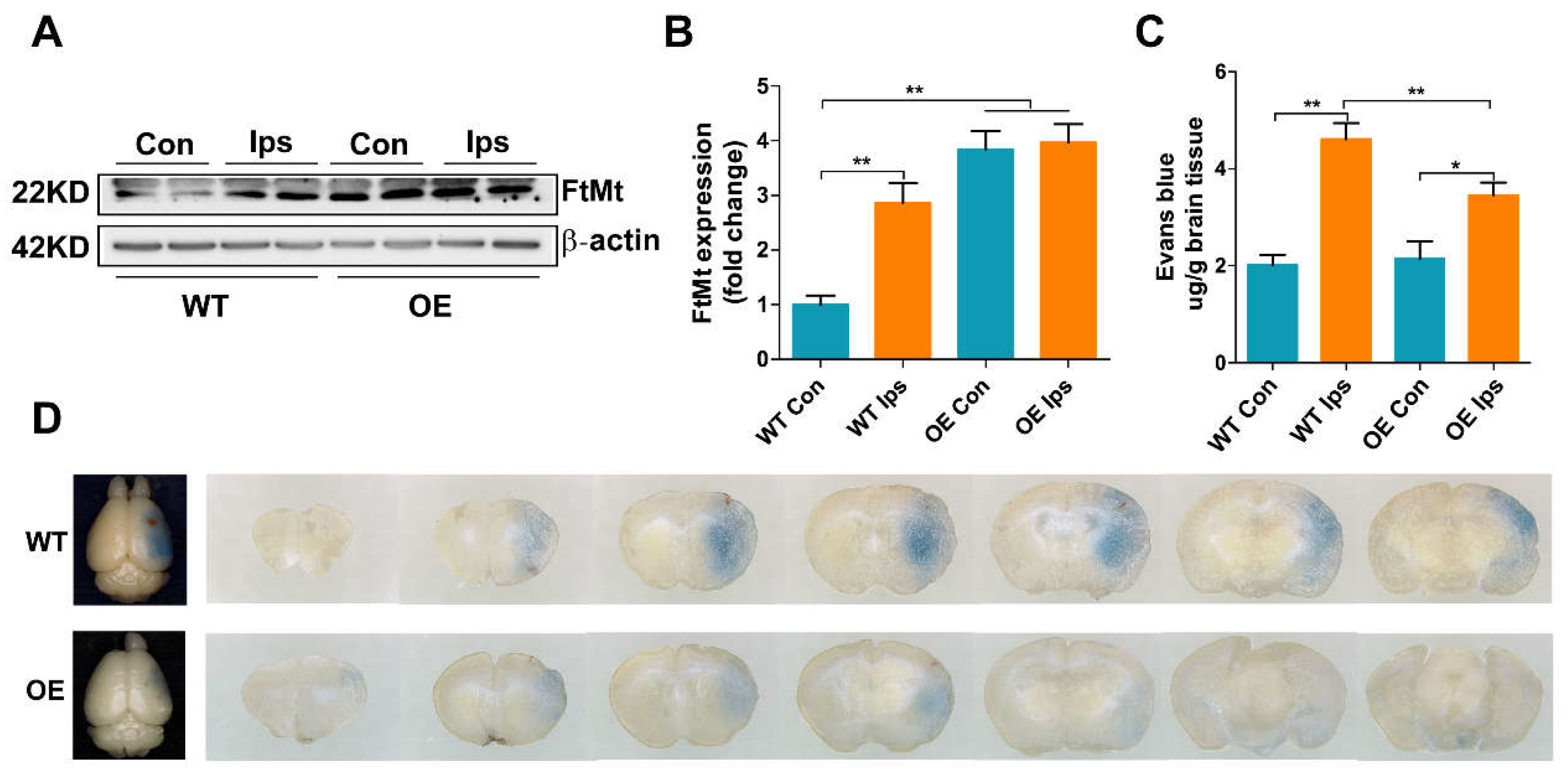
3.1. Overexpression of FtMt Attenuates I/R-Induced BBB Disruption
3.2. Overexpression of FtMt Increases Tight Junctional Integrity in Cerebral I/R
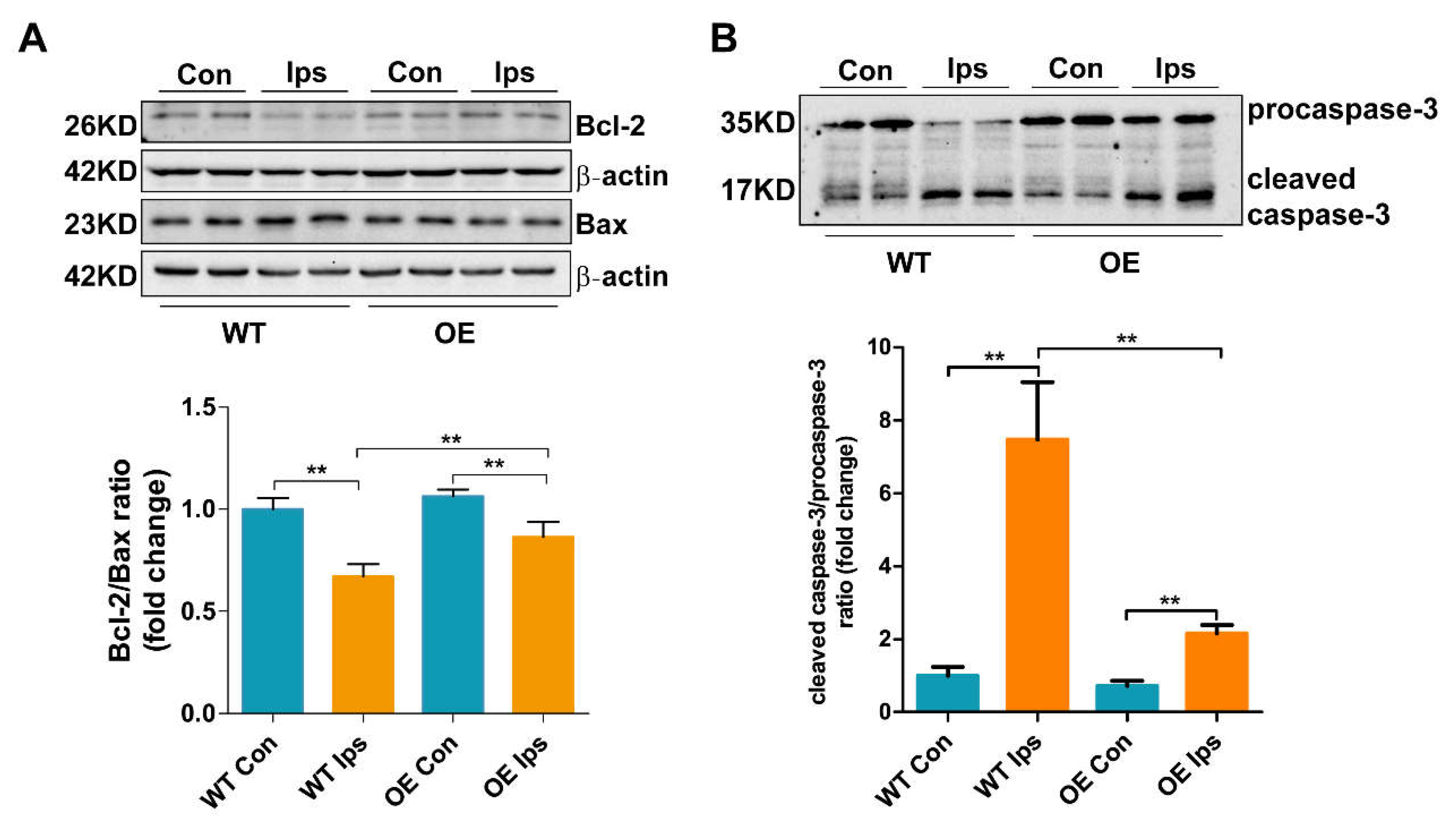
3.3. Overexpression of FtMt Abrogates Endothelial Cell Apoptosis after Cerebral I/R
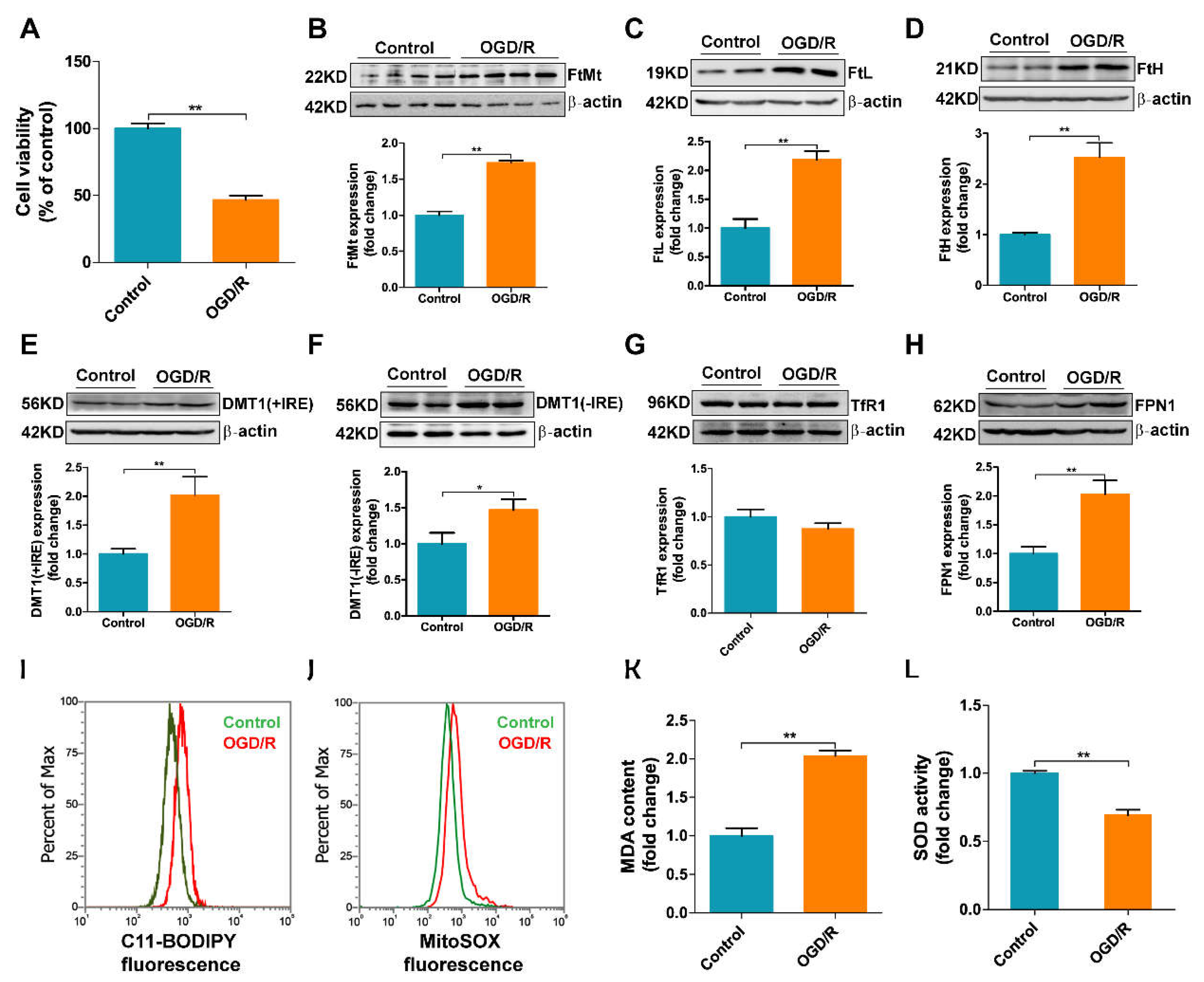
3.4. Overexpression of FtMt Rescues I/R-Induced Iron Dysregulation and Oxidative Stress in BMVECs
3.5. OGD/R Induces Iron Dysregulation and Oxidative Stress in bEnd.3 Cells
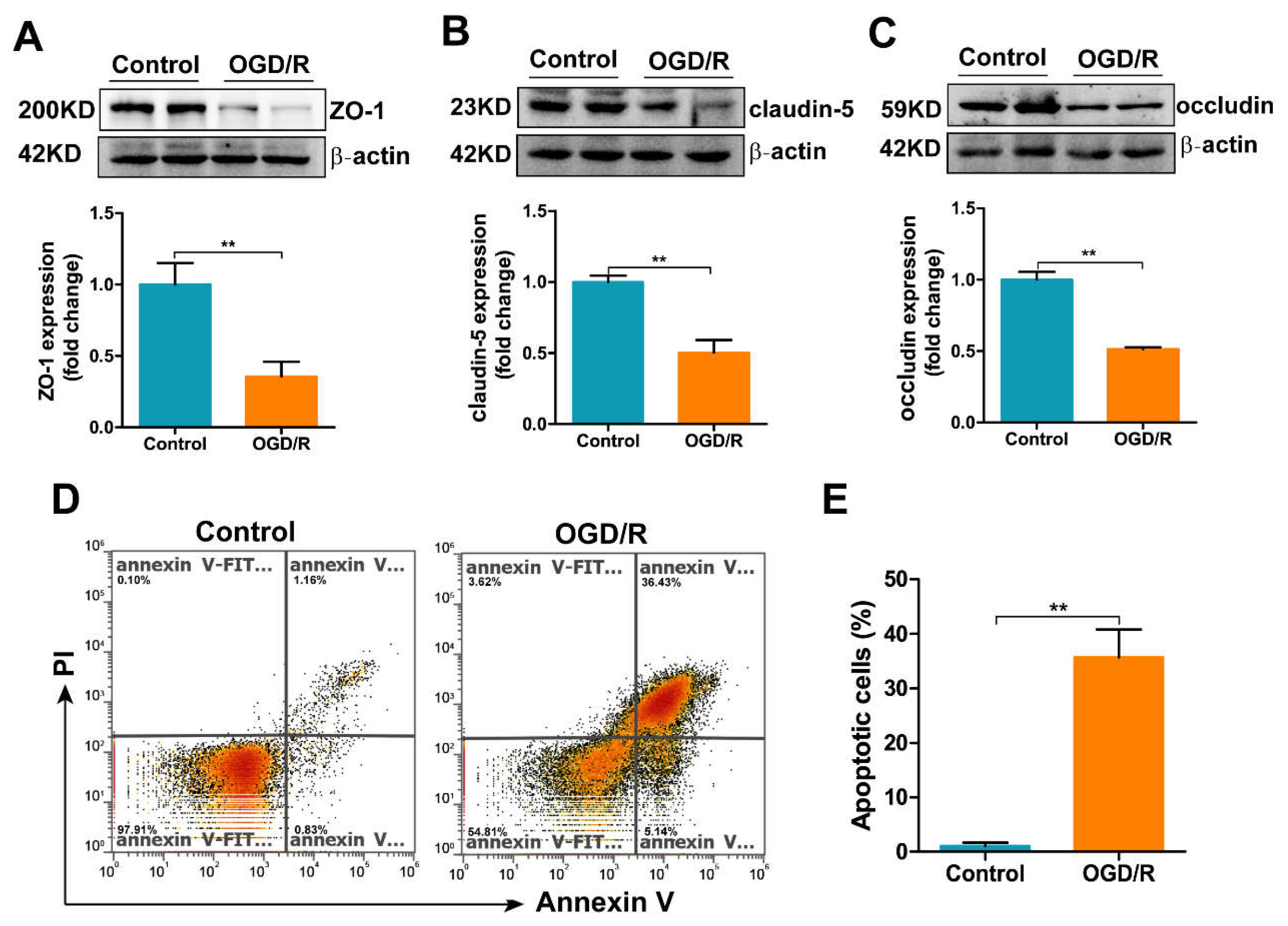
3.6. OGD/R Induces Tight Junction Loss and Apoptosis in bEnd.3 Cells
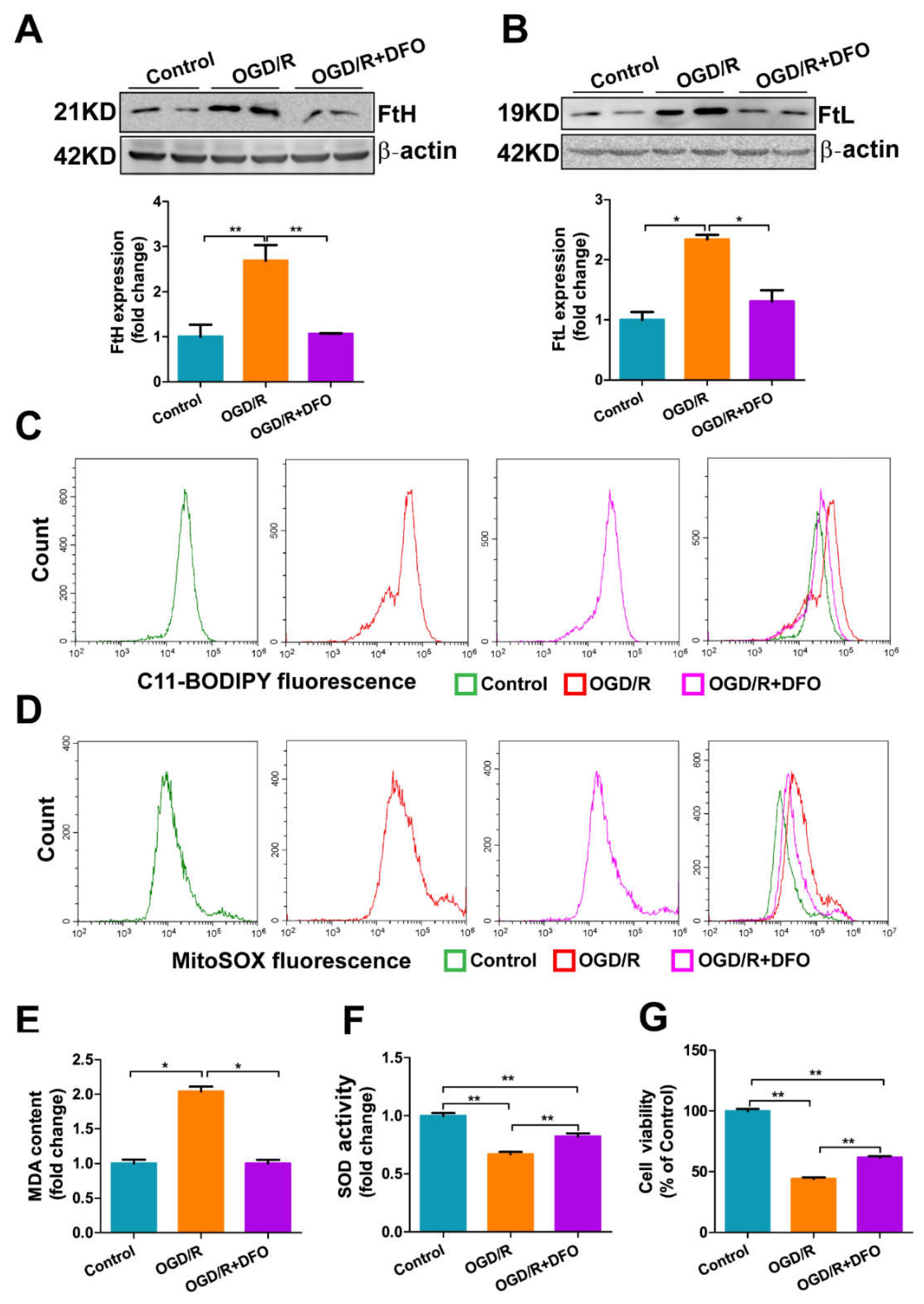
3.7. DFO Treatment Attenuates OGD/R-Induced Oxidative Damage in bEnd.3 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, B.C.V.; De Silva, D.A.; MacLeod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Prim. 2019, 5, 70. [Google Scholar] [CrossRef]
- Xu, S.; Bian, H.; Shu, S.; Xia, S.; Gu, Y.; Zhang, M.; Xu, Y.; Cao, X. AIM2 deletion enhances blood-brain barrier integrity in experimental ischemic stroke. CNS Neurosci. Ther. 2021, 27, 1224–1237. [Google Scholar] [CrossRef]
- Sun, M.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.; Guo, Z. Free Radical Damage in Ischemia Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy Acute Ischemic Stroke Is a Common Cause of Morbidity and Mortality Worldwide. Thrombectomy Are the Main Revascularization Therapies. Oxidative Med. Cell. Longev. 2017, 2018, 3804979. [Google Scholar]
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Li, Y.; Zhong, W.; Jiang, Z.; Tang, X. New progress in the approaches for blood–brain barrier protection in acute ischemic stroke. Brain Res. Bull. 2018, 144, 46–57. [Google Scholar] [CrossRef]
- Kim, K.-A.; Kim, D.; Kim, J.-H.; Shin, Y.-J.; Kim, E.-S.; Akram, M.; Kim, E.-H.; Majid, A.; Baek, S.-H.; Bae, O.-N. Autophagy-mediated occludin degradation contributes to blood–brain barrier disruption during ischemia in bEnd.3 brain endothelial cells and rat ischemic stroke models. Fluids Barriers CNS 2020, 17, 21. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171. [Google Scholar] [CrossRef]
- Amruta, N.; Bix, G. ATN-161 Ameliorates Ischemia/Reperfusion-induced Oxidative Stress, Fibro-inflammation, Mitochondrial damage, and Apoptosis-mediated Tight Junction Disruption in bEnd.3 Cells. Inflammation 2021, 44, 2377–2394. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C.; Anrather, J. Stroke research at a crossroad: Asking the brain for directions. Nat. Neurosci. 2011, 14, 1363–1368. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, Y.; Xiao, Y.; Hua, Z.; Cheng, J.; Jia, J. Hydrogen Sulfide Attenuates Tissue Plasminogen Activator-Induced Cerebral Hemorrhage Following Experimental Stroke. Transl. Stroke Res. 2016, 7, 209–219. [Google Scholar] [CrossRef]
- Keep, R.F.; Zhou, N.; Xiang, J.; Andjelkovic, A.V.; Hua, Y.; Xi, G. Vascular disruption and blood–brain barrier dysfunction in intracerebral hemorrhage. Fluids Barriers CNS 2014, 11, 18. [Google Scholar] [CrossRef] [Green Version]
- Horsch, A.D.; Dankbaar, J.W.; Van Seeters, T.; Niesten, J.M.; Luitse, M.J.; Vos, P.C.; Van Der Schaaf, I.C.; Biessels, G.-J.; Van Der Graaf, Y.; Kappelle, L.J.; et al. Relation between stroke severity, patient characteristics and CT-perfusion derived blood-brain barrier permeability measurements in acute ischemic stroke. Clin. Neuroradiol. 2015, 26, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Zhou, F.; Han, L.; Yang, J.; Fan, H.; Li, S.; Li, J.; Zhang, X.; Wang, X.; Chen, X.; et al. Sodium Tanshinone IIA Sulfonate Enhances Effectiveness Rt-PA Treatment in Acute Ischemic Stroke Patients Associated with Ameliorating Blood-Brain Barrier Damage. Transl. Stroke Res. 2017, 8, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, S.; Zhang, H.; Lee, S.; Kim, H.; Kim, Y.; Won, M.-H.; Kim, Y.-M.; Kwon, Y.-G. CLEC14A deficiency exacerbates neuronal loss by increasing blood-brain barrier permeability and inflammation. J. Neuroinflamm. 2020, 17, 48. [Google Scholar] [CrossRef]
- Pan, J.; Qu, M.; Li, Y.; Wang, L.; Zhang, L.; Wang, Y.; Tang, Y.; Tian, H.-L.; Zhang, Z.; Yang, G.-Y. MicroRNA-126-3p/-5p Overexpression Attenuates Blood-Brain Barrier Disruption in a Mouse Model of Middle Cerebral Artery Occlusion. Stroke 2020, 51, 619–627. [Google Scholar] [CrossRef]
- Ridder, D.A.; Wenzel, J.; Müller, K.; Töllner, K.; Tong, X.-K.; Assmann, J.C.; Stroobants, S.; Weber, T.; Niturad, C.; Fischer, L.; et al. Brain endothelial TAK1 and NEMO safeguard the neurovascular unit. J. Exp. Med. 2015, 212, 1529–1549. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef]
- Drysdale, J.; Arosio, P.; Invernizzi, R.; Cazzola, M.; Volz, A.; Corsi, B.; Biasiotto, G.; Levi, S. Mitochondrial Ferritin: A New Player in Iron Metabolism. Blood Cells Mol. Dis. 2002, 29, 376–383. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.-S.; Su, L.; Zheng, X.; Santambrogio, P.; Gou, Y.-J.; Hao, Q.; Wang, P.-N.; Li, Y.-R.; Zhao, B.-L.; et al. Mitochondrial Ferritin Is a Hypoxia-Inducible Factor 1α-Inducible Gene That Protects from Hypoxia-Induced Cell Death in Brain. Antioxid. Redox Signal. 2019, 30, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Biasiotto, G.; Sanvito, F.; Olivieri, S.; Arosio, P.; Levi, S. Mitochondrial Ferritin Expression in Adult Mouse Tissues. J. Histochem. Cytochem. 2007, 55, 1129–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patt, A.; Horesh, I.R.; Berger, E.M.; Harken, A.H.; Repine, J.E. Iron Depletion or Chelation Reduces Ischemia/Reperfusion-Induced Edema in Gerbil Brains. J. Pediatr. Surg. 1990, 25, 224–228. [Google Scholar] [CrossRef]
- Ding, H.; Yan, C.-Z.; Shi, H.; Zhao, Y.-S.; Chang, S.-Y.; Yu, P.; Wu, W.-S.; Zhao, C.-Y.; Chang, Y.-Z.; Duan, X.-L. Hepcidin Is Involved in Iron Regulation in the Ischemic Brain. PLoS ONE 2011, 6, e25324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Cui, Y.; Ren, Q.; Yan, B.; Zhao, Y.; Yu, P.; Gao, G.; Shi, H.; Chang, S.; Chang, Y.-Z. Mitochondrial ferritin attenuates cerebral ischaemia/reperfusion injury by inhibiting ferroptosis. Cell Death Dis. 2021, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, P.; Wu, Q.; Xie, L.; Cui, Y.; Li, H.; Yu, P.; Chang, Y.-Z. The Construction and Characterization of Mitochondrial Ferritin Overexpressing Mice. Int. J. Mol. Sci. 2017, 18, 1518. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Lin, V.; Davis, W.; Huang, T.; Carranza, A.; Sprague, S.; Reyes, R.; Jimenez, D.; Ding, Y. Preischemic Induction of TNF-Alpha by Physical Exercise Reduces Blood-Brain Barrier Dysfunction in Stroke. J. Cereb. Blood Flow Metab. 2008, 28, 1422–1430. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wu, Q.; Wu, W.; Li, H.; Guo, Y.; Yu, P.; Gao, G.; Shi, Z.; Zhao, B.; Chang, Y.-Z. Mitochondrial Ferritin Deletion Exacerbatesβ-Amyloid-Induced Neurotoxicity in Mice. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tang, X.; Ma, F.; Fan, Y.; Sun, P.; Zhu, T.; Zhang, J.; Hamblin, M.H.; Chen, Y.E.; Yin, K.-J. Endothelium-targeted overexpression of Krüppel-like factor 11 protects the blood-brain barrier function after ischemic brain injury. Brain Pathol. 2020, 30, 746–765. [Google Scholar] [CrossRef]
- Lum, H.; Roebuck, K.A. Oxidant Stress and Endothelial Cell Dysfunction. AM J. Physiol. Cell Physiol. 2001, 280, C719–C741. [Google Scholar] [CrossRef]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Haldar, S.; Tripathi, A.K.; Horback, K.; Wong, J.; Sharma, D.; Beserra, A.; Suda, S.; Anbalagan, C.; Dev, S.; et al. Brain Iron Homeostasis: From Molecular Mechanisms to Clinical Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2014, 20, 1324–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selim, M.H.; Ratan, R.R. The role of iron neurotoxicity in ischemic stroke. Ageing Res. Rev. 2004, 3, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, T. Iron, Oxidative Stress and Early Neurological Deterioration in Ischemic Stroke. Curr. Med. Chem. 2007, 14, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Tuo, Q.-Z.; Lei, P.; Jackman, K.A.; Li, X.-L.; Xiong, H.; Liuyang, Z.-Y.; Roisman, L.C.; Zhang, S.-T.; Ayton, S.; Wang, Q.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.; Sobrino, T.; Castellanos, M.; Nombela, F.; Arenillas, J.F.; Riva, E.; Cristobo, I.; García, M.M.; Vivancos, J.; Serena, J.; et al. Increased Body Iron Stores Are Associated with Poor Outcome After Thrombolytic Treatment in Acute Stroke. Stroke 2007, 38, 90–95. [Google Scholar] [CrossRef] [Green Version]
- You, L.-H.; Yan, C.-Z.; Zheng, B.-J.; Ci, Y.-Z.; Chang, S.-Y.; Yu, P.; Gao, G.-F.; Li, H.-Y.; Dong, T.-Y.; Chang, Y.-Z. Astrocyte hepcidin is a key factor in LPS-induced neuronal apoptosis. Cell Death Dis. 2017, 8, e2676. [Google Scholar] [CrossRef] [Green Version]
- Qian, Z.M.; Shen, X. Brain iron transport and neurodegeneration. Trends Mol. Med. 2001, 7, 103–108. [Google Scholar] [CrossRef]
- Ueno, M.; Chiba, Y.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Fujihara, R. Blood–brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol. 2016, 33, 89–96. [Google Scholar] [CrossRef]
- Kikuchi, K.; Tanaka, E.; Murai, Y.; Tancharoen, S. Clinical Trials in Acute Ischemic Stroke. CNS Drugs 2014, 28, 929–938. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Ren, Q.; Shi, M.; Liu, Y.; Bai, H.; Chang, Y.-Z. Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants 2022, 11, 1257. https://doi.org/10.3390/antiox11071257
Wang P, Ren Q, Shi M, Liu Y, Bai H, Chang Y-Z. Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants. 2022; 11(7):1257. https://doi.org/10.3390/antiox11071257
Chicago/Turabian StyleWang, Peina, Qianqian Ren, Mengtong Shi, Yuanyuan Liu, Huiyuan Bai, and Yan-Zhong Chang. 2022. "Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells" Antioxidants 11, no. 7: 1257. https://doi.org/10.3390/antiox11071257
APA StyleWang, P., Ren, Q., Shi, M., Liu, Y., Bai, H., & Chang, Y.-Z. (2022). Overexpression of Mitochondrial Ferritin Enhances Blood–Brain Barrier Integrity Following Ischemic Stroke in Mice by Maintaining Iron Homeostasis in Endothelial Cells. Antioxidants, 11(7), 1257. https://doi.org/10.3390/antiox11071257