
Blocking of P2X7r Reduces Mitochondrial Stress Induced by Alcohol and Electronic Cigarette Exposure in Brain Microvascular Endothelial Cells


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents/Kits
2.2. Cell Culture and Treatments
2.3. Western Blot Analyses
2.4. Mito-Stress Test in Cultured D3 Cells
2.5. Quantitative RT-PCR
2.6. ATP Detection Assay
2.7. Intracellular Calcium (Ca2+) Assay
2.8. Barrier Function Measurement by Trans-Endothelial Electrical Resistance (TEER)
2.9. Statistical Analysis
3. Results
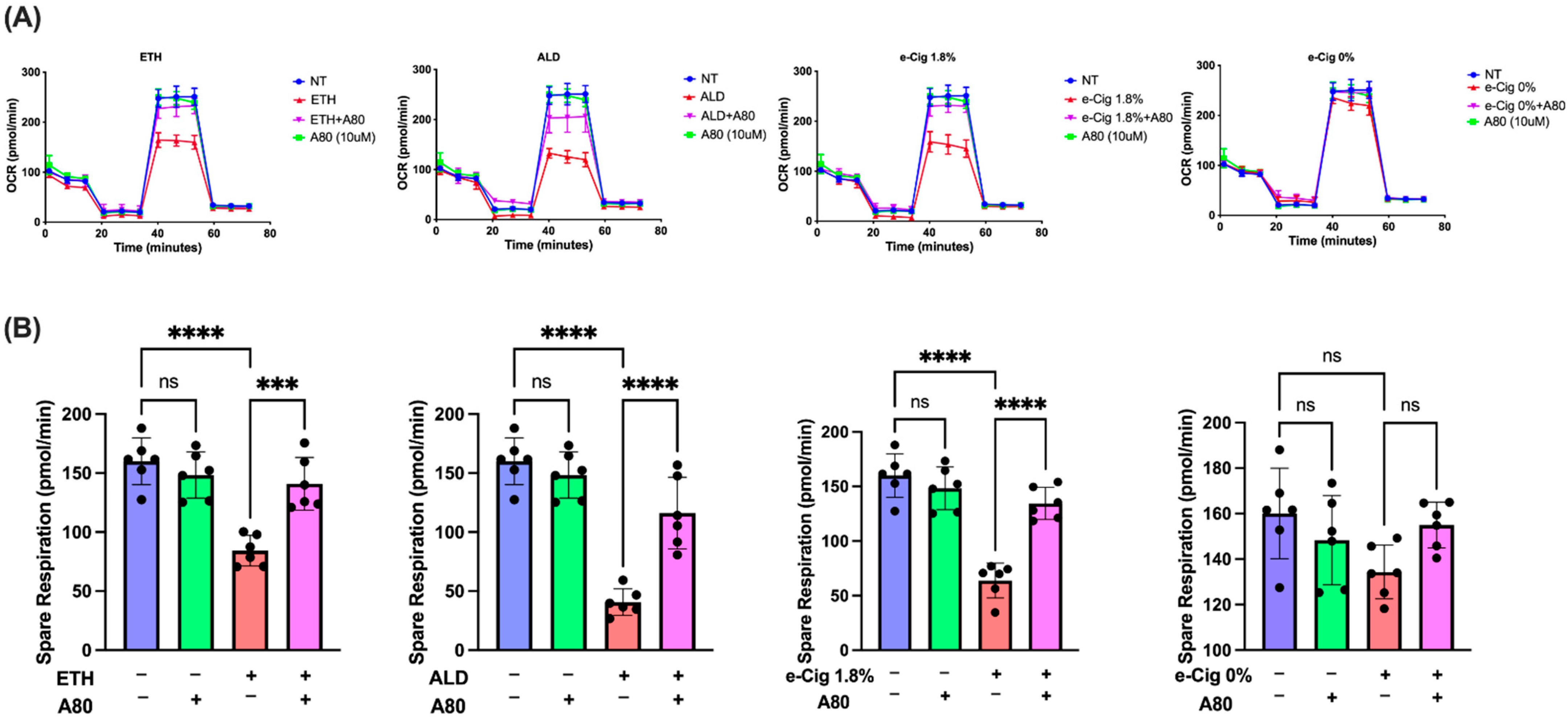
3.1. P2X7r Inhibition Ameliorated Mitochondrial Dysfunction in hBMVECs Exposed with ETH, ALD and 1.8% e-Cig
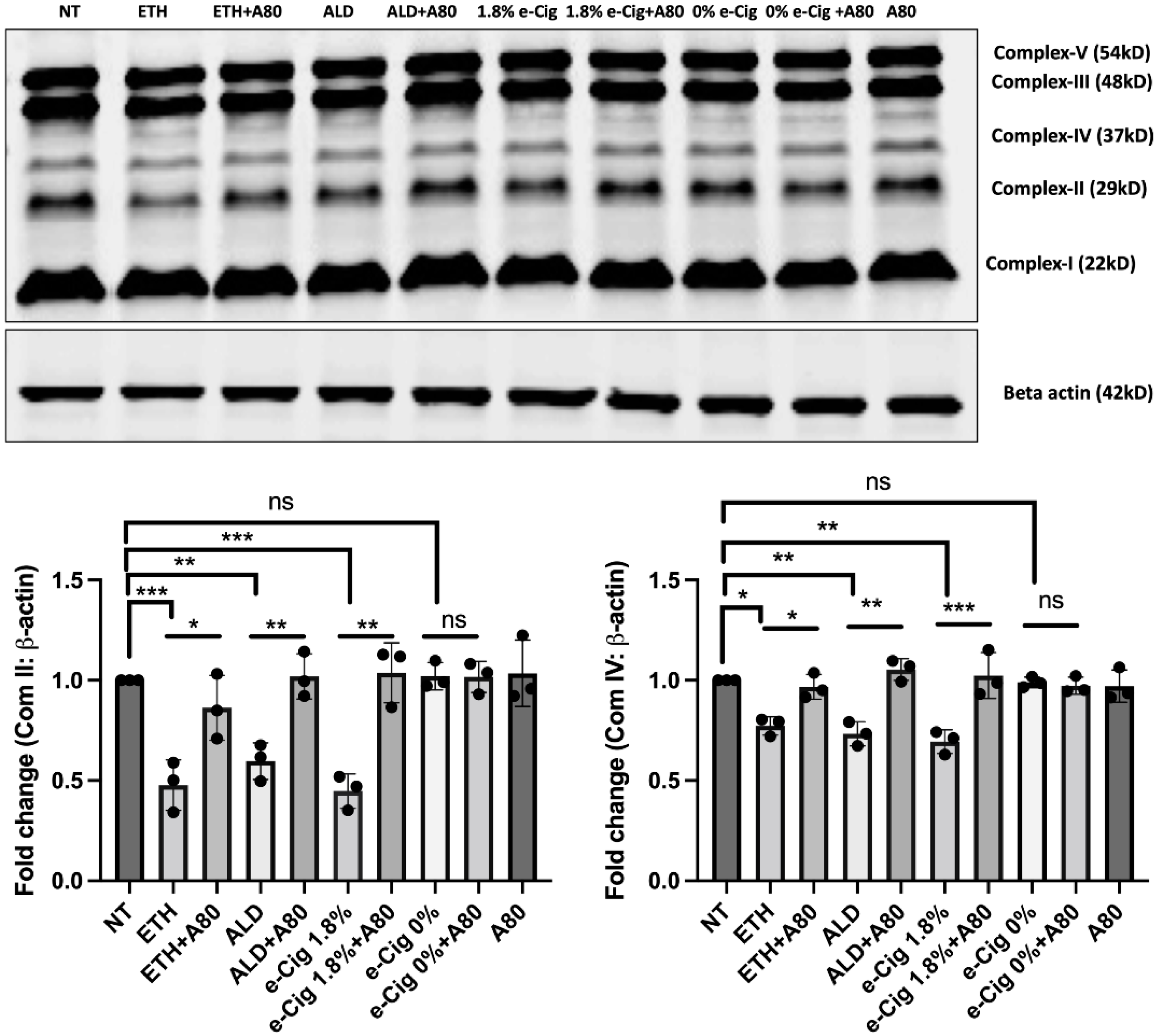
3.2. Reduced Expression of Mitochondrial OXPHOS Markers (Complexes II and IV) after ETH, ALD and 1.8% e-Cig Exposure Were Normalized by P2X7r Inhibition
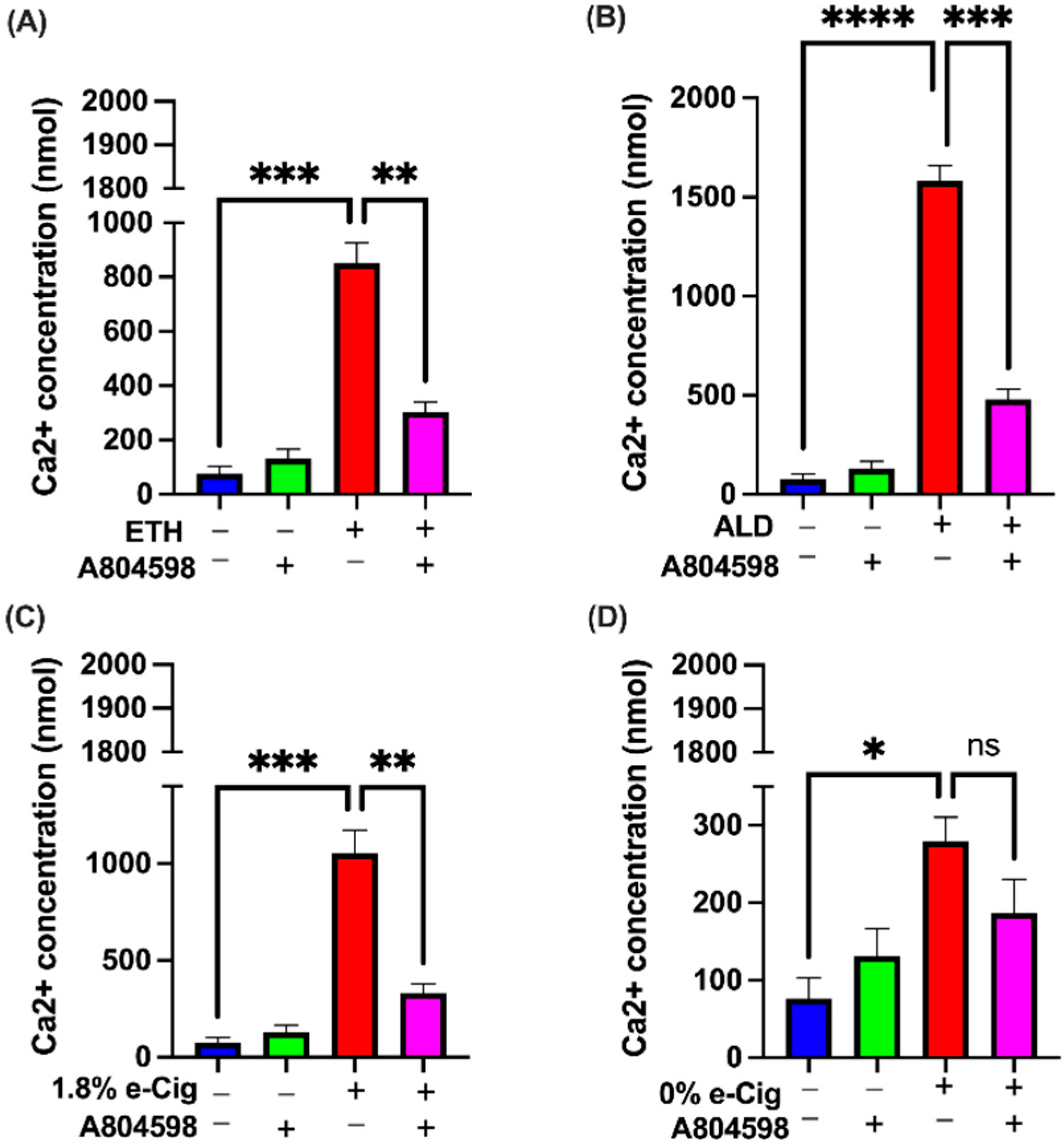
3.3. P2X7r Antagonist Prevented the Intracellular Ca2+ Accumulation Induced by ETH, ALD, and 1.8% e-Cig Exposure
3.4. P2X7r Inhibition Diminished Endoplasmic Reticulum (ER) Stress Caused by ETH, ALD and 1.8% e-Cig in hBMVECs
3.5. P2X7r Inhibition Protected the Mitochondrial Function and Prevented Upregulation of Pro-Apoptotic Factors in Response to ETH, ALD, and 1.8% e-Cig Exposure
3.6. Elevated P2X7r and TRPV1 Gene Expression by ETH, ALD, and 1.8% e-Cig Exposure Was Lowered by P2X7r Antagonist
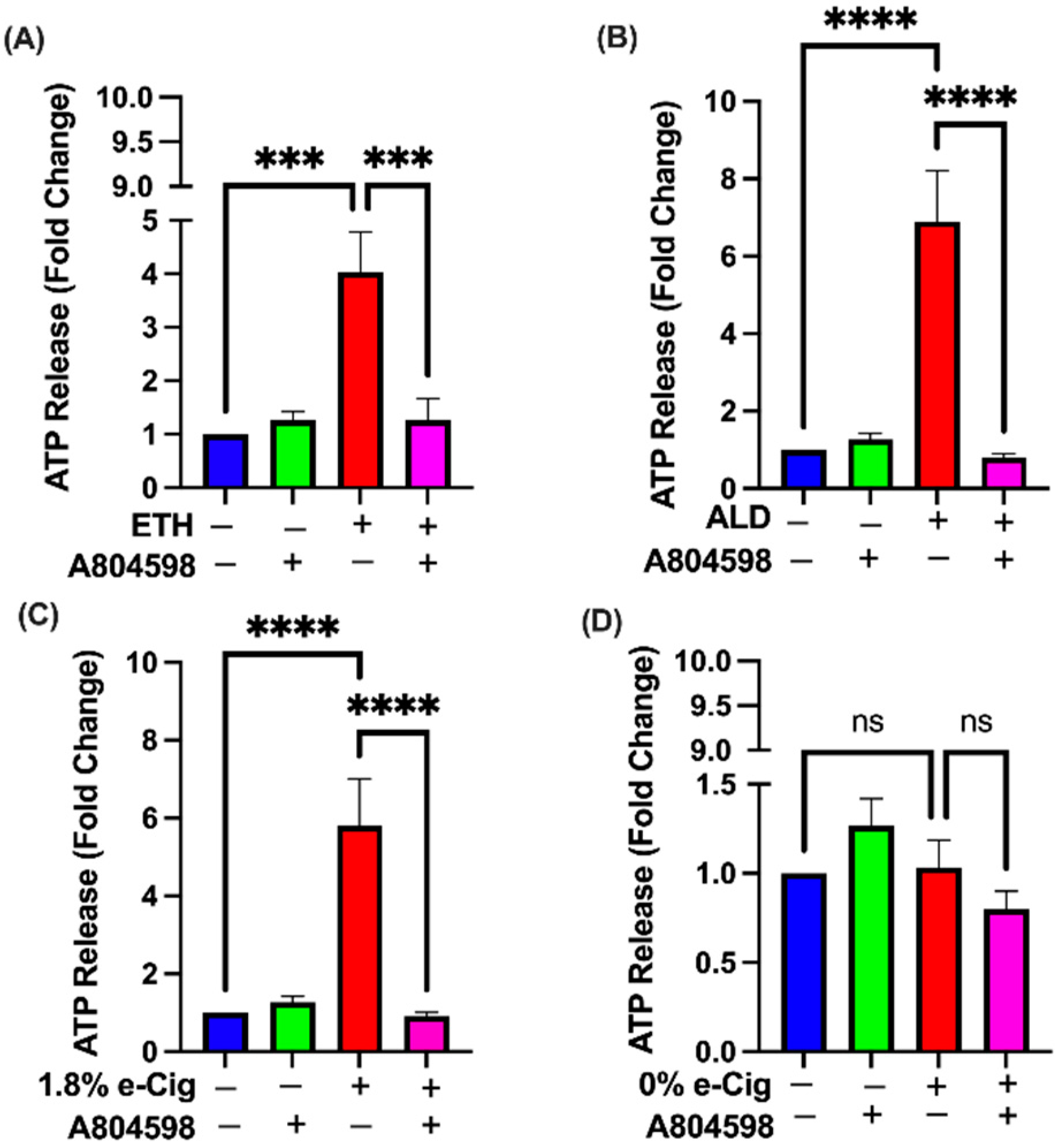
3.7. A804598 Blocks the Extracellular ATP (eATP) Release
3.8. Inhibition of P2X7r Protects the Barrier Tightness against ETH, ALD, and 1.8% e-Cig Exposure in hBMVECs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thrul, J.; Gubner, N.R.; Nguyen, N.; Nguyen, C.; Goodell, E.A.; Holmes, L.M.; Vandrey, R.G.; Ling, P.M. Perceived reward from using cigarettes with alcohol or cannabis and concurrent use: A smartphone-based daily diary study. Addict. Behav. 2021, 114, 106747. [Google Scholar] [CrossRef] [PubMed]
- Okunna, N. A Comparison of Mental and Behavioral Health Risks Factors Associated With Current Dual Use of Electronic Cigarette and Conventional Tobacco Cigarettes With Exclusive Tobacco Cigarette Use and Nonuse Among Adults in the United States. Am. J. Addict. 2021, 30, 138–146. [Google Scholar] [CrossRef]
- Gentzke, A.; Creamer, M.; Cullen, K.A. Vital Signs: Tobacco Products Use among middle and high school students-United States, 2011-2018. Morb. Mortal. Wkly. Rep. 2019, 68, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldt, N.A.; Reichenbach, N.; McGary, H.M.; Persidsky, Y. Effects of Electronic Nicotine Delivery Systems and Cigarettes on Systemic Circulation and Blood-Brain Barrier: Implications for Cognitive Decline. Am. J. Pathol. 2021, 191, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Rump, T.J.; Muneer, P.A.; Szlachetka, A.M.; Lamb, A.; Haorei, C.; Alikunju, S.; Xiong, H.; Keblesh, J.; Liu, J.; Zimmerman, M.C.; et al. Acetyl-l-carnitine protects neuronal function from alcohol-induced oxidative damage in the brain. Free Radic. Biol. Med. 2010, 49, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Heldt, N.A.; Seliga, A.; Winfield, M.; Gajghate, S.; Reichenbach, N.; Yu, X.; Rom, S.; Tenneti, A.; May, D.; Gregory, B.D.; et al. Electronic cigarette exposure disrupts blood-brain barrier integrity and promotes neuroinflammation. Brain Behav. Immun. 2020, 88, 363–380. [Google Scholar] [CrossRef]
- Pimentel, E.; Sivalingam, K.; Doke, M.; Samikkannu, T. Effects of Drugs of Abuse on the Blood-Brain Barrier: A Brief Overview. Front. Neurosci. 2020, 14, 513. [Google Scholar] [CrossRef]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of claudin-5 and occludin by Rho kinase in brain endothelial cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, Y.; Zuo, L.; Hu, W.L.; Jiang, T. Increase of blood-brain barrier leakage is related to cognitive decline in vascular mild cognitive impairment. BMC Neurol. 2021, 21, 159. [Google Scholar] [CrossRef]
- Rao, R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front. Biosci. 2008, 13, 7210–7226. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Moreira, P.I. Oxidative Stress: A Major Player in Cerebrovascular Alterations Associated to Neurodegenerative Events. Front. Physiol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haorah, J.; Ramirez, S.H.; Floreani, N.; Gorantla, S.; Morsey, B.; Persidsky, Y. Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic. Biol. Med. 2008, 45, 1542–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, S.H.; Potula, R.; Fan, S.; Eidem, T.; Papugani, A.; Reichenbach, N.; Dykstra, H.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; et al. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J. Cereb. Blood Flow Metab. 2009, 29, 1933–1945. [Google Scholar] [CrossRef]
- Haorah, J.; Heilman, D.; Knipe, B.; Chrastil, J.; Leibhart, J.; Ghorpade, A.; Miller, D.W.; Persidsky, Y. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol. Clin. Exp. Res. 2005, 29, 999–1009. [Google Scholar] [CrossRef]
- Jin, L.; Lorkiewicz, P.; Malovichko, M.V.; Bhatnagar, A.; Srivastava, S.; Conklin, D.J. Acetaldehyde Induces an Endothelium-Dependent Relaxation of Superior Mesenteric Artery: Potential Role in Postprandial Hyperemia. Front. Physiol. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Fang, P.; Mai, J.T.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Mekala, N.K.; Kurdys, J.; Depuydt, M.M.; Vazquez, E.J.; Rosca, M.G. Apoptosis inducing factor deficiency causes retinal photoreceptor degeneration. The protective role of the redox compound methylene blue. Redox Biol. 2019, 20, 107–117. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Oliveira, C.R. Oxidative Stress and Drugs of Abuse: An Update. Mini-Rev. Org. Chem. 2013, 10, 321–334. [Google Scholar] [CrossRef]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Dosch, M.; Gerber, J.; Jebbawi, F.; Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int. J. Mol. Sci. 2018, 19, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Introduction to Purinergic Signalling in the Brain. Adv. Exp. Med. Biol. 2020, 1202, 1–12. [Google Scholar] [CrossRef]
- Wilkaniec, A.; Cieslik, M.; Murawska, E.; Babiec, L.; Gassowska-Dobrowolska, M.; Palasz, E.; Jesko, H.; Adamczyk, A. P2X7 Receptor is Involved in Mitochondrial Dysfunction Induced by Extracellular Alpha Synuclein in Neuroblastoma SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 3959. [Google Scholar] [CrossRef]
- Sarti, A.C.; Vultaggio-Poma, V.; Falzoni, S.; Missiroli, S.; Giuliani, A.L.; Boldrini, P.; Bonora, M.; Faita, F.; Di Lascio, N.; Kusmic, C.; et al. Mitochondrial P2X7 Receptor Localization Modulates Energy Metabolism Enhancing Physical Performance. Function 2021, 2, zqab005. [Google Scholar] [CrossRef]
- Zhao, H.L.; Zhang, X.; Dai, Z.Q.; Feng, Y.; Li, Q.; Zhang, J.H.; Liu, X.; Chen, Y.J.; Feng, H. P2X7 Receptor Suppression Preserves Blood-Brain Barrier through Inhibiting RhoA Activation after Experimental Intracerebral Hemorrhage in Rats. Sci. Rep. 2016, 6, 23286. [Google Scholar] [CrossRef]
- Kiss, F.; Pohoczky, K.; Szallasi, A.; Helyes, Z. Transient Receptor Potential (TRP) Channels in Head-and-Neck Squamous Cell Carcinomas: Diagnostic, Prognostic, and Therapeutic Potentials. Int. J. Mol. Sci. 2020, 21, 6374. [Google Scholar] [CrossRef]
- Donnelly-Roberts, D.L.; Namovic, M.T.; Surber, B.; Vaidyanathan, S.X.; Perez-Medrano, A.; Wang, Y.; Carroll, W.A.; Jarvis, M.F. [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacology 2009, 56, 223–229. [Google Scholar] [CrossRef]
- Taidi, Z.; Mansfield, K.J.; Sana-Ur-Rehman, H.; Moore, K.H.; Liu, L. Protective Effect of Purinergic P2X7 Receptor Inhibition on Acrolein-Induced Urothelial Cell Damage. Front. Physiol. 2022, 13, 885545. [Google Scholar] [CrossRef]
- Vu, K.; Weksler, B.; Romero, I.; Couraud, P.O.; Gelli, A. Immortalized human brain endothelial cell line HCMEC/D3 as a model of the blood-brain barrier facilitates in vitro studies of central nervous system infection by Cryptococcus neoformans. Eukaryot. Cell 2009, 8, 1803–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haorah, J.; Knipe, B.; Leibhart, J.; Ghorpade, A.; Persidsky, Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J. Leukoc. Biol. 2005, 78, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, D.L.; Zuluaga-Ramirez, V.; Gajghate, S.; Reichenbach, N.L.; Polyak, B.; Persidsky, Y.; Rom, S. miR-98 reduces endothelial dysfunction by protecting blood-brain barrier (BBB) and improves neurological outcomes in mouse ischemia/reperfusion stroke model. J. Cereb. Blood Flow Metab. 2020, 40, 1953–1965. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Fan, S.; Dykstra, H.; Rom, S.; Mercer, A.; Reichenbach, N.L.; Gofman, L.; Persidsky, Y. Inhibition of glycogen synthase kinase 3beta promotes tight junction stability in brain endothelial cells by half-life extension of occludin and claudin-5. PLoS ONE 2013, 8, e55972. [Google Scholar] [CrossRef]
- Kleerekooper, I.; Chua, S.; Foster, P.J.; Trip, S.A.; Plant, G.T.; Petzold, A.; Patel, P.; Consortium, U.B.E.V. Associations of Alcohol Consumption and Smoking With Disease Risk and Neurodegeneration in Individuals With Multiple Sclerosis in the United Kingdom. JAMA Netw. Open 2022, 5, e220902. [Google Scholar] [CrossRef]
- Desler, C.; Hansen, T.L.; Frederiksen, J.B.; Marcker, M.L.; Singh, K.K.; Juel Rasmussen, L. Is There a Link between Mitochondrial Reserve Respiratory Capacity and Aging? J. Aging Res. 2012, 2012, 192503. [Google Scholar] [CrossRef] [Green Version]
- Deniaud, A.; el Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Garcia, J.J.; Martinez-Banaclocha, H.; Angosto-Bazarra, D.; De Torre-Minguela, C.; Baroja-Mazo, A.; Alarcon-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martin-Sanchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 2711. [Google Scholar] [CrossRef] [Green Version]
- Freire, D.; Reyes, R.E.; Baghram, A.; Davies, D.L.; Asatryan, L. P2X7 Receptor Antagonist A804598 Inhibits Inflammation in Brain and Liver in C57BL/6J Mice Exposed to Chronic Ethanol and High Fat Diet. J. Neuroimmune Pharmacol. 2019, 14, 263–277. [Google Scholar] [CrossRef]
- Stolwijk, J.A.; Matrougui, K.; Renken, C.W.; Trebak, M. Impedance analysis of GPCR-mediated changes in endothelial barrier function: Overview and fundamental considerations for stable and reproducible measurements. Pflügers Arch. Eur. J. Physiol. 2015, 467, 2193–2218. [Google Scholar] [CrossRef] [Green Version]
- Sadikot, R.T.; Bedi, B.; Li, J.; Yeligar, S.M. Alcohol-induced mitochondrial DNA damage promotes injurious crosstalk between alveolar epithelial cells and alveolar macrophages. Alcohol 2019, 80, 65–72. [Google Scholar] [CrossRef]
- Song, B.J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, E.K.; Grantham, E.K.; Warden, A.S.; Harris, R.A. Neuroimmune signaling in alcohol use disorder. Pharmacol. Biochem. Behav. 2019, 177, 34–60. [Google Scholar] [CrossRef]
- Moyzis, A.; Gustafsson, A.B. Multiple recycling routes: Canonical vs. non-canonical mitophagy in the heart. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 797–809. [Google Scholar] [CrossRef]
- Nowak, A.J.; Relja, B. The Impact of Acute or Chronic Alcohol Intake on the NF-kappaB Signaling Pathway in Alcohol-Related Liver Disease. Int. J. Mol. Sci. 2020, 21, 9407. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: A microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Haorah, J.; Ramirez, S.H.; Schall, K.; Smith, D.; Pandya, R.; Persidsky, Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J. Neurochem. 2007, 101, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Le Dare, B.; Victoni, T.; Bodin, A.; Vlach, M.; Vene, E.; Loyer, P.; Lagente, V.; Gicquel, T. Ethanol upregulates the P2X7 purinergic receptor in human macrophages. Fundam. Clin. Pharmacol. 2019, 33, 63–74. [Google Scholar] [CrossRef]
- Le Dare, B.; Ferron, P.J.; Gicquel, T. The Purinergic P2X7 Receptor-NLRP3 Inflammasome Pathway: A New Target in Alcoholic Liver Disease? Int. J. Mol. Sci. 2021, 22, 2139. [Google Scholar] [CrossRef]
- Kass, G.E.; Orrenius, S. Calcium signaling and cytotoxicity. Environ. Health Perspect. 1999, 107 (Suppl. S1), 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Kadio, B.; Yaya, S.; Basak, A.; Dje, K.; Gomes, J.; Mesenge, C. Calcium role in human carcinogenesis: A comprehensive analysis and critical review of literature. Cancer Metastasis Rev. 2016, 35, 391–411. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic reticulum Ca(2+) handling in excitable cells in health and disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Noh, K.T.; Park, Y.M.; Cho, S.G.; Choi, E.J. GSK-3beta-induced ASK1 stabilization is crucial in LPS-induced endotoxin shock. Exp. Cell Res. 2011, 317, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. p38 Mitogen-activated protein kinase regulates Bax translocation in cyanide-induced apoptosis. Toxicol. Sci. 2003, 75, 99–107. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Nussinov, R.; Ma, B. Release of Cytochrome C from Bax Pores at the Mitochondrial Membrane. Sci. Rep. 2017, 7, 2635. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Balaban, R.S. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 2009, 1787, 1334–1341. [Google Scholar] [CrossRef] [Green Version]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T.; Moncada, S. Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 643–647. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira-Giacomelli, A.; Petiz, L.L.; Andrejew, R.; Turrini, N.; Silva, J.B.; Sack, U.; Ulrich, H. Role of P2X7 Receptors in Immune Responses During Neurodegeneration. Front. Cell. Neurosci. 2021, 15, 662935. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekala, N.; Gheewala, N.; Rom, S.; Sriram, U.; Persidsky, Y. Blocking of P2X7r Reduces Mitochondrial Stress Induced by Alcohol and Electronic Cigarette Exposure in Brain Microvascular Endothelial Cells. Antioxidants 2022, 11, 1328. https://doi.org/10.3390/antiox11071328
Mekala N, Gheewala N, Rom S, Sriram U, Persidsky Y. Blocking of P2X7r Reduces Mitochondrial Stress Induced by Alcohol and Electronic Cigarette Exposure in Brain Microvascular Endothelial Cells. Antioxidants. 2022; 11(7):1328. https://doi.org/10.3390/antiox11071328
Chicago/Turabian StyleMekala, Naveen, Nishi Gheewala, Slava Rom, Uma Sriram, and Yuri Persidsky. 2022. "Blocking of P2X7r Reduces Mitochondrial Stress Induced by Alcohol and Electronic Cigarette Exposure in Brain Microvascular Endothelial Cells" Antioxidants 11, no. 7: 1328. https://doi.org/10.3390/antiox11071328