
Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Obesity, Lack of Physical Activity, Poor Diet, and Insulin Resistance Are Major Risk Factors for Pediatric T2D
1.2. The Role of Postprandial Oxidative Stress (POS) in T2D
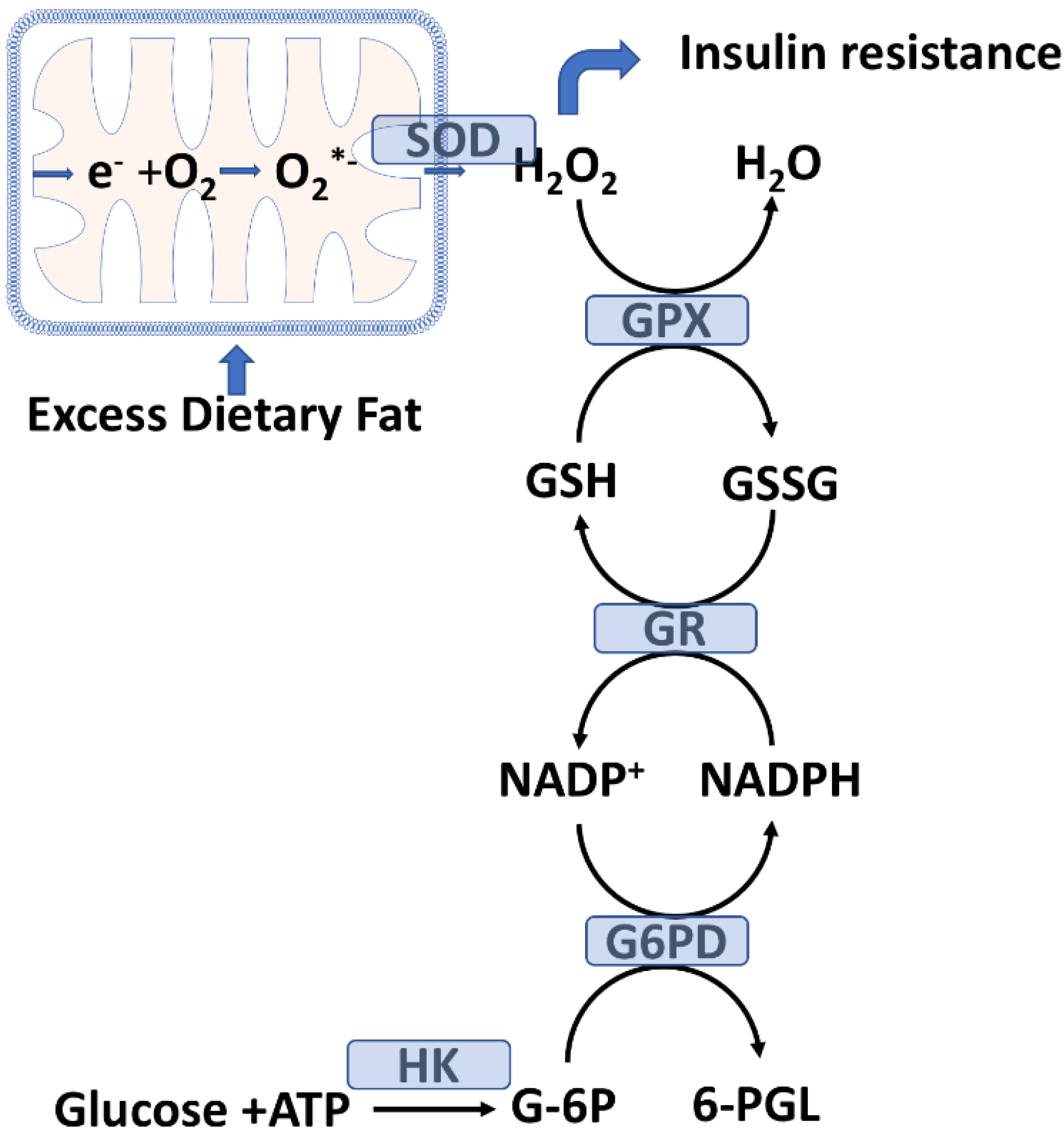
1.3. Dietary Fat Level Is a Key Determinant of Plasma-POS
1.4. Dietary Fat Type and Dietary Antioxidants Influence Plasma-POS
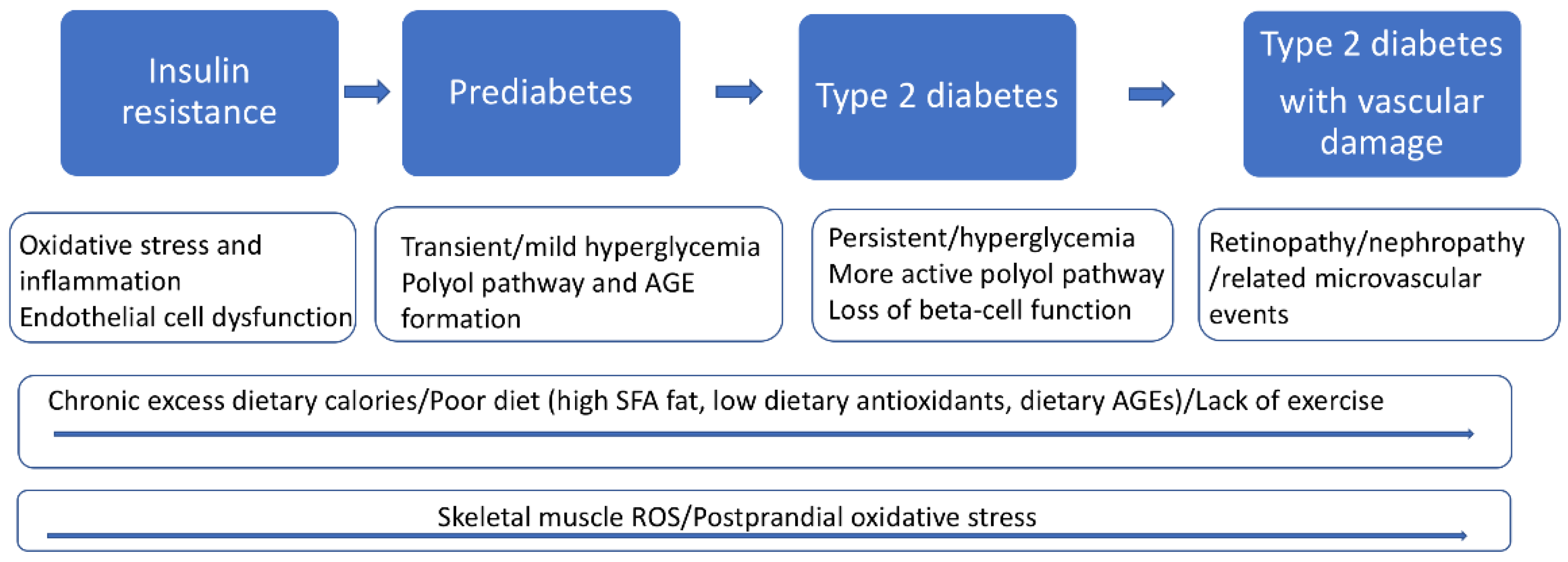
2. The Pathophysiology of T2D
2.1. Skeletal Muscle Insulin Resistance Is a Primary Defect in T2D
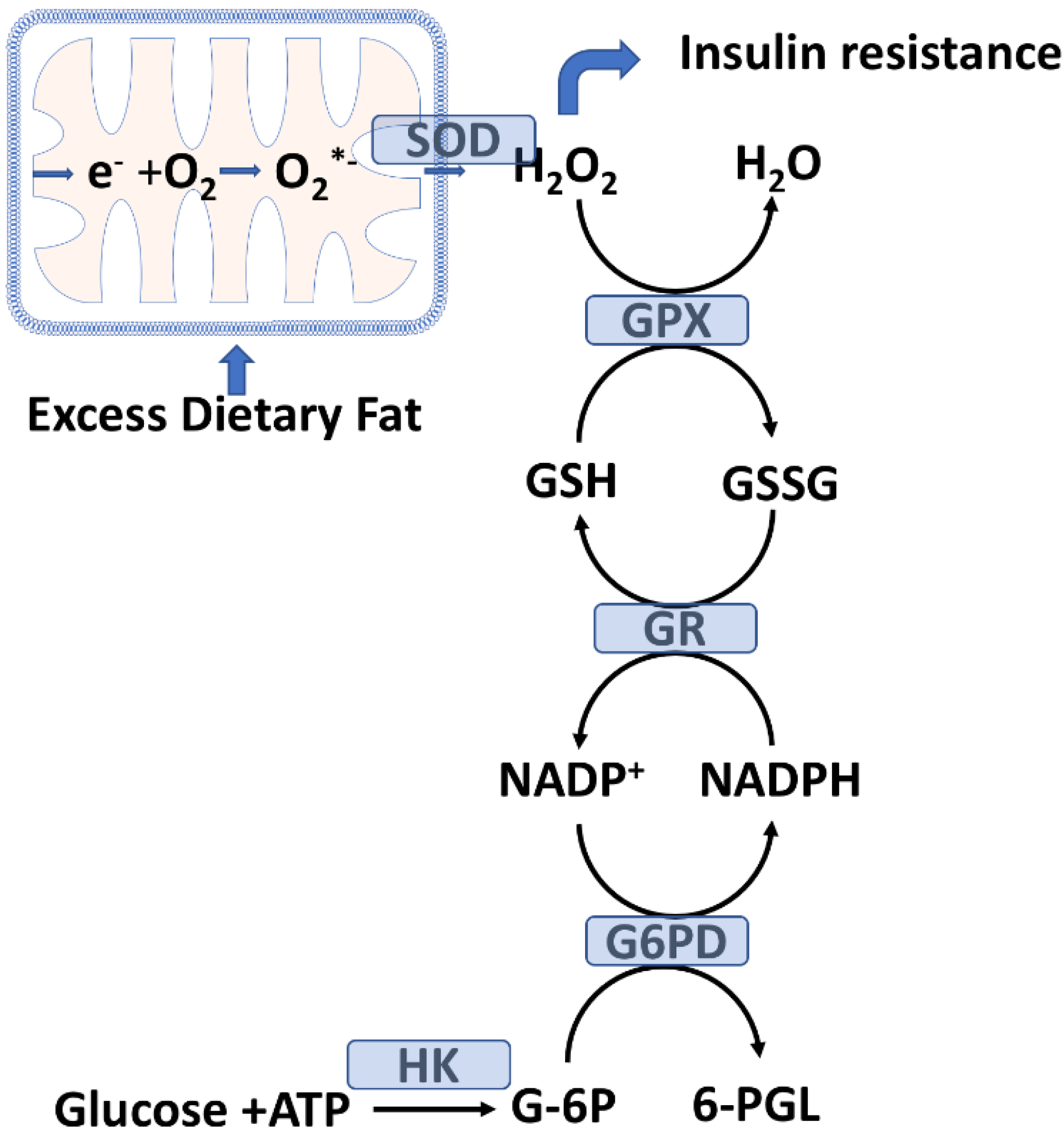
2.2. Skeletal Muscle Glutathione Peroxidase System and Oxidative Stress
2.3. Mitochondrial H2O2 Emission as a Potential Initiating Event for Insulin Resistance
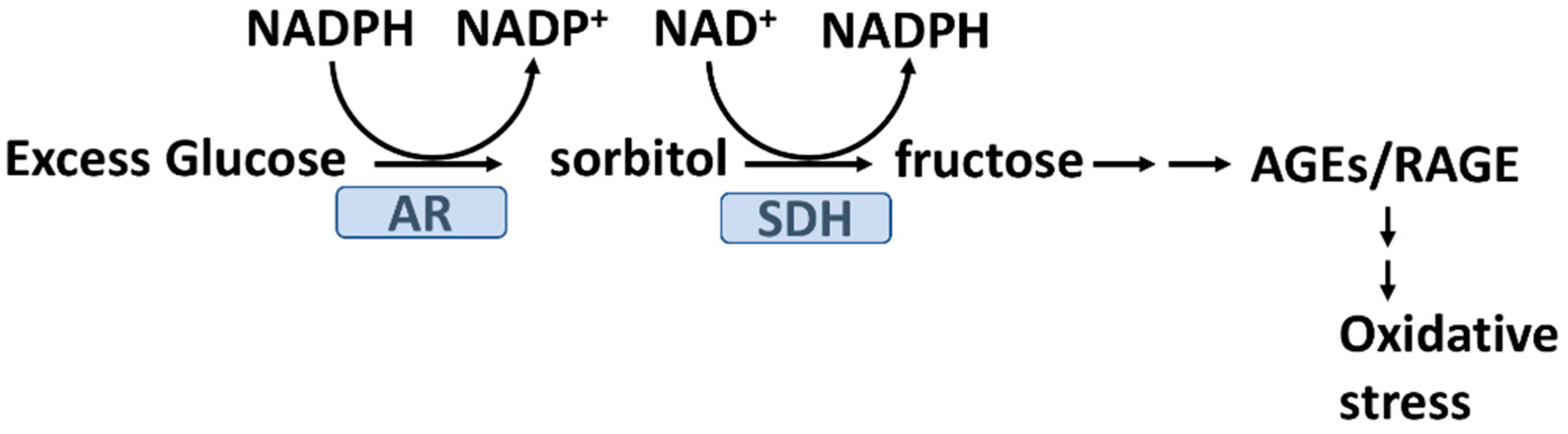
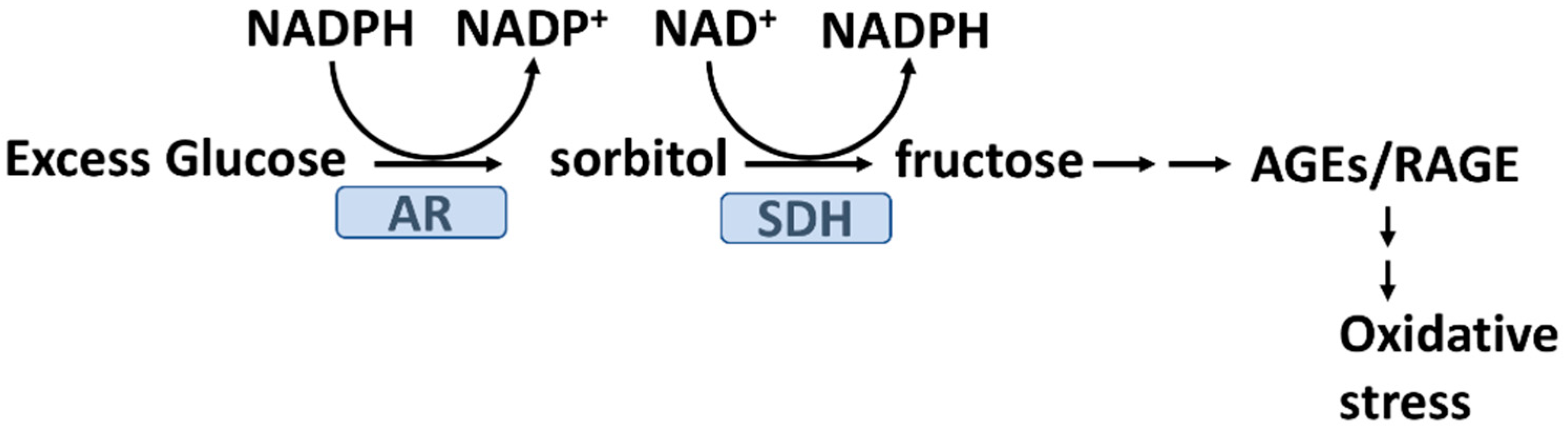
2.4. Hyperglycemia and Glycation Induced Oxidative Stress
2.5. Advanced Glycation End Products (AGEs) and Oxidative Stress in T2D
2.6. AGEs, Skeletal Muscle GLUT4 Expression, and Insulin Resistance
2.7. Vascular Endothelial Cells, Oxidative Stress, and T2D
3. System Medicine and a Future Redoxomics Approach to Pediatric T2D
3.1. Genomics and T2D
3.1.1. Genomics and Stratification of Adult T2D Patients
3.1.2. Genomics May Help Stratify Youth Onset T2D Patients
3.1.3. Genomics and Oxidative Stress
3.2. Metabolomics, Oxidative Stress, and T2D
3.3. Proteomics and T2D
3.3.1. Protein Glycation and Oxidation in T2D
3.3.2. The Proximity Ligation Assay, T2D, and Metformin Response Stratification
4. System Medicine, Nutrigenomics and T2D
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, W.; Dall, T. Economic costs of diabetes in the U.S. in 2017. Diabetes Care 2018, 41, 917–928. [Google Scholar] [CrossRef] [Green Version]
- CDC. Type 2 Diabetes. Available online: https://www.cdc.gov/diabetes/basics/type2.html (accessed on 9 February 2022).
- Mayer-Davis, E.J.; Kahkoska, A.R.; Jefferies, C.; Dabelea, D.; Balde, N.; Gong, C.X.; Aschner, P.; Craig, M.E. ISPAD Clinical Practice Consensus Guidelines 2018: Definition, Epidemiology, and Classification of Diabetes in Children and Adolescents. Pediatric Diabetes 2018, 19 (Suppl. 27), 7–19. [Google Scholar] [CrossRef] [PubMed]
- Pulgaron, E.R.; Delamater, A.M. Obesity and Type 2 Diabetes in Children: Epidemiology and Treatment. Curr. Diabetes Rep. 2014, 14, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andes, L.J.; Cheng, Y.J.; Rolka, D.B.; Gregg, E.W.; Imperatore, G. Prevalence of Prediabetes Among Adolescents and Young Adults in the United States, 2005–2016. JAMA Pediatric 2020, 174, e194498. [Google Scholar] [CrossRef] [PubMed]
- 1 in 5 Adolescents and 1 in 4 Young Adults Now Living with Prediabete. Available online: https://www.cdc.gov/media/releases/2019/p1202-diabetes.html (accessed on 2 May 2022).
- Imperatore, G.; Boyle, J.P.; Thompson, T.J.; Case, D.; Dabelea, D.; Hamman, R.F.; Lawrence, J.M.; Liese, A.D.; Liu, L.L.; Mayer-Davis, E.J.; et al. Projections of type 1 and type 2 diabetes burden in the U.S. population aged <20 years through 2050: Dynamic modeling of incidence, mortality, and population growth. Diabetes Care 2012, 35, 2515–2520. [Google Scholar] [CrossRef] [Green Version]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm—2018 executive summary. Endocr. Pract. 2018, 24, 91–120. [Google Scholar] [CrossRef] [Green Version]
- Mechanick, J.I.; Garber, A.J.; Grunberger, G.; Handelsman, Y.; Garvey, W.T. Dysglycemia-based chronic disease: An american association of clinical endocrinologists position statement. Endocr. Pract. 2018, 24, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53 (Suppl. 1), S110–S118. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, S.; Cocchia, N.; Landolfi, F.; Ciani, F.; Iorio, E.L. Redoxomics and Oxidative Stress: From the Basic Research to the Clinical Practice. Free Rad. Dis. 2016, 149–169. [Google Scholar] [CrossRef] [Green Version]
- Dansinger, M. Type 2 Diabetes in Children. 2021. Available online: www.webmd.com/diabetes/type-2-diabetes-in-children (accessed on 2 May 2022).
- Frohnert, B.I.; Jacobs, D.R.; Steinberger, J.; Moran, A.; Steffen, L.M.; Sinaiko, A.R. Relation between serum free fatty acids and adiposity, insulin resistance, and cardiovascular risk factors from adolescence to adulthood. Diabetes 2013, 62, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int. J. Clin. Pract. 2006, 60, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottero, B.; Gargiulo, S.; Russo, I.; Barale, C.; Poli, G.; Cavalot, F. Postprandial Dysmetabolism and Oxidative Stress in Type 2 Diabetes: Pathogenetic Mechanisms and Therapeutic Strategies. Med. Res. Rev. 2015, 35, 968–1031. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Calvagno, S.; Mauceri, B.; Misseri, M.; Tsami, A.; Vecchio, C.; Mastrosimone, G.; Di Pino, A.; Maiorca, D.; Judica, A.; et al. Effects of antioxidants on postprandial oxidative stress and endothelial dysfunction in subjects with impaired glucose tolerance and type 2 diabetes. Eur. J. Nutr. 2010, 49, 409–416. [Google Scholar] [CrossRef]
- Ottum, M.S.; Mistry, A.M. Advanced glycation end-products: Modifiable environmental factors profoundly mediate insulin resistance. J. Clin. Biochem. Nutr. 2015, 57, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Götting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [Green Version]
- Bloomer, R.J.; Kabir, M.M.; Marshall, K.E.; Canale, R.E.; Farney, T.M. Postprandial oxidative stress in response to dextrose and lipid meals of differing size. Lipids Health Dis. 2010, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Perez-Martinez, P.; Garcia-Quintana, J.M.; Yubero-Serrano, E.M.; Tasset-Cuevas, I.; Tunez, I.; Garcia-Rios, A.; Delgado-Lista, J.; Marin, C.; Perez-Jimenez, F.; Roche, H.M.; et al. Postprandial oxidative stress is modified by dietary fat: Evidence from a human intervention study. Clin. Sci. 2010, 119, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Yubero-Serrano, E.M.; Garcia-Rios, A.; Delgado-Lista, J.; Delgado-Casado, N.; Perez-Martinez, P.; Rodriguez-Cantalejo, F.; Fuentes, F.; Cruz-Teno, C.; Tunez, I.; Tasset-Cuevas, I.; et al. Postprandial effects of the Mediterranean diet on oxidant and antioxidant status in elderly men and women. J. Am. Geriatr. Soc. 2011, 59, 938–940. [Google Scholar] [CrossRef]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and nutritional approaches for prevention and management of type 2 diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef] [Green Version]
- Georgoulis, M.; Kontogianni, M.D.; Yiannakouris, N. Mediterranean Diet and Diabetes: Prevention and Treatment. Nutrients 2014, 6, 1406–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billingsley, H.E.; Carbone, S. The antioxidant potential of the Mediterranean diet in patients at high cardiovascular risk: An in-depth review of the PREDIMED. Nutr. Diabetes 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Velázquez-López, L.; Santiago-Díaz, G.; Nava-Hernández, J.; Muñoz-Torres, A.V.; Medina-Bravo, P.; Torres-Tamayo, M. Mediterranean-style diet reduces metabolic syndrome components in obese children and adolescents with obesity. BMC Pediatrics 2014, 14, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, W.L.; Pham, T.; Mohiuddin, S.S. Biochemistry, Antioxidants. In StatPearls; 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK541064/ (accessed on 2 May 2022).
- Rajan, A.S. Regulation of Glucose Uptake. Available online: https://www.medscape.org/viewarticle/438374 (accessed on 2 May 2022).
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Barrett, E.J.; Liu, Z. The endothelial cell: An “early responder” in the development of insulin resistance. Rev. Endocr. Metab. Disord. 2013, 14, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaraldi, T.P.; Mudaliar, S.; Barzin, A.; Macievic, J.A.; Edelman, S.V.; Park, K.S.; Henry, R.R. Skeletal muscle GLUT1 transporter protein expression and basal leg glucose uptake are reduced in type 2 diabetes. J. Clin. Endocrinol. Metab. 2005, 90, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [Green Version]
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. 2), S135–S148. [Google Scholar] [CrossRef] [Green Version]
- Hudish, L.I.; Reusch, J.E.; Sussel, L. β Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J. Clin. Investig. 2019, 129, 4001–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- İhsan, B.; Türkan, Y.; Handan, A. Is Oxidative Stress a Consequence of Hyperglycemia? Or is Hyperglycemia the Consequence of Oxidative Stress? Or are Both Caused by Insulin Resistance? Int. Arch. Endocrinol. Clin. Res. 2021, 7, 023. [Google Scholar] [CrossRef]
- Kyselová, P.; Zourek, M.; Rusavý, Z.; Trefil, L.; Racek, J. Hyperinsulinemia and oxidative stress. Physiol. Res. 2002, 51, 591–595. [Google Scholar] [PubMed]
- Cline, G.W.; Petersen, K.F.; Krssak, M.; Shen, J.; Hundal, R.S.; Trajanoski, Z.; Inzucchi, S.; Dresner, A.; Rothman, D.L.; Shulman, G.I. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N. Engl. J. Med. 1999, 341, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Rothman, D.L.; Shulman, R.G.; Shulman, G.I. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 89, 1069–1075. [Google Scholar] [CrossRef]
- Yang, H.C.; Wu, Y.H.; Yen, W.C.; Liu, H.Y.; Hwang, T.L.; Stern, A.; Chiu, D.T. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef] [Green Version]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Mahmoud, A.A.; Nor El-Din, A.K. Glucose-6-Phosphate Dehydrogenase Activity and Protein Oxidative Modification in Patients with Type 2 Diabetes Mellitus. J. Biomark. 2013, 2013, 430813. [Google Scholar] [CrossRef] [Green Version]
- Heymann, A.D.; Cohen, Y.; Chodick, G. Glucose-6-phosphate dehydrogenase deficiency and type 2 diabetes. Diabetes Care 2012, 35, e58. [Google Scholar] [CrossRef] [Green Version]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.; Dratz, E. Selenium and non-selenium glutathione peroxidase activities in selected ocular and non-ocular rat tissues. Exp. Eye Res. 1982, 35, 405–412. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.S.; Kim, M.; Youn, B.S.; Lee, N.S.; Park, J.W.; Lee, I.K.; Lee, Y.S.; Kim, J.B.; Cho, Y.M.; Lee, H.K.; et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol. Cell. Biol. 2009, 29, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, V.M.; Rocha, M.; Herance, R.; Hernandez-Mijares, A. Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr. Pharm. Des. 2011, 17, 3947–3958. [Google Scholar] [CrossRef]
- Tagi, V.M.; Giannini, C.; Chiarelli, F. Insulin Resistance in Children. Front. Endocrinol. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef]
- Bai, J.; Cederbaum, A.I. Mitochondrial catalase and oxidative injury. Biol. Signals Recept. 2001, 10, 189–199. [Google Scholar] [CrossRef]
- Quindry, J.; Stone, W.; King, J.; Broeder, C. The effects of acute exercise on neutrophils and plasma oxidative stress. Med. Sci. Sports Exerc. 2003, 35, 1139–1145. [Google Scholar] [CrossRef]
- Zhang, Q.; Monroe, M.E.; Schepmoes, A.A.; Clauss, T.R.; Gritsenko, M.A.; Meng, D.; Petyuk, V.A.; Smith, R.D.; Metz, T.O. Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J. Proteome Res. 2011, 10, 3076–3088. [Google Scholar] [CrossRef] [Green Version]
- Pu, L.J.; Shen, Y.; Lu, L.; Zhang, R.Y.; Zhang, Q.; Shen, W.F. Increased blood glycohemoglobin A1c levels lead to overestimation of arterial oxygen saturation by pulse oximetry in patients with type 2 diabetes. Cardiovasc. Diabetol. 2012, 11, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indyk, D.; Bronowicka-Szydełko, A.; Gamian, A.; Kuzan, A. Advanced glycation end products and their receptors in serum of patients with type 2 diabetes. Sci. Rep. 2021, 11, 13264. [Google Scholar] [CrossRef] [PubMed]
- Goyal, R.; Singhai, M.; Faizy, A.F. Glutathione peroxidase activity in obese and nonobese diabetic patients and role of hyperglycemia in oxidative stress. J. Midlife Health 2011, 2, 72–76. [Google Scholar] [CrossRef] [PubMed]
- González de Vega, R.; Fernández-Sánchez, M.L.; Fernández, J.C.; Álvarez Menéndez, F.V.; Sanz-Medel, A. Selenium levels and Glutathione peroxidase activity in the plasma of patients with type II diabetes mellitus. J. Trace Elem. Med. Biol. 2016, 37, 44–49. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Salazar, J.; Navarro, C.; Ortega, Á.; Nava, M.; Morillo, D.; Torres, W.; Hernández, M.; Cabrera, M.; Angarita, L.; Ortiz, R.; et al. Advanced Glycation End Products: New Clinical and Molecular Perspectives. Int. J. Environ. Res. Public Health 2021, 18, 7236. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma Proteins Modified by Advanced Glycation End Products (AGEs) Reveal Site-specific Susceptibilities to Glycemic Control in Patients with Type 2 Diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef] [Green Version]
- Rammos, G.; Peppes, V.; Zakopoulos, N. Transient insulin resistance in normal subjects: Acute hyperglycemia inhibits endothelial-dependent vasodilatation in normal subjects. Metab. Syndr. Relat. Disord. 2008, 6, 159–170. [Google Scholar] [CrossRef]
- Kida, T.; Oku, H.; Osuka, S.; Horie, T.; Ikeda, T. Hyperglycemia-induced VEGF and ROS production in retinal cells is inhibited by the mTOR inhibitor, rapamycin. Sci. Rep. 2021, 11, 1885. [Google Scholar] [CrossRef]
- Bakker, W.; Eringa, E.C.; Sipkema, P.; van Hinsbergh, V.W. Endothelial dysfunction and diabetes: Roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 2009, 335, 165–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweet, I.R.; Gilbert, M.; Maloney, E.; Hockenbery, D.M.; Schwartz, M.W.; Kim, F. Endothelial inflammation induced by excess glucose is associated with cytosolic glucose 6-phosphate but not increased mitochondrial respiration. Diabetologia 2009, 52, 921–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gero, D. Hyperglycemia-Induced Endothelial Dysfunction. In Endothelial Dysfunction—Old Concepts and New Challenges; IntechOpen: London, UK, 2007; pp. 179–206. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model. Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Brand, I. Kinetics of glutathione peroxidase. Biochim. Biophys. Acta 1969, 191, 541–549. [Google Scholar] [CrossRef]
- Aragno, M.; Mastrocola, R. Dietary Sugars and Endogenous Formation of Advanced Glycation Endproducts: Emerging Mechanisms of Disease. Nutrients 2017, 9, 385. [Google Scholar] [CrossRef] [Green Version]
- Levi, B.; Werman, M.J. Long-term fructose consumption accelerates glycation and several age-related variables in male rats. J. Nutr. 1998, 128, 1442–1449. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Mapstone, M.; Connors, E.; Jacobson, M.; Monuki, E.S.; Malik, S.; Macciardi, F.; Federoff, H.J. Systems healthcare: A holistic paradigm for tomorrow. BMC Syst. Biol. 2017, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Kussmann, M.; Morine, M.J.; Hager, J.; Sonderegger, B.; Kaput, J. Perspective: A systems approach to diabetes research. Front. Genet. 2013, 4, 205. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, M.; Nestor, C.E.; Zhang, H.; Barabási, A.L.; Baranzini, S.; Brunak, S.; Chung, K.F.; Federoff, H.J.; Gavin, A.C.; Meehan, R.R.; et al. Modules, networks and systems medicine for understanding disease and aiding diagnosis. Genome Med. 2014, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Mardinoglu, A.; Nielsen, J. Systems medicine and metabolic modelling. J. Intern. Med. 2012, 271, 142–154. [Google Scholar] [CrossRef]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The Need for Multi-Omics Biomarker Signatures in Precision Medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Beissbarth, T.; Berthold, M.R.; Blüthgen, N.; Burmeister, Y.; Dammann, O.; Deutsch, A.; Feuerhake, F.; Franke, A.; Hasenauer, J.; et al. Whither systems medicine? Exp. Mol. Med. 2018, 50, e453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, M.; Glusman, G.; Brogaard, K.; Price, N.D.; Hood, L. P4 medicine: How systems medicine will transform the healthcare sector and society. Pers. Med. 2013, 10, 565–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, L.; Flores, M. A personal view on systems medicine and the emergence of proactive P4 medicine: Predictive, preventive, personalized and participatory. New Biotechnol. 2012, 29, 613–624. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Qin, S.; Jin, X.; Jin, L.; Gu, W.; Mu, Y. Insights Into Genome-Wide Association Study for Diabetes: A Bibliometric and Visual Analysis From 2001 to 2021. Front. Endocrinol. 2022, 13, 817620. [Google Scholar] [CrossRef]
- Polfus, L.M.; Darst, B.F.; Highland, H.; Sheng, X.; Ng, M.C.Y.; Below, J.E.; Petty, L.; Bien, S.; Sim, X.; Wang, W.; et al. Genetic discovery and risk characterization in type 2 diabetes across diverse populations. HGG Adv. 2021, 2, 100029. [Google Scholar] [CrossRef]
- Xue, A.; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; McRae, A.F.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 2941. [Google Scholar] [CrossRef] [Green Version]
- Udler, M.S.; McCarthy, M.I.; Florez, J.C.; Mahajan, A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr. Rev. 2019, 40, 1500–1520. [Google Scholar] [CrossRef] [Green Version]
- Ahlqvist, E.; Storm, P.; Käräjämäki, A.; Martinell, M.; Dorkhan, M.; Carlsson, A.; Vikman, P.; Prasad, R.B.; Aly, D.M.; Almgren, P.; et al. Novel subgroups of adult-onset diabetes and their association with outcomes: A data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018, 6, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Nadeau, K.J.; Anderson, B.J.; Berg, E.G.; Chiang, J.L.; Chou, H.; Copeland, K.C.; Hannon, T.S.; Huang, T.T.; Lynch, J.L.; Powell, J.; et al. Youth-Onset Type 2 Diabetes Consensus Report: Current Status, Challenges, and Priorities. Diabetes Care 2016, 39, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.L.; Khan, G.F.; Magwire, M.M.; Tabor, C.L.; Mackay, T.F.; Anholt, R.R. Genome-wide association analysis of oxidative stress resistance in Drosophila melanogaster. PLoS ONE 2012, 7, e34745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, M.; Wang, H.; Xu, S.W.; Yang, L.H.; Chen, W.; Zhao, S.X.; Shen, H.; Liu, Q.; Yang, R.M.; Wang, J. Variants in oxidative stress-related genes affect the chemosensitivity through Nrf2-mediated signaling pathway in biliary tract cancer. EBioMedicine 2019, 48, 143–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabatabaei-Malazy, O.; Khodaeian, M.; Bitarafan, F.; Larijani, B.; Amoli, M.M. Polymorphisms of Antioxidant Genes as a Target for Diabetes Management. Int. J. Mol. Cell Med. 2017, 6, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.W.; Lin, T.K.; Weng, S.W.; Liou, C.W. Mitochondrial DNA variants in the pathogenesis of type 2 diabetes—Relevance of asian population studies. Rev. Diabetes Stud. 2009, 6, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silzer, T.; Barber, R.; Sun, J.; Pathak, G.; Johnson, L.; O’Bryant, S.; Phillips, N. Circulating mitochondrial DNA: New indices of type 2 diabetes-related cognitive impairment in Mexican Americans. PLoS ONE 2019, 14, e0213527. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Litt, L.; Segal, M.R.; Kelly, M.J.; Pelton, J.G.; Kim, M. Metabolomics of oxidative stress in recent studies of endogenous and exogenously administered intermediate metabolites. Int. J. Mol. Sci. 2011, 12, 6469–6501. [Google Scholar] [CrossRef] [Green Version]
- Bloomgarden, Z. Diabetes and branched-chain amino acids: What is the link? J. Diabetes 2018, 10, 350–352. [Google Scholar] [CrossRef] [Green Version]
- Zhenyukh, O.; González-Amor, M.; Rodrigues-Diez, R.R.; Esteban, V.; Ruiz-Ortega, M.; Salaices, M.; Mas, S.; Briones, A.M.; Egido, J. Branched-chain amino acids promote endothelial dysfunction through increased reactive oxygen species generation and inflammation. J. Cell. Mol. Med. 2018, 22, 4948–4962. [Google Scholar] [CrossRef]
- Wang, Q.; Holmes, M.V.; Davey Smith, G.; Ala-Korpela, M. Genetic Support for a Causal Role of Insulin Resistance on Circulating Branched-Chain Amino Acids and Inflammation. Diabetes Care 2017, 40, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.J.; Larson, M.G.; Vasan, R.S.; Cheng, S.; Rhee, E.P.; McCabe, E.; Lewis, G.D.; Fox, C.S.; Jacques, P.F.; Fernandez, C.; et al. Metabolite profiles and the risk of developing diabetes. Nat. Med. 2011, 17, 448–453. [Google Scholar] [CrossRef]
- Lauber, C.; Gerl, M.J.; Klose, C.; Ottosson, F.; Melander, O.; Simons, K. Lipidomic risk scores are independent of polygenic risk scores and can predict incidence of diabetes and cardiovascular disease in a large population cohort. PLoS Biol. 2022, 20, e3001561. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Z.; Gerszten, R.E. Metabolomics and Proteomics in Type 2 Diabetes. Circ. Res. 2020, 126, 1613–1627. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Wagner, A.H. Role of protein carbonylation in diabetes. J. Inherit. Metab. Dis. 2018, 41, 29–38. [Google Scholar] [CrossRef]
- Bollineni, R.C.; Fedorova, M.; Blüher, M.; Hoffmann, R. Carbonylated plasma proteins as potential biomarkers of obesity induced type 2 diabetes mellitus. J. Proteome Res. 2014, 13, 5081–5093. [Google Scholar] [CrossRef]
- Fu, J.; Luo, Y.; Mou, M.; Zhang, H.; Tang, J.; Wang, Y.; Zhu, F. Advances in Current Diabetes Proteomics: From the Perspectives of Label- free Quantification and Biomarker Selection. Curr. Drug Targets 2020, 21, 34–54. [Google Scholar] [CrossRef]
- Zhong, W.; Edfors, F.; Gummesson, A.; Bergström, G.; Fagerberg, L.; Uhlén, M. Next generation plasma proteome profiling to monitor health and disease. Nat. Commun. 2021, 12, 2493. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, C.; Ruff, D.; Kirvell, S.; Johnson, G.; Dhillon, H.S.; Bustin, S.A. Proximity assays for sensitive quantification of proteins. Biomol. Detect. Quantif. 2015, 4, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Weibrecht, I.; Leuchowius, K.J.; Clausson, C.M.; Conze, T.; Jarvius, M.; Howell, W.M.; Kamali-Moghaddam, M.; Söderberg, O. Proximity ligation assays: A recent addition to the proteomics toolbox. Expert Rev. Proteom. 2010, 7, 401–409. [Google Scholar] [CrossRef]
- Consortium, R. Impact of Insulin and Metformin Versus Metformin Alone on β-Cell Function in Youth With Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes. Diabetes Care 2018, 41, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. Supporting Healthier School Meals. Available online: www.diabetes.org/blog/supporting-healthier-school-meals (accessed on 20 April 2022).
- Glycemic. What Is Glycemic Index? Available online: health.clevelandclinic.org/glycemic-index/ (accessed on 20 April 2022).
- Berná, G.; Oliveras-López, M.J.; Jurado-Ruíz, E.; Tejedo, J.; Bedoya, F.; Soria, B.; Martín, F. Nutrigenetics and nutrigenomics insights into diabetes etiopathogenesis. Nutrients 2014, 6, 5338–5369. [Google Scholar] [CrossRef] [Green Version]
- Felisbino, K.; Granzotti, J.G.; Bello-Santos, L.; Guiloski, I.C. Nutrigenomics in Regulating the Expression of Genes Related to Type 2 Diabetes Mellitus. Front. Physiol. 2021, 12, 699220. [Google Scholar] [CrossRef]
- Kang, J.X. Nutrigenomics and systems biology. J. Nutr. Nutr. 2012, 5, I–II. [Google Scholar] [CrossRef] [PubMed]
- Shahwan, M.; Alhumaydhi, F.; Ashraf, G.M.; Hasan, P.M.Z.; Shamsi, A. Role of polyphenols in combating Type 2 Diabetes and insulin resistance. Int. J. Biol. Macromol. 2022, 206, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Miao, M. Dietary polyphenols modulate starch digestion and glycaemic level: A review. Crit. Rev. Food Sci. Nutr. 2020, 60, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.A.; Keske, M.A.; Wadley, G.D. Effects of Vitamin C Supplementation on Glycemic Control and Cardiovascular Risk Factors in People With Type 2 Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis of Randomized Controlled Trials. Diabetes Care 2021, 44, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care 2011, 34, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, R.V. GlyNAC (Glycine and N-Acetylcysteine) Supplementation Improves Impaired Mitochondrial Fuel Oxidation and Lowers Insulin Resistance in Patients with Type 2 Diabetes: Results of a Pilot Study. Antioxidants 2022, 11, 154. [Google Scholar] [CrossRef]
- Georgetown. What Is Systems Medicine? Available online: https://systemsmedicine.georgetown.edu/georgetown-difference/what-is-system-medicine/ (accessed on 2 May 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alu, S.N.; Los, E.A.; Ford, G.A.; Stone, W.L. Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies. Antioxidants 2022, 11, 1336. https://doi.org/10.3390/antiox11071336
Alu SN, Los EA, Ford GA, Stone WL. Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies. Antioxidants. 2022; 11(7):1336. https://doi.org/10.3390/antiox11071336
Chicago/Turabian StyleAlu, Stephanie N., Evan A. Los, George A. Ford, and William L. Stone. 2022. "Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies" Antioxidants 11, no. 7: 1336. https://doi.org/10.3390/antiox11071336
APA StyleAlu, S. N., Los, E. A., Ford, G. A., & Stone, W. L. (2022). Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies. Antioxidants, 11(7), 1336. https://doi.org/10.3390/antiox11071336