
Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH2-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Kinetics Measurement of DCF Fluorescence (Cell-Free)
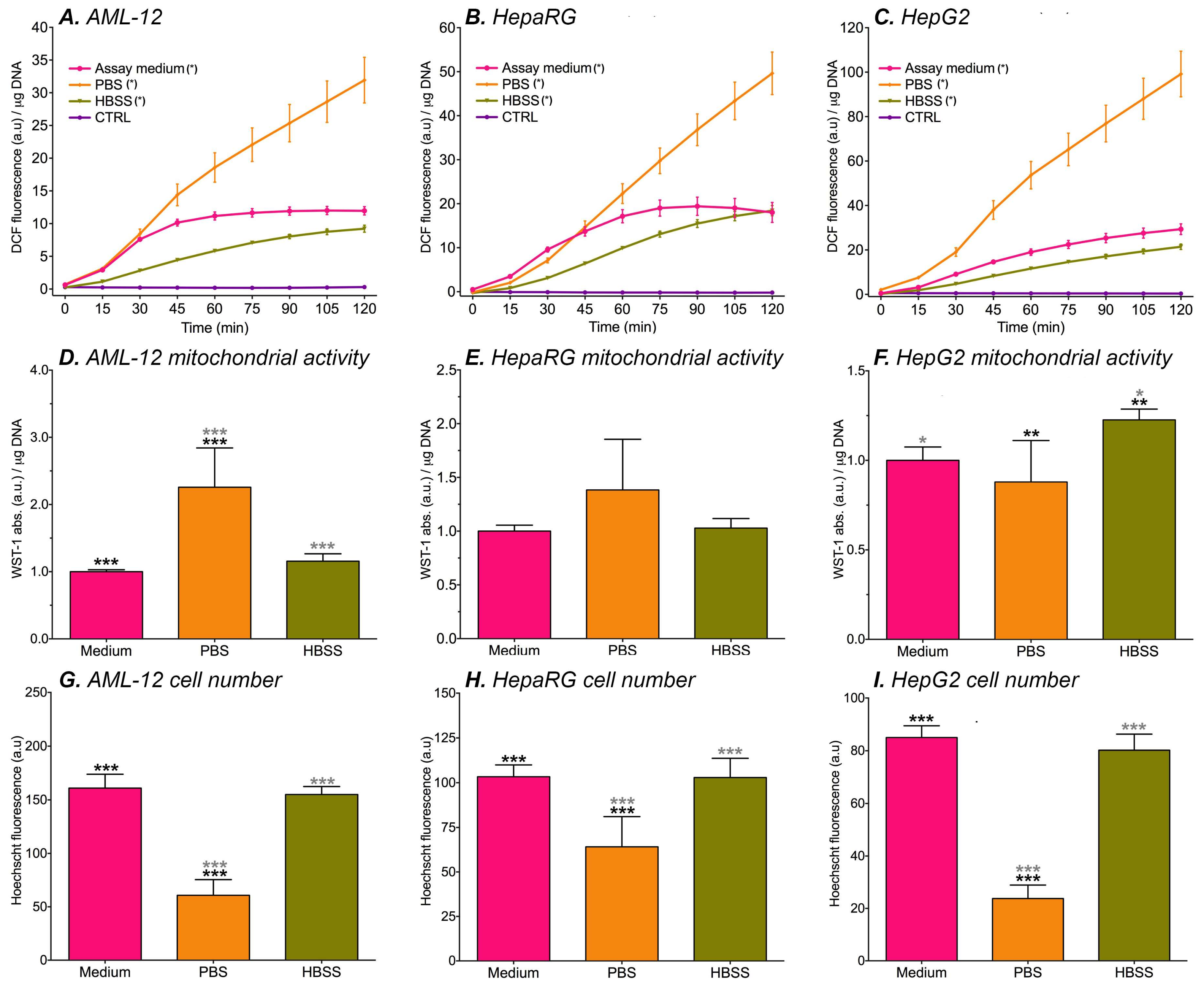
2.4. Kinetics Measurement of DCF Fluorescence in Hepatocytes after Physiological Buffer Exposure
2.5. Kinetics Measurement of DCF Fluorescence in Hepatocytes after ETC Inhibition
2.6. DNA Quantification
2.7. Statistical Analysis
3. Results and Discussion
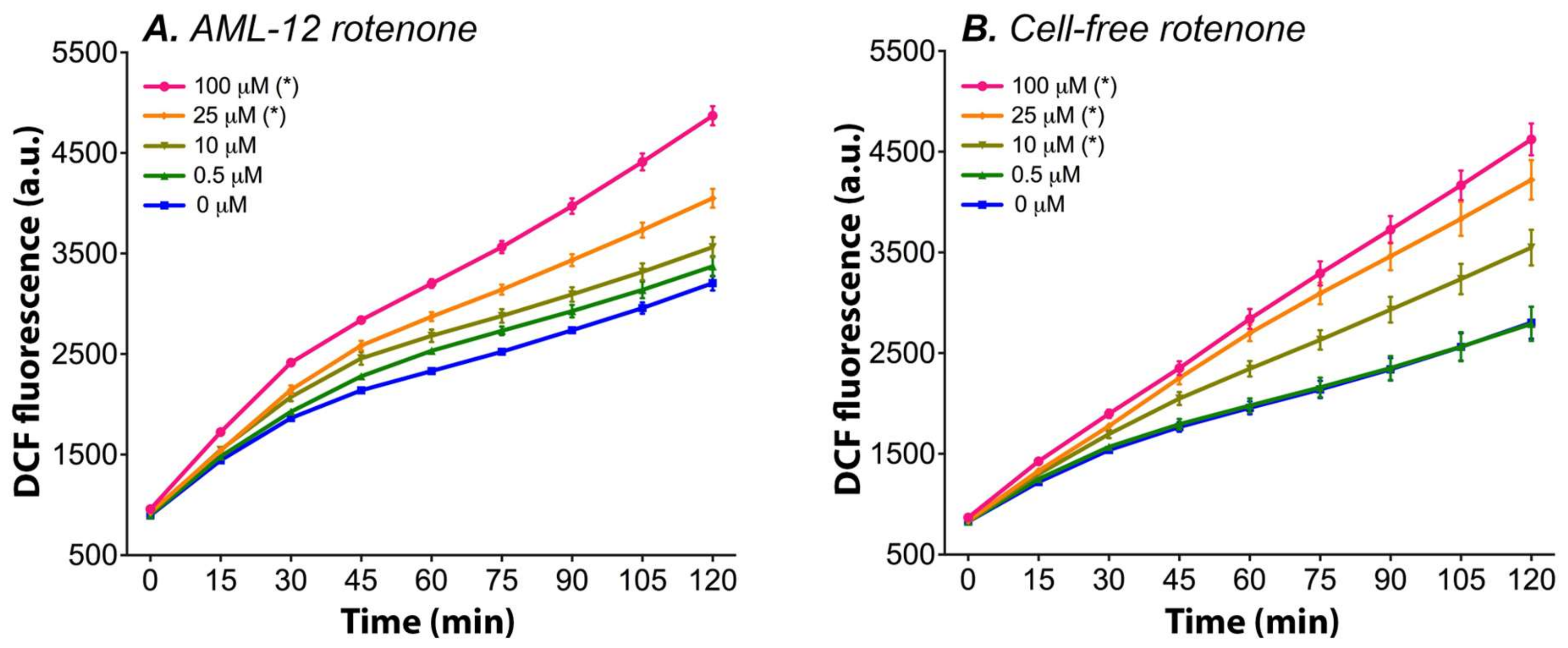
3.1. Rotenone Dose-Dependently Induces DCFH2-DA → DCF Conversion in Cell Culture Medium
3.2. DCF Formation in Cultured Hepatocytes
3.3. The DCFH2-DA → DCF Conversion Is Mediated by Mitochondrial Complex Inhibitors in Cell-Free Assay Medium
3.4. Optimization of the DCFH2-DA Assay System
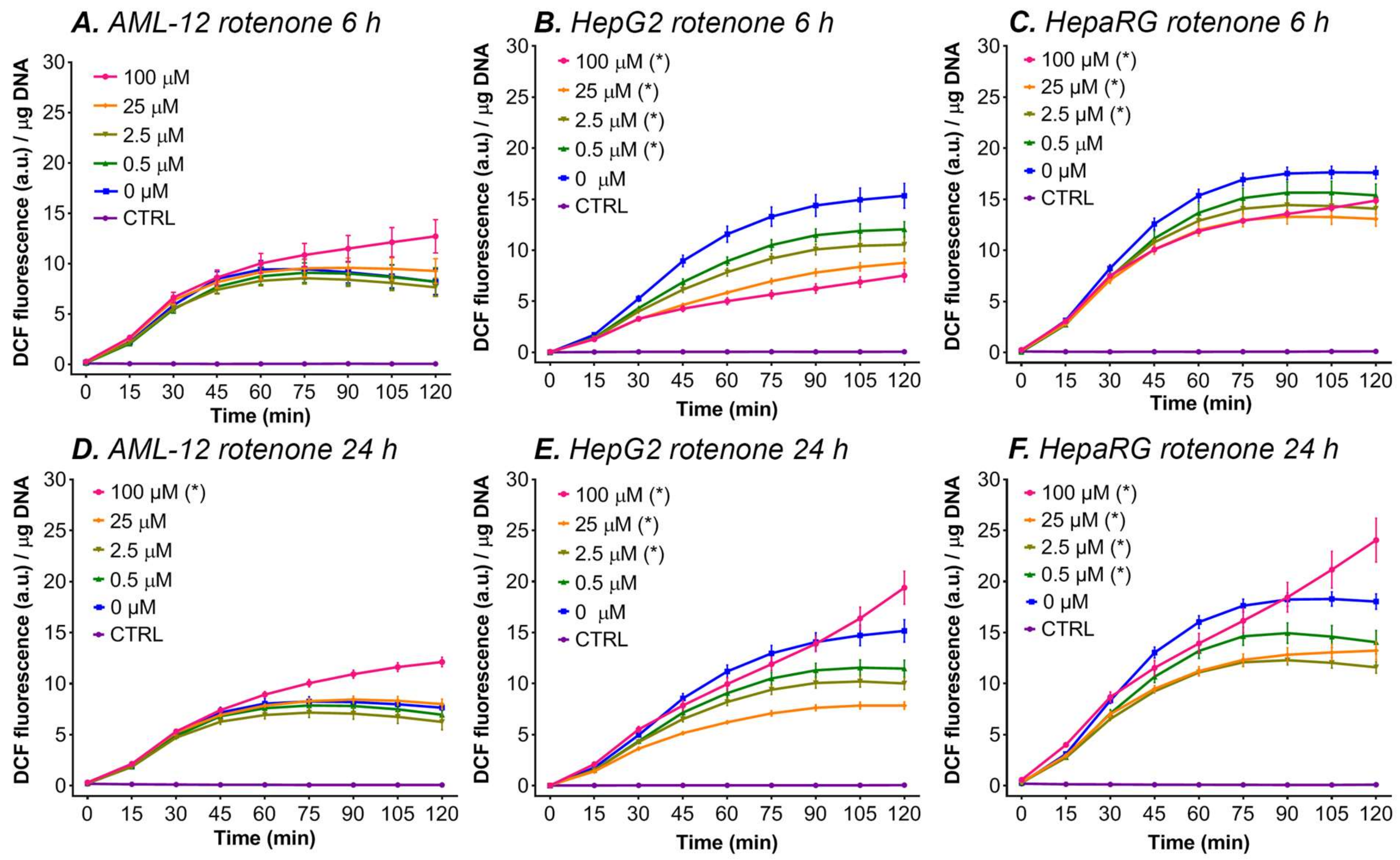
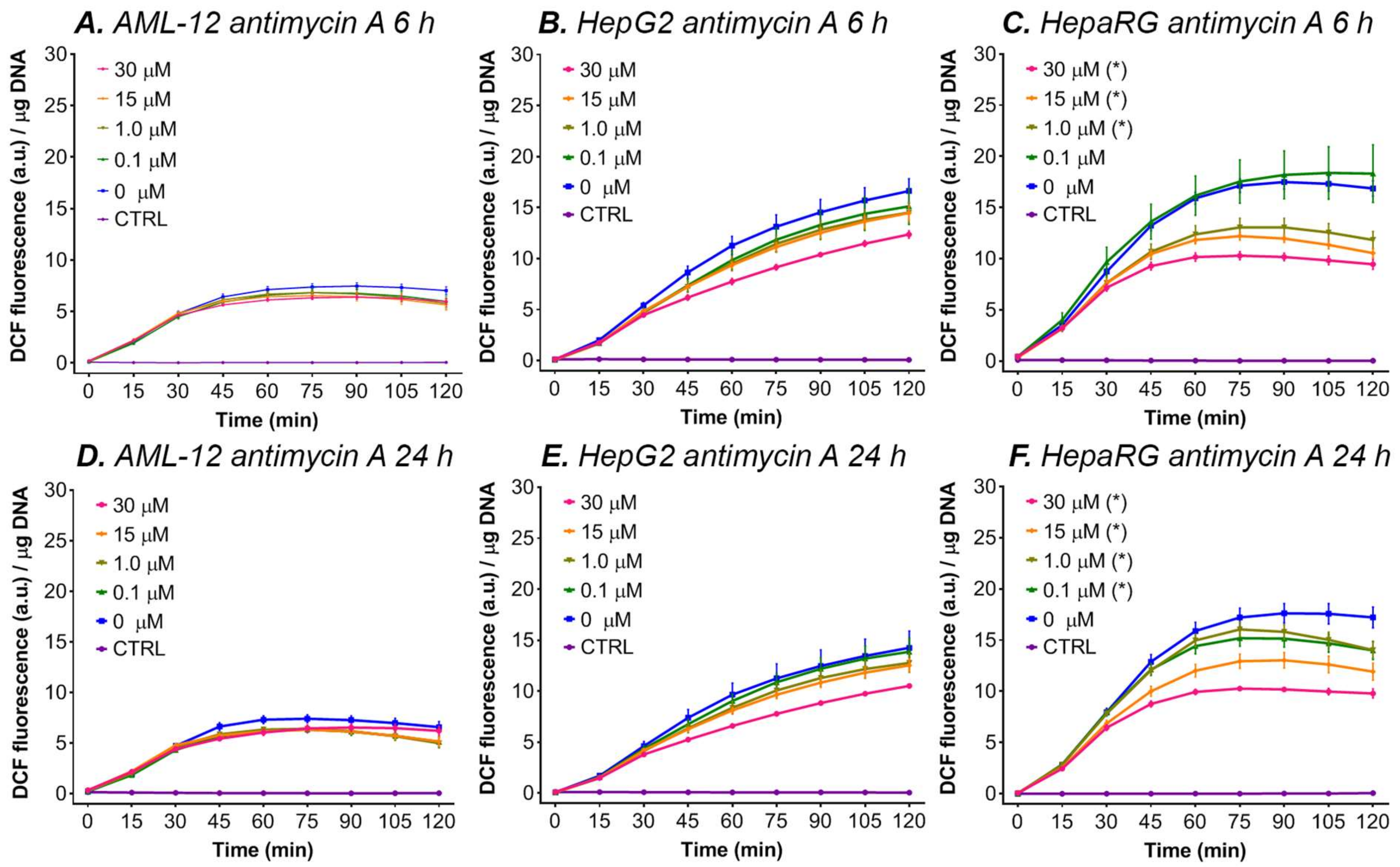
3.5. DCF Fluorescence and ETC Inhibition in Hepatocyte Cell Lines
3.5.1. Rotenone
- (I).
- At the lower rotenone concentrations, the cells are able to withstand the formation of mitochondrial ROS by upregulating their antioxidant enzyme systems (e.g., superoxide dismutase and catalase), but at higher concentrations (i.e., 100 μM rotenone) and longer incubation periods the antioxidant systems can no longer compensate [43], resulting in an increase in ∆flu. An increase in the antioxidative capacity not only effectively protects the cells from the formed ROS but might also induce a shift in the cellular redox state to a more reduced state, which might explain the drop in DCF formation at lower concentrations and shorter incubation periods with ETC inhibitors (Figure 3).
- (II).
- At lower rotenone concentrations, the cells stop using the ETC as their main energy source and meet their ATP demand by switching to glycolysis. Since most cancer cells have a strong predisposition for aerobic glycolysis for ATP production, this theory is supported by the fact that a decrease in fluorescence was measured in the two cancer-derived cell lines (HepG2 and HepaRG) and not in non-transformed AML-12 cells [98]. The increase in DCF formation at 100 μM rotenone in HepG2 and HepaRG cells can be explained by the fact that glycolysis alone is not sufficient to meet the cellular energy demand and that the cancer cells are partially respiring through the ETC (i.e., Warburg metabolism). At this rotenone concentration, the rest capacity of the ETC is blocked, resulting in ROS formation, and thus an increase in ∆flu.
3.5.2. Antimycin A
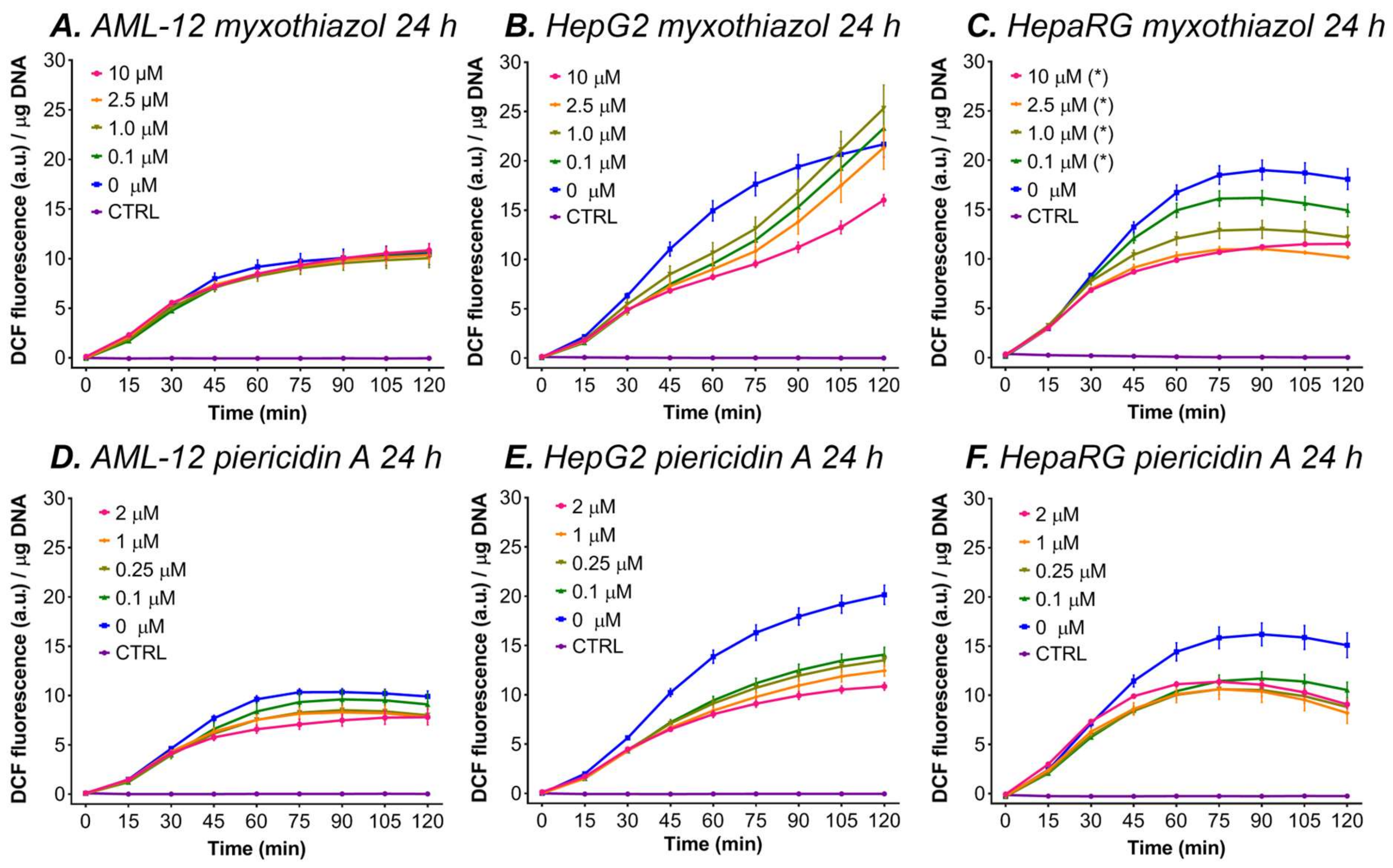
3.5.3. Myxothiazol and Piericidin A
3.5.4. Rotenone in Combination with Antimycin A
4. Concluding Remarks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Control of reactive oxygen species production in contracting skeletal muscle. Antioxid. Redox Signal. 2011, 15, 2477–2486. [Google Scholar] [CrossRef]
- Bai, H.; Zhang, W.; Qin, X.J.; Zhang, T.; Wu, H.; Liu, J.Z.; Hai, C.X. Hydrogen peroxide modulates the proliferation/quiescence switch in the liver during embryonic development and posthepatectomy regeneration. Antioxid. Redox Signal. 2015, 22, 921–937. [Google Scholar] [CrossRef]
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Reiniers, M.J.; van Golen, R.F.; van Gulik, T.M.; Heger, M. Reactive oxygen and nitrogen species in steatotic hepatocytes: A molecular perspective on the pathophysiology of ischemia-reperfusion injury in the fatty liver. Antioxid. Redox Signal. 2014, 21, 1119–1142. [Google Scholar] [CrossRef] [Green Version]
- Van Golen, R.F.; Reiniers, M.J.; Vrisekoop, N.; Zuurbier, C.J.; Olthof, P.B.; van Rheenen, J.; van Gulik, T.M.; Parsons, B.J.; Heger, M. The mechanisms and physiological relevance of glycocalyx degradation in hepatic ischemia/reperfusion injury. Antioxid. Redox Signal. 2014, 21, 1098–1118. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sharma, S.; Verma, S.; Kapoor, M.; Saini, A.; Nehru, B. Alzheimer’s disease like pathology induced six weeks after aggregated amyloid-beta injection in rats: Increased oxidative stress and impaired long-term memory with anxiety-like behavior. Neurol. Res. 2016, 38, 838–850. [Google Scholar] [CrossRef]
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef]
- Kloek, J.J.; Marechal, X.; Roelofsen, J.; Houtkooper, R.H.; van Kuilenburg, A.B.; Kulik, W.; Bezemer, R.; Neviere, R.; van Gulik, T.M.; Heger, M. Cholestasis is associated with hepatic microvascular dysfunction and aberrant energy metabolism before and during ischemia-reperfusion. Antioxid. Redox Signal. 2012, 17, 1109–1123. [Google Scholar] [CrossRef]
- Van Golen, R.F.; van Gulik, T.M.; Heger, M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev. 2012, 23, 69–84. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Wu, J.; Xiao, Q.; Zhang, N.; Xue, C.; Leung, A.W.; Zhang, H.; Tang, Q.J.; Xu, C. Palmatine hydrochloride mediated photodynamic inactivation of breast cancer MCF-7 cells: Effectiveness and mechanism of action. Photodiagnosis Photodyn. Ther. 2016, 15, 133–138. [Google Scholar] [CrossRef]
- Diao, Q.X.; Zhang, J.Z.; Zhao, T.; Xue, F.; Gao, F.; Ma, S.M.; Wang, Y. Vitamin E promotes breast cancer cell proliferation by reducing ROS production and p53 expression. Eur. Rev. Med. Pharmacol. Sci 2016, 20, 2710–2717. [Google Scholar]
- Manti, S.; Marseglia, L.; D’Angelo, G.; Cuppari, C.; Cusumano, E.; Arrigo, T.; Gitto, E.; Salpietro, C. “Cumulative Stress”: The Effects of Maternal and Neonatal Oxidative Stress and Oxidative Stress-Inducible Genes on Programming of Atopy. Oxid. Med. Cell. Longev. 2016, 2016, 8651820. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef]
- Wardman, P. Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects. Free Radic. Biol. Med. 2007, 43, 995–1022. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., II; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Zielonka, J.; Kalyanaraman, B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free Radic. Biol. Med. 2018, 128, 3–22. [Google Scholar] [CrossRef]
- Gomes, A.; Fernandes, E.; Lima, J.L. Fluorescence probes used for detection of reactive oxygen species. J. Biochem. Biophys. Methods 2005, 65, 45–80. [Google Scholar] [CrossRef]
- Wrona, M.; Patel, K.; Wardman, P. Reactivity of 2’,7’-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms toward carbonate, nitrogen dioxide, and hydroxyl radicals. Free Radic. Biol. Med. 2005, 38, 262–270. [Google Scholar] [CrossRef]
- Afri, M.; Frimer, A.A.; Cohen, Y. Active oxygen chemistry within the liposomal bilayer. Part IV: Locating 2’,7’-dichlorofluorescein (DCF), 2’,7’-dichlorodihydrofluorescein (DCFH) and 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) in the lipid bilayer. Chem. Phys. Lipids 2004, 131, 123–133. [Google Scholar] [CrossRef]
- LeBel, C.P.; Ischiropoulos, H.; Bondy, S.C. Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem. Res. Toxicol. 1992, 5, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Reiniers, M.J.; van Golen, R.F.; Bonnet, S.; Broekgaarden, M.; van Gulik, T.M.; Egmond, M.R.; Heger, M. Preparation and Practical Applications of 2’,7’-Dichlorodihydrofluorescein in Redox Assays. Anal. Chem. 2017, 89, 3853–3857. [Google Scholar] [CrossRef]
- Wrona, M.; Patel, K.B.; Wardman, P. The roles of thiol-derived radicals in the use of 2’,7’-dichlorodihydrofluorescein as a probe for oxidative stress. Free Radic. Biol. Med. 2008, 44, 56–62. [Google Scholar] [CrossRef]
- Burkitt, M.J.; Wardman, P. Cytochrome C is a potent catalyst of dichlorofluorescin oxidation: Implications for the role of reactive oxygen species in apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Reiniers, M.J.; de Haan, L.R.; Reeskamp, L.F.; Broekgaarden, M.; van Golen, R.F.; Heger, M. Analysis and Optimization of Conditions for the Use of 2’,7’-Dichlorofluorescein Diacetate in Cultured Hepatocytes. Antioxidants 2021, 10, 674. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.S.; Zhou, Z.; Kennedy, A.R. Adaptation of the dichlorofluorescein assay for detection of radiation-induced oxidative stress in cultured cells. Radiat. Res. 2003, 160, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Augusto, O.; Brigelius-Flohe, R.; Dennery, P.A.; Kalyanaraman, B.; Ischiropoulos, H.; Mann, G.E.; Radi, R.; Roberts, L.J., 2nd; Vina, J.; et al. Even free radicals should follow some rules: A guide to free radical research terminology and methodology. Free Radic. Biol. Med. 2015, 78, 233–235. [Google Scholar] [CrossRef]
- Bonini, M.G.; Rota, C.; Tomasi, A.; Mason, R.P. The oxidation of 2’,7’-dichlorofluorescin to reactive oxygen species: A self-fulfilling prophesy? Free Radic. Biol. Med. 2006, 40, 968–975. [Google Scholar] [CrossRef]
- Yazdani, M. Concerns in the application of fluorescent probes DCDHF-DA, DHR 123 and DHE to measure reactive oxygen species in vitro. Toxicol. Vitro 2015, 30, 578–582. [Google Scholar] [CrossRef]
- Yazdani, M.; Paulsen, R.E.; Gjoen, T.; Hylland, K. Reactive oxygen species and cytotoxicity in rainbow trout hepatocytes: Effects of medium and incubation time. Bull. Environ. Contam. Toxicol. 2015, 94, 193–198. [Google Scholar] [CrossRef]
- Allard, C.; De Lamirande, G.; Cantero, A. Mitochondrial population of mammalian cells. II. Variation in the mitochondrial population of the average rat liver cell during regeneration; use of the mitochondrion as a unit of measurement. Cancer Res. 1952, 12, 580–583. [Google Scholar]
- Bhogal, R.H.; Weston, C.J.; Curbishley, S.M.; Bhatt, A.N.; Adams, D.H.; Afford, S.C. Variable responses of small and large human hepatocytes to hypoxia and hypoxia/reoxygenation (H-R). FEBS Lett. 2011, 585, 935–941. [Google Scholar] [CrossRef] [Green Version]
- De Pedro, N.; Cautain, B.; Melguizo, A.; Vicente, F.; Genilloud, O.; Pelaez, F.; Tormo, J.R. Mitochondrial complex I inhibitors, acetogenins, induce HepG2 cell death through the induction of the complete apoptotic mitochondrial pathway. J. Bioenerg. Biomembr. 2013, 45, 153–164. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, X.; Chen, R.; Zhang, H. Radiosensitioring the boundaries of additivity: Mixtures of NADH: Quinone oxidoreductase inhibitortoma HepG2 cells to X-ray radiation. J. Radiat. Res. 2012, 53, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Boyd, J.; Saksena, A.; Patrone, J.B.; Williams, H.N.; Boggs, N.; Le, H.; Theodore, M. Exploring the boundaries of additivity: Mixtures of NADH: Quinone oxidoreductase inhibitors. Chem. Res. Toxicol. 2011, 24, 1242–1250. [Google Scholar] [CrossRef]
- Cuello, S.; Goya, L.; Madrid, Y.; Campuzano, S.; Pedrero, M.; Bravo, L.; Cámara, C.; Ramos, S. Molecular mechanisms of methylmercury-induced cell death in human hepg2 cells. Food Chem. Toxicol. 2010, 48, 1405–1411. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Ahmad, J.; Farshori, N.N.; Saquib, Q.; Jahan, S.; Kashyap, M.P.; Ahamed, M.; Musarrat, J.; Al-Khedhairy, A.A. Rotenone-induced oxidative stress and apoptosis in human liver HepG2 cells. Mol. Cell. Biochem. 2013, 384, 59–69. [Google Scholar] [CrossRef]
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]
- Isenberg, J.S.; Klaunig, J.E. Role of the mitochondrial membrane permeability transition (MPT) in rotenone-induced apoptosis in liver cells. Toxicol. Sci. 2000, 53, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Von Montfort, C.; Matias, N.; Fernandez, A.; Fucho, R.; Conde de la Rosa, L.; Martinez-Chantar, M.L.; Mato, J.M.; Machida, K.; Tsukamoto, H.; Murphy, M.P.; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012, 57, 852–859. [Google Scholar] [CrossRef] [Green Version]
- Farfan Labonne, B.E.; Gutierrez, M.; Gomez-Quiroz, L.E.; Konigsberg Fainstein, M.; Bucio, L.; Souza, V.; Flores, O.; Ortiz, V.; Hernandez, E.; Kershenobich, D.; et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599–609. [Google Scholar] [CrossRef]
- Busk, M.; Boutilier, R.G. Metabolic arrest and its regulation in anoxic eel hepatocytes. Physiol. Biochem. Zool. 2005, 78, 926–936. [Google Scholar] [CrossRef]
- Berthiaume, F.; MacDonald, A.D.; Kang, Y.H.; Yarmush, M.L. Control analysis of mitochondrial metabolism in intact hepatocytes: Effect of interleukin-1beta and interleukin-6. Metab. Eng. 2003, 5, 108–123. [Google Scholar] [CrossRef]
- Zhang, J.G.; Nicholls-Grzemski, F.A.; Tirmenstein, M.A.; Fariss, M.W. Vitamin E succinate protects hepatocytes against the toxic effect of reactive oxygen species generated at mitochondrial complexes I and III by alkylating agents. Chem. Biol. Interact. 2001, 138, 267–284. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, A.; Barja, G. Sites and mechanisms responsible for the low rate of free radical production of heart mitochondria in the long-lived pigeon. Mech. Ageing Dev. 1997, 98, 95–111. [Google Scholar] [CrossRef]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M.; Ghelli, A.; Crimi, M.; Estornell, E.; Fato, R.; Lenaz, G. Complex I and complex III of mitochondria have common inhibitors acting as ubiquinone antagonists. Biochem. Biophys. Res. Commun. 1993, 190, 1090–1096. [Google Scholar] [CrossRef]
- Keipert, S.; Ost, M.; Johann, K.; Imber, F.; Jastroch, M.; van Schothorst, E.M.; Keijer, J.; Klaus, S. Skeletal muscle mitochondrial uncoupling drives endocrine cross-talk through the induction of FGF21 as a myokine. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E469–E482. [Google Scholar] [CrossRef]
- Young, T.A.; Cunningham, C.C.; Bailey, S.M. Reactive oxygen species production by the mitochondrial respiratory chain in isolated rat hepatocytes and liver mitochondria: Studies using myxothiazol. Arch. Biochem. Biophys. 2002, 405, 65–72. [Google Scholar] [CrossRef]
- Shiryaeva, A.; Arkadyeva, A.; Emelyanova, L.; Sakuta, G.; Morozov, V. Superoxide anion production by the mitochondrial respiratory chain of hepatocytes of rats with experimental toxic hepatitis. J. Bioenerg. Biomembr. 2009, 41, 379–385. [Google Scholar] [CrossRef]
- Nobes, C.D.; Brown, G.C.; Olive, P.N.; Brand, M.D. Non-ohmic proton conductance of the mitochondrial inner membrane in hepatocytes. J. Biol. Chem. 1990, 265, 12903–12909. [Google Scholar] [CrossRef]
- Johnson, J.E., Jr.; Choksi, K.; Widger, W.R. NADH-Ubiquinone oxidoreductase: Substrate-dependent oxygen turnover to superoxide anion as a function of flavin mononucleotide. Mitochondrion 2003, 3, 97–110. [Google Scholar] [CrossRef]
- Lambert, A.J.; Brand, M.D. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 2004, 279, 39414–39420. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef]
- Ohnishi, S.T.; Shinzawa-Itoh, K.; Ohta, K.; Yoshikawa, S.; Ohnishi, T. New insights into the superoxide generation sites in bovine heart NADH-ubiquinone oxidoreductase (Complex I): The significance of protein-associated ubiquinone and the dynamic shifting of generation sites between semiflavin and semiquinone radicals. Biochim. Biophys. Acta 2010, 1797, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- King, M.S.; Sharpley, M.S.; Hirst, J. Reduction of hydrophilic ubiquinones by the flavin in mitochondrial nadh:Ubiquinone oxidoreductase (complex i) and production of reactive oxygen species. Biochemistry 2009, 48, 2053–2062. [Google Scholar] [CrossRef] [Green Version]
- Salaheldin, T.A.; Loutfy, S.A.; Ramadan, M.A.; Youssef, T.; Mousa, S.A. Ir-enhanced photothermal therapeutic effect of graphene magnetite nanocomposite on human liver cancer hepg2 cell model. Int. J. Nanomed. 2019, 14, 4397–4412. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, R.; Nibourg, G.A.; van der Hoeven, T.V.; Ackermans, M.T.; Hakvoort, T.B.; van Gulik, T.M.; Lamers, W.H.; Elferink, R.P.; Chamuleau, R.A. The heparg cell line is suitable for bioartificial liver application. Int. J. Biochem. Cell Biol. 2011, 43, 1483–1489. [Google Scholar] [CrossRef]
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2’,7’-Dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: Forty years of application and controversy. Free Radic. Res. 2010, 44, 587–604. [Google Scholar] [CrossRef]
- Reiniers, M.J.; van Golen, R.F.; van Gulik, T.M.; Heger, M. 2’,7’-Dichlorofluorescein is not a probe for the detection of reactive oxygen and nitrogen species. J. Hepatol. 2012, 56, 1214–1216. [Google Scholar] [CrossRef]
- Reiniers, M.J.; de Haan, L.R.; Reeskamp, L.F.; Broekgaarden, M.; Hoekstra, R.; van Golen, R.F.; Heger, M. Optimal Use of 2’,7’-Dichlorofluorescein Diacetate in Cultured Hepatocytes. Methods Mol. Biol. 2022, 2451, 721–747. [Google Scholar] [CrossRef]
- Oparka, M.; Walczak, J.; Malinska, D.; van Oppen, L.; Szczepanowska, J.; Koopman, W.J.H.; Wieckowski, M.R. Quantifying ROS levels using CM-H2DCFDA and HyPer. Methods 2016, 109, 3–11. [Google Scholar] [CrossRef]
- Schulz, S.; Schmitt, S.; Wimmer, R.; Aichler, M.; Eisenhofer, S.; Lichtmannegger, J.; Eberhagen, C.; Artmann, R.; Tookos, F.; Walch, A.; et al. Progressive stages of mitochondrial destruction caused by cell toxic bile salts. Biochim. Biophys. Acta 2013, 1828, 2121–2133. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.M.; Steer, C.J. Mitochondrial membrane perturbations in cholestasis. J. Hepatol. 2000, 32, 135–141. [Google Scholar] [CrossRef]
- Sokol, R.J.; Straka, M.S.; Dahl, R.; Devereaux, M.W.; Yerushalmi, B.; Gumpricht, E.; Elkins, N.; Everson, G. Role of oxidant stress in the permeability transition induced in rat hepatic mitochondria by hydrophobic bile acids. Pediatr. Res. 2001, 49, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Biasi, F.; Bosco, M.; Chiappino, I.; Chiarpotto, E.; Lanfranco, G.; Ottobrelli, A.; Massano, G.; Donadio, P.P.; Vaj, M.; Andorno, E.; et al. Oxidative damage in human liver transplantation. Free Radic. Biol. Med. 1995, 19, 311–317. [Google Scholar] [CrossRef]
- Jaeschke, H. Role of reactive oxygen species in hepatic ischemia-reperfusion injury and preconditioning. J. Investig. Surg. 2003, 16, 127–140. [Google Scholar] [CrossRef]
- Toth-Zsamboki, E.; Horvath, E.; Vargova, K.; Pankotai, E.; Murthy, K.; Zsengeller, Z.; Barany, T.; Pek, T.; Fekete, K.; Kiss, R.G.; et al. Activation of poly(ADP-ribose) polymerase by myocardial ischemia and coronary reperfusion in human circulating leukocytes. Mol. Med. 2006, 12, 221–228. [Google Scholar] [CrossRef]
- Wen, J.J.; Garg, N.J. Mitochondrial generation of reactive oxygen species is enhanced at the Q(o) site of the complex III in the myocardium of Trypanosoma cruzi-infected mice: Beneficial effects of an antioxidant. J. Bioenerg. Biomembr. 2008, 40, 587–598. [Google Scholar] [CrossRef]
- Oei, G.T.; Heger, M.; van Golen, R.F.; Alles, L.K.; Flick, M.; van der Wal, A.C.; van Gulik, T.M.; Hollmann, M.W.; Preckel, B.; Weber, N.C. Reduction of cardiac cell death after helium postconditioning in rats: Transcriptional analysis of cell death and survival pathways. Mol. Med. 2015, 20, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.; Bonacasa, B.; Fenoy, F.J.; Salom, M.G. Reactive oxygen and nitrogen species in the renal ischemia/reperfusion injury. Curr. Pharm. Des. 2013, 19, 2776–2794. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Baust, J.M.; Buehring, G.C.; Campbell, L.; Elmore, E.; Harbell, J.W.; Nims, R.W.; Price, P.; Reid, Y.A.; Simione, F. Best practices in cell culture: An overview. Vitro Cell. Dev. Biol. Anim. 2017, 53, 669–672. [Google Scholar] [CrossRef]
- Stock Solutions. Common buffers, media, and stock solutions; John and Wiley and Sons: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Wieczorowska-Tobis, K.; Polubinska, A.; Breborowicz, A.; Oreopoulos, D.G. A comparison of the biocompatibility of phosphate-buffered saline and dianeal 3.86% in the rat model of peritoneal dialysis. Adv. Perit. Dial. 2001, 17, 42–46. [Google Scholar] [PubMed]
- Trumpower, B.L. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar] [CrossRef]
- Degli Esposti, M. Inhibitors of NADH-ubiquinone reductase: An overview. Biochim. Biophys. Acta 1998, 1364, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Fenical, W. The unique chemistry and biology of the piericidins. J. Antibiot. 2016, 69, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Leonhardt, H.; Gordon, L.; Livingston, R. Acid-base equilibriums of fluorescein and 2’, 7’-dichlorofluorescein in their ground and fluorescent states. J. Phys. Chem. 1971, 75, 245–249. [Google Scholar] [CrossRef]
- Zhu, H.; Bannenberg, G.L.; Moldeus, P.; Shertzer, H.G. Oxidation pathways for the intracellular probe 2’,7’-dichlorofluorescein. Arch. Toxicol. 1994, 68, 582–587. [Google Scholar] [CrossRef]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Tetz, L.M.; Kamau, P.W.; Cheng, A.A.; Meeker, J.D.; Loch-Caruso, R. Troubleshooting the dichlorofluorescein assay to avoid artifacts in measurement of toxicant-stimulated cellular production of reactive oxidant species. J. Pharmacol. Toxicol. Methods 2013, 67, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Boulton, S.; Anderson, A.; Swalwell, H.; Henderson, J.R.; Manning, P.; Birch-Machin, M.A. Implications of using the fluorescent probes, dihydrorhodamine 123 and 2’,7’-dichlorodihydrofluorescein diacetate, for the detection of UVA-induced reactive oxygen species. Free Radic. Res. 2011, 45, 139–146. [Google Scholar] [CrossRef]
- Bolling, B.W.; Chen, Y.Y.; Kamil, A.G.; Oliver Chen, C.Y. Assay dilution factors confound measures of total antioxidant capacity in polyphenol-rich juices. J. Food Sci. 2012, 77, H69–H75. [Google Scholar] [CrossRef] [Green Version]
- Thayyullathil, F.; Chathoth, S.; Hago, A.; Patel, M.; Galadari, S. Rapid reactive oxygen species (ROS) generation induced by curcumin leads to caspase-dependent and -independent apoptosis in L929 cells. Free Radic. Biol. Med. 2008, 45, 1403–1412. [Google Scholar] [CrossRef]
- Reistad, T.; Mariussen, E.; Fonnum, F. The effect of a brominated flame retardant, tetrabromobisphenol-A, on free radical formation in human neutrophil granulocytes: The involvement of the MAP kinase pathway and protein kinase C. Toxicol. Sci. 2005, 83, 89–100. [Google Scholar] [CrossRef] [Green Version]
- White, E.Z.; Pennant, N.M.; Carter, J.R.; Hawsawi, O.; Odero-Marah, V.; Hinton, C.V. Serum deprivation initiates adaptation and survival to oxidative stress in prostate cancer cells. Sci. Rep. 2020, 10, 12505. [Google Scholar] [CrossRef]
- Chiao, C.; Zhang, Y.; Kaufman, D.G.; Kaufmann, W.K. Phenobarbital modulates the type of cell death by rat hepatocytes during deprivation of serum in vitro. Hepatology 1995, 22, 297–303. [Google Scholar] [CrossRef]
- Korystov, Y.N.; Shaposhnikova, V.V.; Korystova, A.F.; Emel’yanov, M.O. Detection of reactive oxygen species induced by radiation in cells using the dichlorofluorescein assay. Radiat. Res. 2007, 168, 226–232. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Liu, X.; Harriman, J.F.; Schnellmann, R.G. Cytoprotective properties of novel nonpeptide calpain inhibitors in renal cells. J. Pharmacol. Exp. Ther. 2002, 302, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, H.; Lemasters, J.J. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 2003, 125, 1246–1257. [Google Scholar] [CrossRef]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Royall, J.A.; Ischiropoulos, H. Evaluation of 2’,7’-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch. Biochem. Biophys. 1993, 302, 348–355. [Google Scholar] [CrossRef]
- Hempel, S.L.; Buettner, G.R.; O’Malley, Y.Q.; Wessels, D.A.; Flaherty, D.M. Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2’,7’-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2’,7’-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic. Biol. Med. 1999, 27, 146–159. [Google Scholar] [CrossRef]
- Swift, L.M.; Sarvazyan, N. Localization of dichlorofluorescin in cardiac myocytes: Implications for assessment of oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H982–H990. [Google Scholar] [CrossRef]
- Myhre, O.; Andersen, J.M.; Aarnes, H.; Fonnum, F. Evaluation of the probes 2’,7’-dichlorofluorescin diacetate, luminol, and lucigenin as indicators of reactive species formation. Biochem. Pharmacol. 2003, 65, 1575–1582. [Google Scholar] [CrossRef]
- Keller, A.; Mohamed, A.; Drose, S.; Brandt, U.; Fleming, I.; Brandes, R.P. Analysis of dichlorodihydrofluorescein and dihydrocalcein as probes for the detection of intracellular reactive oxygen species. Free Radic. Res. 2004, 38, 1257–1267. [Google Scholar] [CrossRef]
- Hafer, K.; Iwamoto, K.S.; Schiestl, R.H. Refinement of the dichlorofluorescein assay for flow cytometric measurement of reactive oxygen species in irradiated and bystander cell populations. Radiat. Res. 2008, 169, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Kurz, T.; Brunk, U.T.; Nilsson, S.E.; Frennesson, C.I. What does the commonly used DCF test for oxidative stress really show? Biochem. J. 2010, 428, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Roper, M.G. Measurement of DCF fluorescence as a measure of reactive oxygen species in murine islets of Langerhans. Anal. Methods 2014, 6, 3019–3024. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, D.; Asaduzzaman, M.; Young, F. Real time monitoring and quantification of reactive oxygen species in breast cancer cell line MCF-7 by 2’,7’-dichlorofluorescin diacetate (DCFDA) assay. J. Pharmacol. Toxicol. Methods 2018, 94, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.; Monico, D.A.; Tedesco, A.C. Implications of dichlorofluorescein photoinstability for detection of UVA-induced oxidative stress in fibroblasts and keratinocyte cells. Photochem. Photobiol. Sci. 2020, 19, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weijer, R.; Broekgaarden, M.; Kos, M.; van Vught, R.; Rauws, E.A.J.; Breukink, E.; van Gulik, T.M.; Storm, G.; Heger, M. Enhancing photodynamic therapy of refractory solid cancers: Combining second-generation photosensitizers with multi-targeted liposomal delivery. J. Photochem. Photobiol. C Photochem. Rev. 2015, 23, 103–131. [Google Scholar] [CrossRef]
- Lee, C.; Nam, J.S.; Lee, C.G.; Park, M.; Yoo, C.M.; Rhee, H.W.; Seo, J.K.; Kwon, T.H. Analysing the mechanism of mitochondrial oxidation-induced cell death using a multifunctional iridium(III) photosensitiser. Nat. Commun. 2021, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; Janes, M.S.; Pehar, M.; Monette, J.S.; Ross, M.F.; Hagen, T.M.; Murphy, M.P.; Beckman, J.S. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. USA 2006, 103, 15038–15043. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Antunes, F.; Eaton, J.W.; Brunk, U.T. Lysosomal enzymes promote mitochondrial oxidant production, cytochrome c release and apoptosis. Eur. J. Biochem. 2003, 270, 3778–3786. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Liu, X.; LeCluyse, E.L.; Brouwer, K.R.; Gan, L.S.; Lemasters, J.J.; Stieger, B.; Meier, P.J.; Brouwer, K.L. Biliary excretion in primary rat hepatocytes cultured in a collagen-sandwich configuration. Am. J. Physiol. 1999, 277, G12–G21. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Khademi, F.; Zal, F.; Sardarian, A.R.; Amini, F. In Vitro Analysis of CsA-Induced Hepatotoxicity in HepG2 Cell Line: Oxidative Stress and alpha2 and beta1 Integrin Subunits Expression. Hepat. Mon. 2013, 13, e11447. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Pang, L.; Cong, H.; Shen, Y.; Yu, B. Application and design of esterase-responsive nanoparticles for cancer therapy. Drug Deliv. 2019, 26, 416–432. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Ogawa, Y.; Afify, A.S.; Kageyama, Y.; Okada, T.; Okuno, H.; Yoshii, Y.; Nose, T. Difference between cancer cells and the corresponding normal tissue in view of stereoselective hydrolysis of synthetic esters. Biochim. Biophys. Acta 1995, 1243, 300–308. [Google Scholar] [CrossRef]
- George, S.; Abrahamse, H. Redox Potential of Antioxidants in Cancer Progression and Prevention. Antioxidants 2020, 9, 1156. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unbuffered | pH = 6.0 | pH = 7.4 | pH = 9.0 | |
|---|---|---|---|---|
| Rotenone | ||||
| 0 μM | 2395 ± 534 | 514 ± 45 | 2407 ± 332 | 1717 ± 306 |
| 0.5 μM | 2363 ± 532 | 509 ± 49 | 2361 ± 346 | 1710 ± 319 |
| 10 μM | 3136 ± 551 # | 627 ± 62 # | 2809 ± 384 | 2288 ± 361 # |
| 25 μM | 3846 ± 604 # | 621 ± 45 # | 3238 ± 330 # | 2713 ± 370 # |
| 100 μM | 3996 ± 365 # | 658 ± 40 # | 4088 ± 333 # | 3294 ± 352 # |
| Antimycin A | ||||
| 0 μM | 2938 ± 331 | 727 ± 17 | 2340 ± 215 | 1520 ± 239 |
| 0.1 μM | 2736 ± 388 | 677 ± 55 | 2314 ± 271 | 1453 ± 256 |
| 1.0 μM | 2677 ± 442 | 712 ± 68 | 2351 ± 355 | 1369 ± 227 |
| 15 μM | 2277 ± 285 # | 985 ± 97 # | 2299 ± 213 | 1218 ± 113 # |
| 30 μM | 2342 ± 284 # | 1211 ± 87 # | 2335 ± 225 | 1263 ± 89 # |
| Myxothiazol | ||||
| 0 μM | 1962 ± 363 | 696 ± 201 | 1898 ± 140 | 1519 ± 232 |
| 0.1 μM | 1901 ± 376 | 673 ± 203 | 1741 ± 141 | 1443 ± 232 |
| 1.0 μM | 1928 ± 336 | 705 ± 207 | 1760 ± 170 | 1540 ± 280 |
| 2.5 μM | 2158 ± 392 | 736 ± 218 | 1846 ± 211 | 1753 ± 342 |
| 10 μM | 2631 ± 484 # | 698 ± 183 | 2030 ± 145 | 2325 ± 391 # |
| Piericidin A | ||||
| 0 μM | 2072 ± 176 | 124 ± 8.7 | 2311 ± 269 | 1123 ± 298 |
| 0.1 μM | 2065 ± 170 | 127 ± 8.7 | 2296 ± 276 | 1071 ± 330 |
| 0.5 μM | 2000 ± 184 | 137 ± 7.1 # | 2295 ± 303 | 1122 ± 368 |
| 1.0 μM | 2042 ± 174 | 155 ± 9.2 # | 2402 ± 326 | 1179 ± 343 |
| 2.0 μM | 2331 ± 146 # | 184 ± 9.6 # | 2702 ± 303 # | 1778 ± 375 # |
| Solvents † | Ethanol | DMSO | Methanol | |
|---|---|---|---|---|
| 0.0% | 2646 ± 381 | 3005 ± 310 | 2884 ± 185 | |
| 0.2% | 2033 ± 372 | 2466 ± 314 # | 2546 ± 183 # | |
| 0.5% | 1902 ± 369 | 2275 ± 333 # | 2434 ± 198 # | |
| 1.0% | 1807 ± 384 # | 2148 ± 295 # | 2345 ± 179 # | |
| 2.0% | 1738 ± 390 # | 1979 ± 286 # | 2342 ± 168 # | |
| 4.0% | 1848 ± 374 # | 2066 ± 232 # | 2299 ± 149 # | |
| SerumBuffers‡ | FCS | FCS-HI | BSA | |
| 0.0% | 10 ± 5 | 17 ± 4 | −12 ± 5 | |
| 1.0% | 564 ± 57 # | 502 ± 65 # | 328 ± 27 # | |
| 2.5% | 930 ± 101 # | 719 ± 147 # | 303 ± 36 # | |
| 5.0% | 873 ± 144 # | 781 ± 185 # | 157 ± 25 # | |
| 7.5% | 607 ± 105 # | 610 ± 186 # | 127 ± 18 # | |
| 10.0% | 425 ± 61 # | 475 ± 155 # | 147 ± 14 # | |
| Buffers‡ | TRIS | HEPES | HEPES [25 mM] | |
| 5 mM | 10.3 ± 14.8 | 4.4 ± 14.3 | pH = 6.0 | −8.9 ± 8.1 |
| 10 mM | 3.8 ± 13.8 | 1.5 ± 15.1 | pH = 7.4 | −14.4 ± 4.9 |
| 25 mM | 1.6 ± 14.6 | 2.1 ± 14.4 | pH = 9.0 | 117.0 ± 40.0 a |
| Culture medium | WE | DMEM | RPMI | DMEM/F12 |
| 2340 ± 215 c | 4017 ± 471 b | 1592 ± 140 c | 1834 ± 150 b | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Haan, L.R.; Reiniers, M.J.; Reeskamp, L.F.; Belkouz, A.; Ao, L.; Cheng, S.; Ding, B.; van Golen, R.F.; Heger, M. Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH2-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status. Antioxidants 2022, 11, 1424. https://doi.org/10.3390/antiox11081424
de Haan LR, Reiniers MJ, Reeskamp LF, Belkouz A, Ao L, Cheng S, Ding B, van Golen RF, Heger M. Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH2-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status. Antioxidants. 2022; 11(8):1424. https://doi.org/10.3390/antiox11081424
Chicago/Turabian Stylede Haan, Lianne R., Megan J. Reiniers, Laurens F. Reeskamp, Ali Belkouz, Lei Ao, Shuqun Cheng, Baoyue Ding, Rowan F. van Golen, and Michal Heger. 2022. "Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH2-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status" Antioxidants 11, no. 8: 1424. https://doi.org/10.3390/antiox11081424
APA Stylede Haan, L. R., Reiniers, M. J., Reeskamp, L. F., Belkouz, A., Ao, L., Cheng, S., Ding, B., van Golen, R. F., & Heger, M. (2022). Experimental Conditions That Influence the Utility of 2′7′-Dichlorodihydrofluorescein Diacetate (DCFH2-DA) as a Fluorogenic Biosensor for Mitochondrial Redox Status. Antioxidants, 11(8), 1424. https://doi.org/10.3390/antiox11081424