
Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response

 , ,
, ,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of Stable Knockdown and Overexpressed Cell Lines
2.3. Determination of Cellular Bioenergetics
2.4. H2DCF-DA Assay
2.5. Cell Viability
2.6. Lipid Peroxidation Assay
2.7. Luciferase Reporter Assays
2.8. RNA Extraction, Purification and cDNA Conversion
2.9. Real-Time Quantitative PCR
2.10. Protein Extraction and Quantification
2.11. Immunoblotting
2.12. RNA Sequencing
3. Results
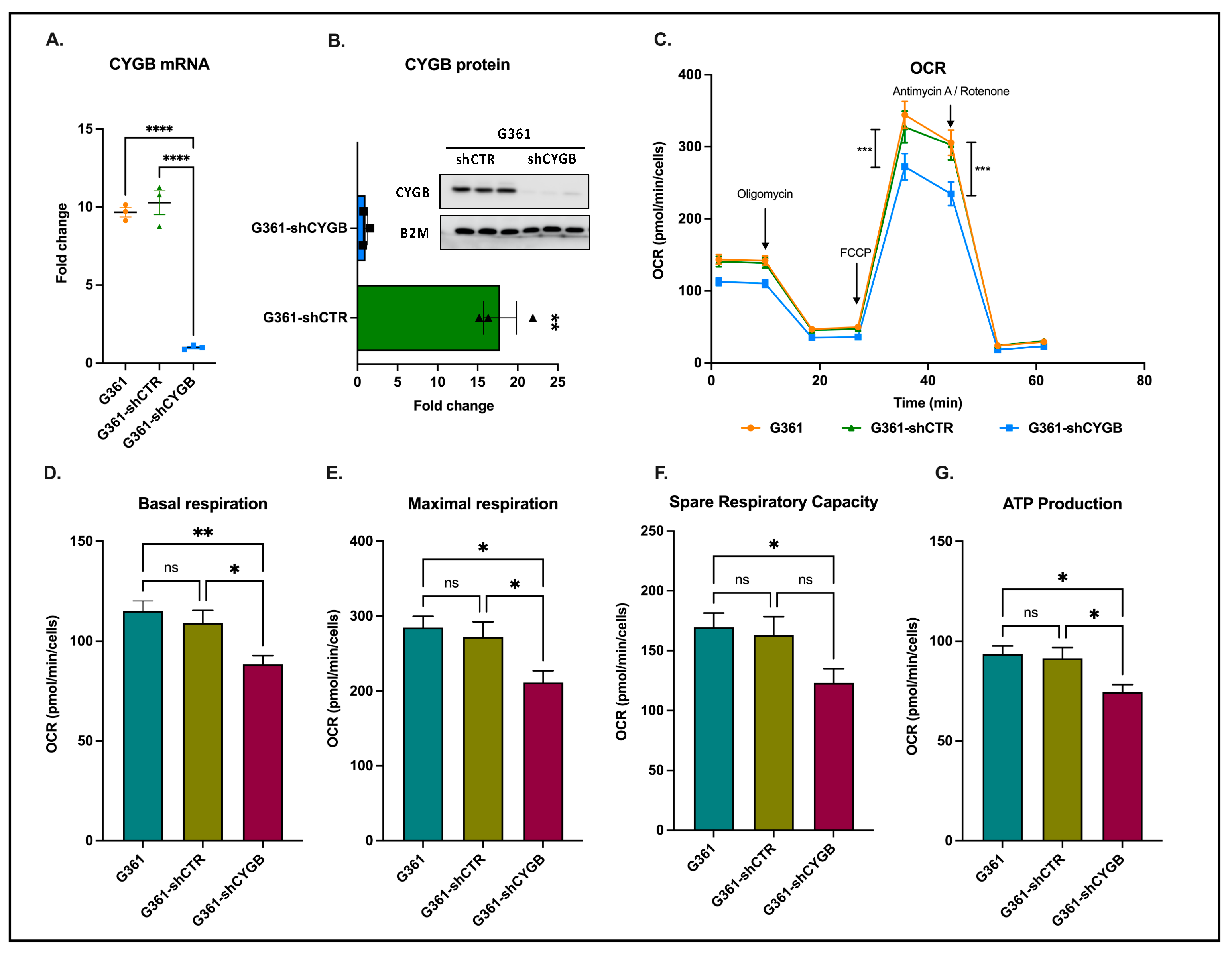
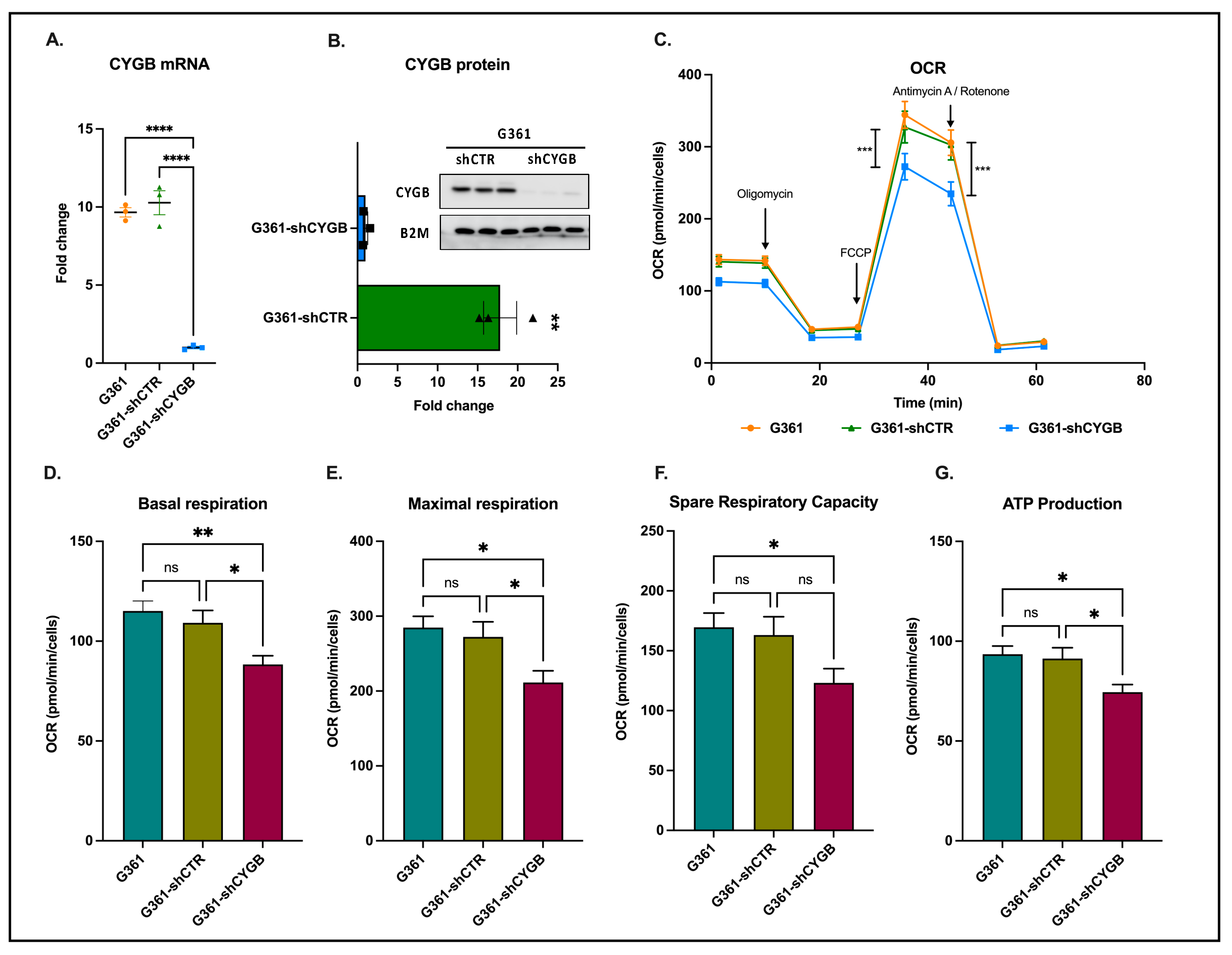
3.1. CYGB Knockdown Influences Cellular Bioenergetics
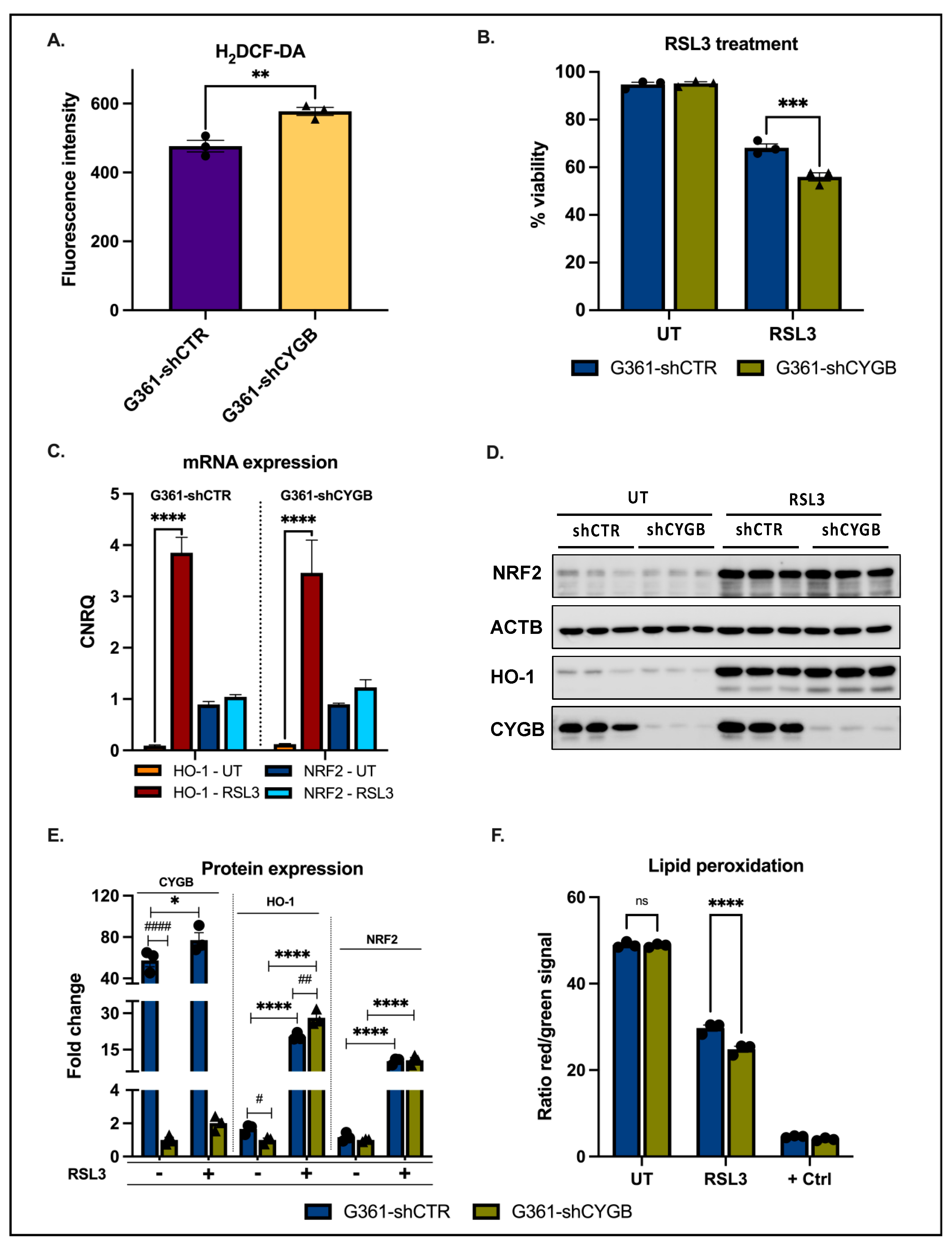
3.2. CYGB Expression Affects ROS Homeostasis and Ferroptosis Sensitivity
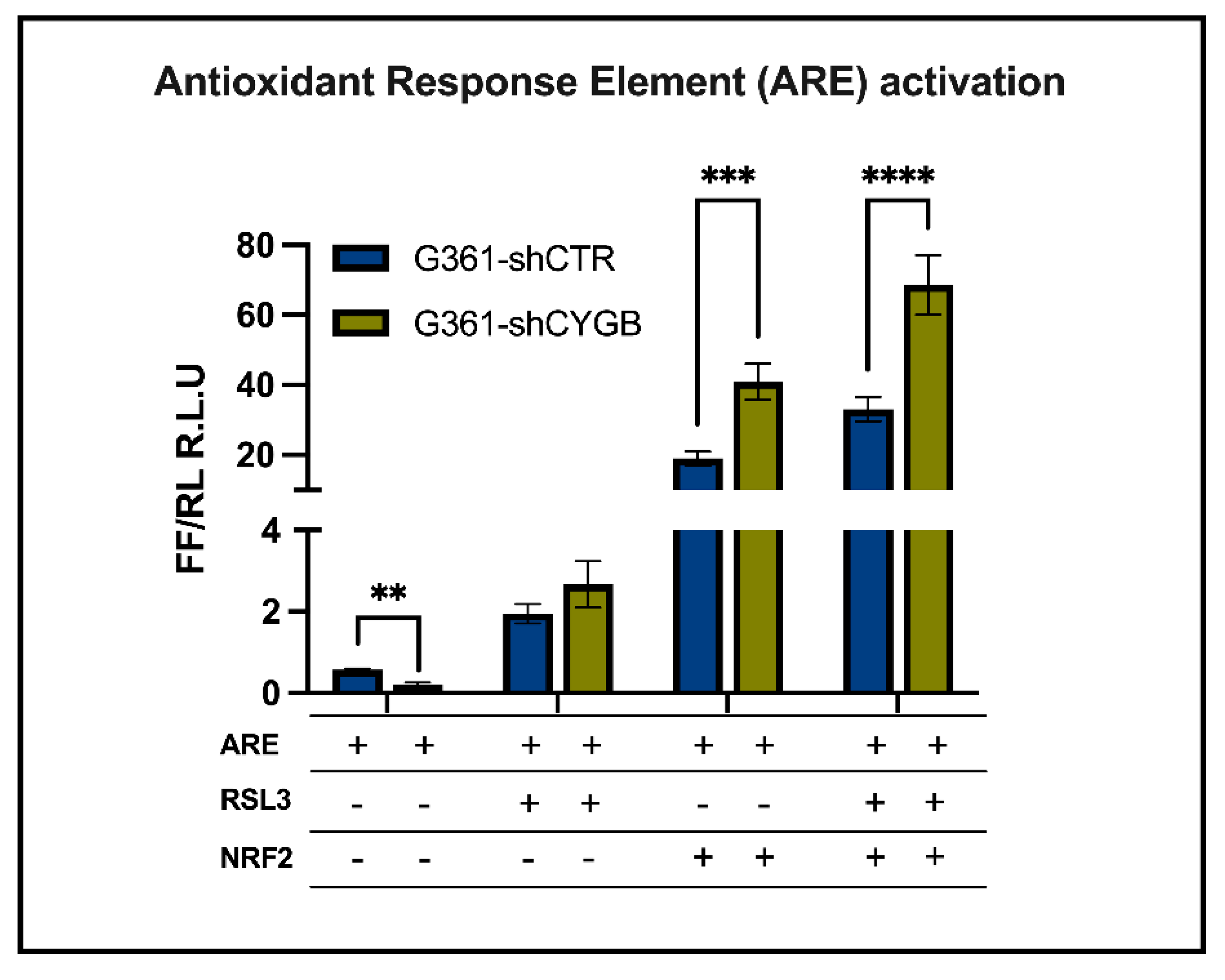
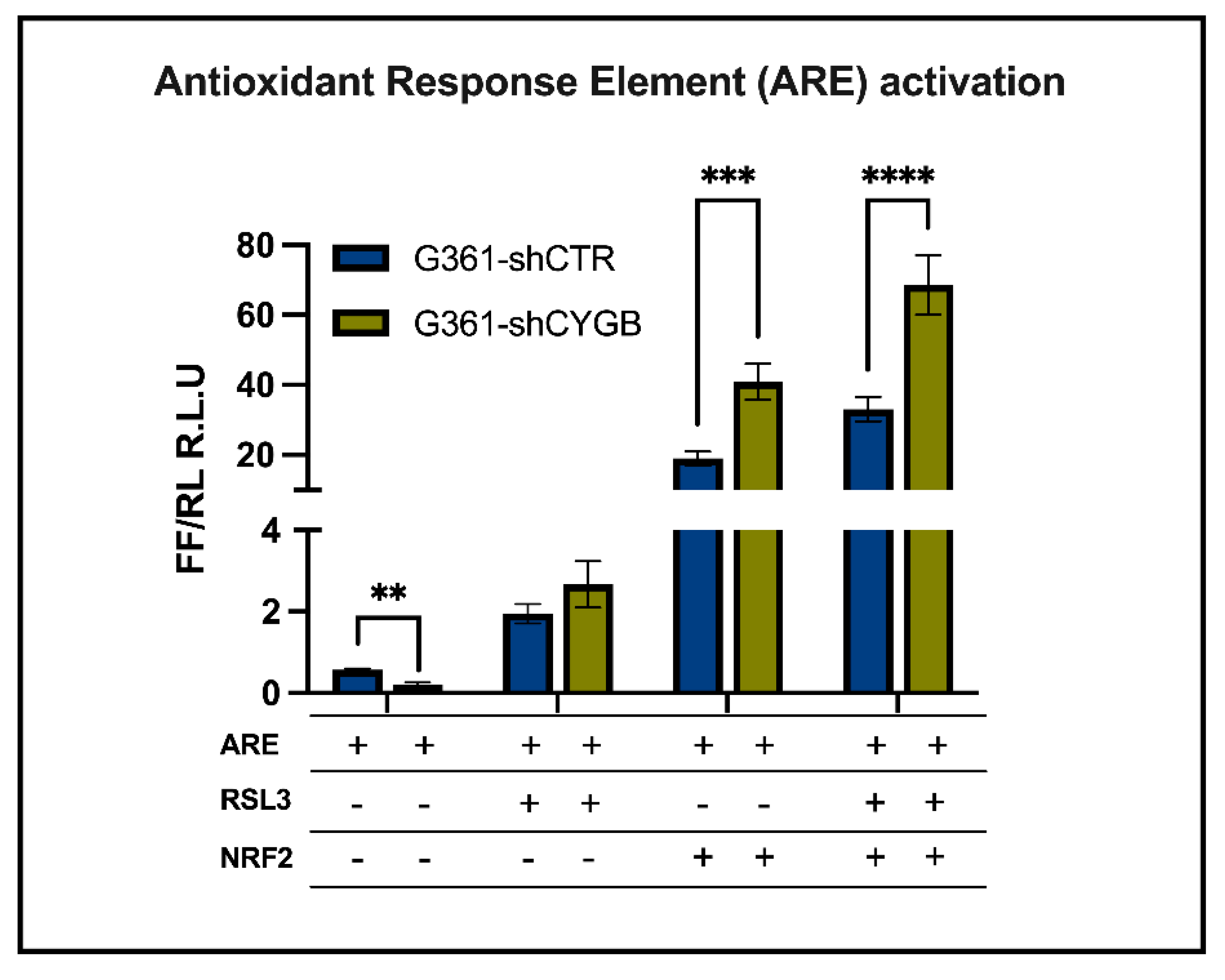
3.3. CYGB-Dependent Antioxidant Response Element-Driven Luciferase Activity Is Increased upon RSL3 Treatment and NRF2 Overexpression
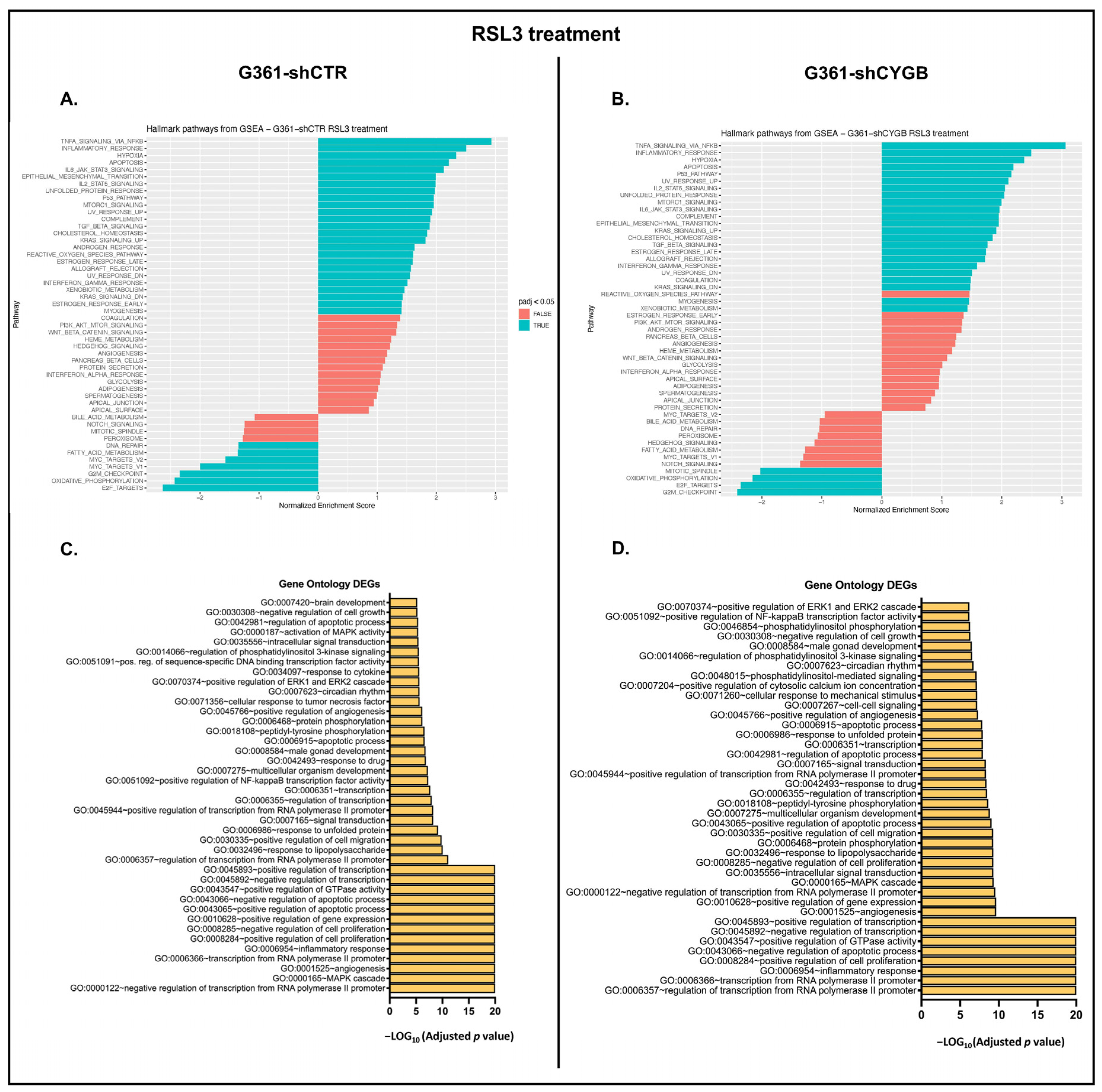
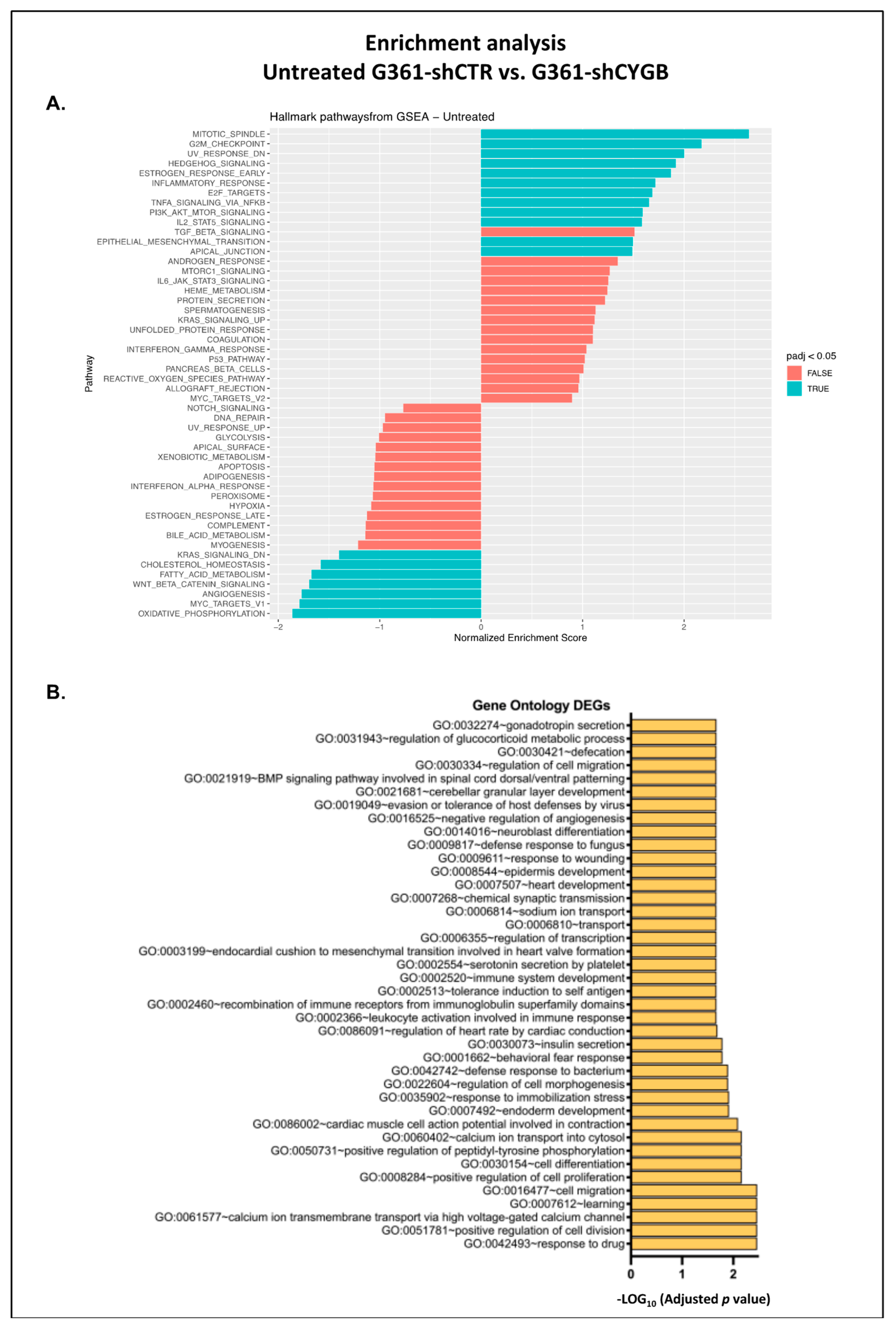
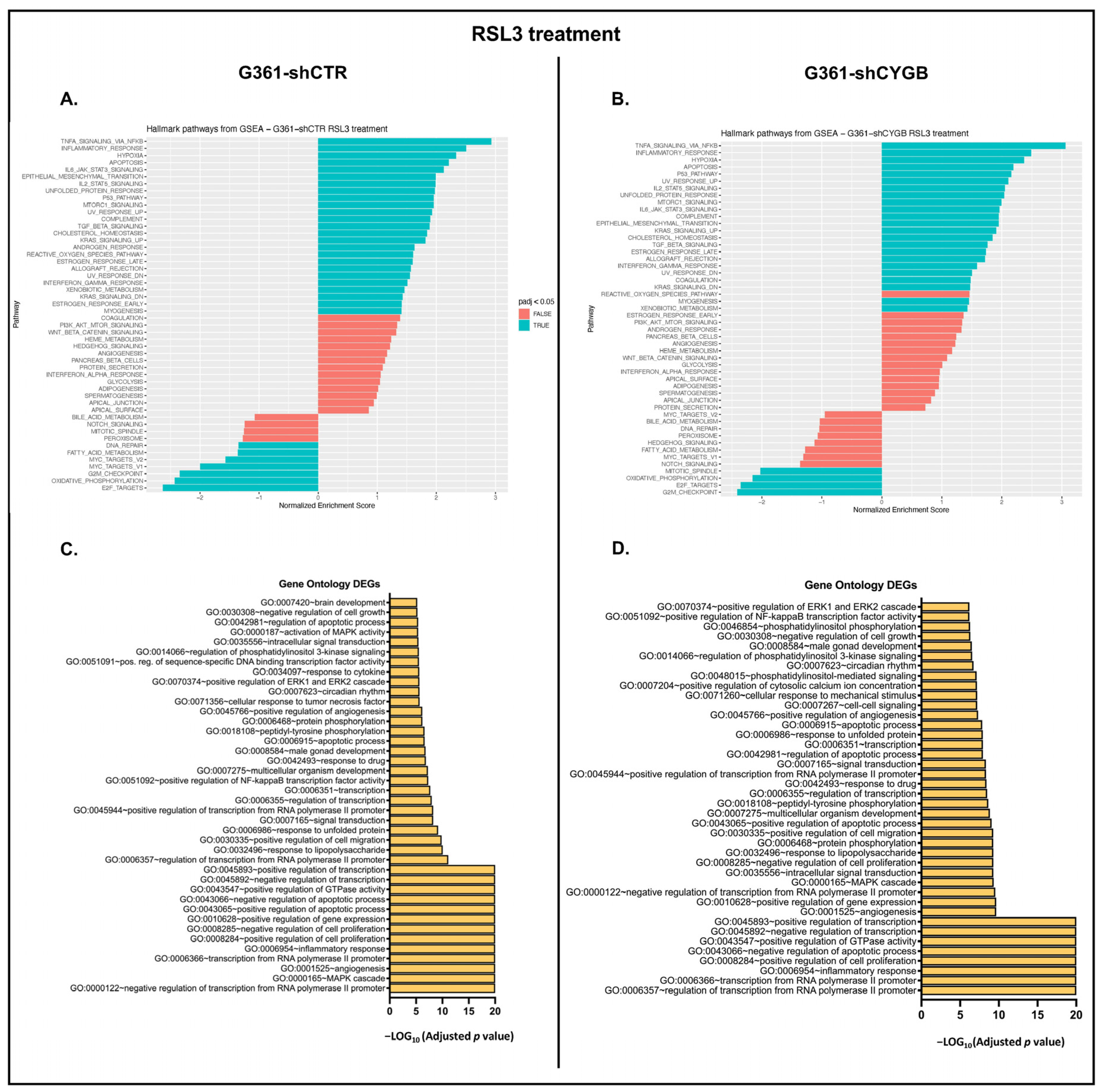
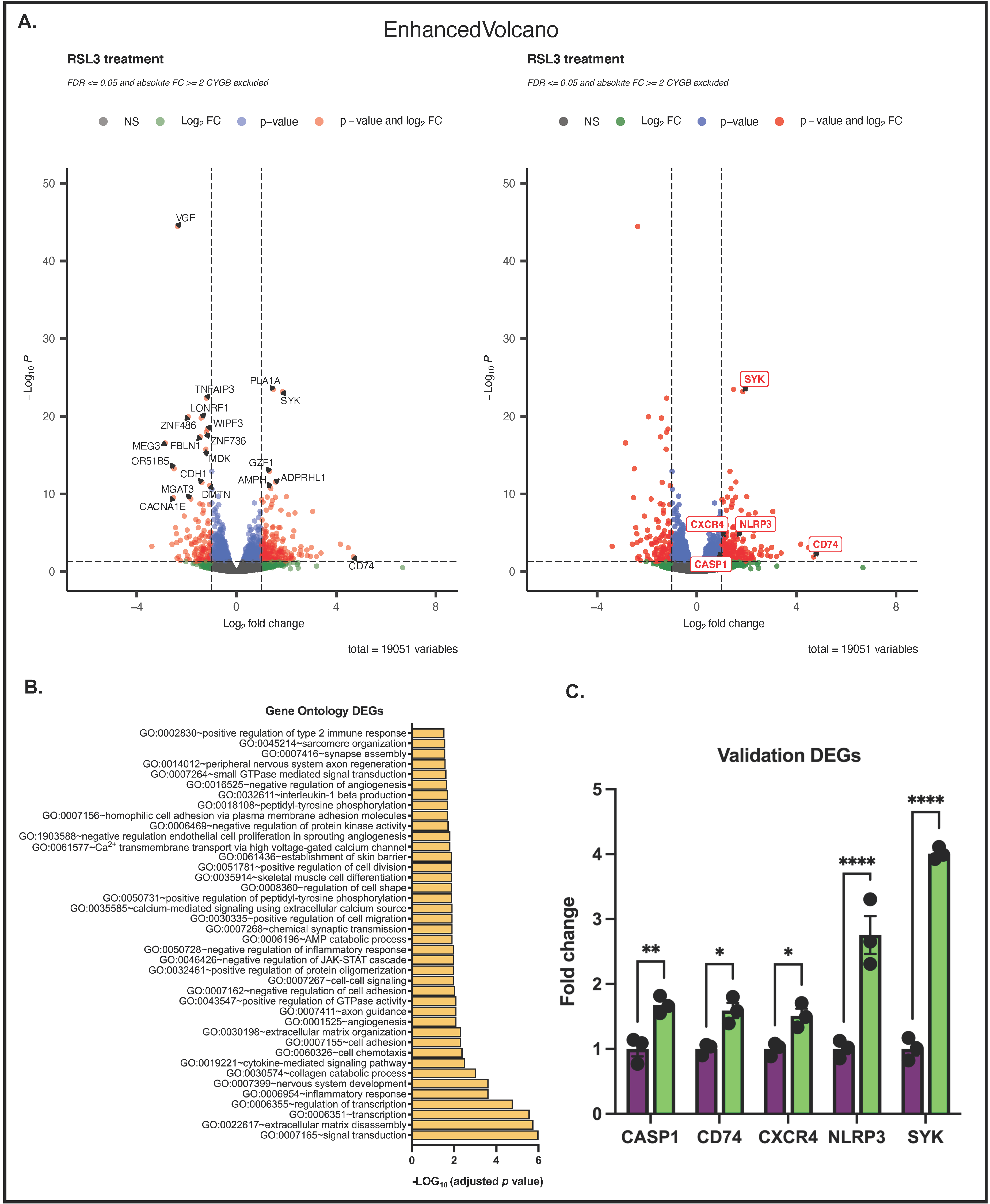
3.4. RNA Sequencing Analysis of RSL3-Treated G361-shCTR and G361-shCYGB versus Control
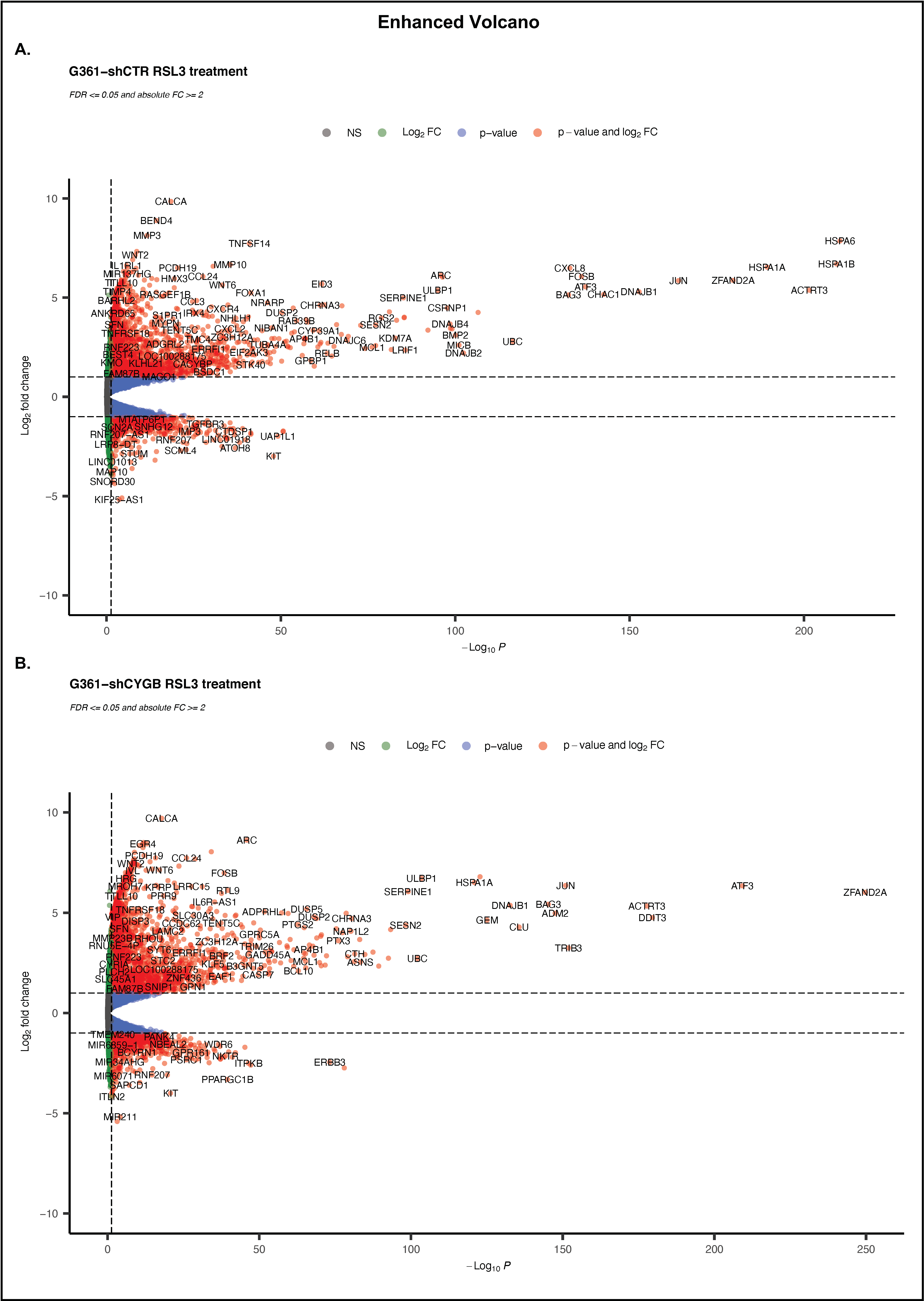
3.5. Differences between RSL3-Treated G361-shCTR and G361-shCYGB Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Sample, A.; He, Y.Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef]
- Curti, B.D.; Faries, M.B. Recent advances in the treatment of melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef]
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of immunotherapy with targeted therapy: Theory and practice in metastatic melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Moreira, A.; Heinzerling, L.; Bhardwaj, N.; Friedlander, P. Current melanoma treatments: Where do we stand? Cancers 2021, 13, 221. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Zweier, J.L.; Ilangovan, G. Regulation of nitric oxide metabolism and vascular tone by cytoglobin. Antioxid. Redox Signal. 2020, 32, 1172–1187. [Google Scholar] [CrossRef]
- Reeder, B.J.; Ukeri, J. Strong modulation of nitrite reductase activity of cytoglobin by disulfide bond oxidation: Implications for nitric oxide homeostasis. Nitric Oxide 2018, 72, 16–23. [Google Scholar] [CrossRef]
- Zweier, J.L.; Hemann, C.; Kundu, T.; Ewees, M.G.; Khaleel, S.A.; Samouilov, A.; Ilangovan, G.; El-Mahdy, M.A. Cytoglobin has potent superoxide dismutase function. Proc. Natl. Acad. Sci. USA 2021, 118, e2105053118. [Google Scholar] [CrossRef]
- Tejero, J.; Kapralov, A.A.; Baumgartner, M.P.; Sparacino-Watkins, C.E.; Anthonymutu, T.S.; Vlasova, I.I.; Camacho, C.J.; Gladwin, M.T.; Bayir, H.; Kagan, V.E. Peroxidase activation of cytoglobin by anionic phospholipids: Mechanisms and consequences. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Randi, E.B.; Vervaet, B.; Tsachaki, M.; Porto, E.; Vermeylen, S.; Lindenmeyer, M.T.; Thuy, L.T.T.; Cohen, C.D.; Devuyst, O.; Kistler, A.D.; et al. The antioxidative role of cytoglobin in podocytes: Implications for a role in chronic kidney disease. Antioxid. Redox Signal. 2020, 32, 1155–1171. [Google Scholar] [CrossRef]
- De Backer, J.; Maric, D.; Bosman, M.; Dewilde, S.; Hoogewijs, D. A reliable set of reference genes to normalize oxygen-dependent cytoglobin gene expression levels in melanoma. Sci. Rep. 2021, 11, 10879. [Google Scholar] [CrossRef]
- Fujita, Y.; Koinuma, S.; De Velasco, M.A.; Bolz, J.; Togashi, Y.; Terashima, M.; Hayashi, H.; Matsuo, T.; Nishio, K. Melanoma transition is frequently accompanied by a loss of cytoglobin expression in melanocytes: A novel expression site of cytoglobin. PLoS ONE 2014, 9, e94772. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Stastny, V.; Okumura, N.; Girard, L.; Xie, Y.; Prinsen, C.; Thunnissen, F.B.; Wistuba, I.I.; Czerniak, B.; Frenkel, E.; et al. Cytoglobin, the newest member of the globin family, functions as a tumor suppressor gene. Cancer Res. 2008, 68, 7448–7456. [Google Scholar] [CrossRef] [Green Version]
- McRonald, F.E.; Liloglou, T.; Xinarianos, G.; Hill, L.; Rowbottom, L.; Langan, J.E.; Ellis, A.; Shaw, J.M.; Field, J.K.; Risk, J.M. Down-regulation of the cytoglobin gene, located on 17q25, in tylosis with oesophageal cancer (TOC): Evidence for trans-allele repression. Hum. Mol. Genet. 2006, 15, 1271–1277. [Google Scholar] [CrossRef]
- Shaw, R.J.; Omar, M.M.; Rokadiya, S.; Kogera, F.A.; Lowe, D.; Hall, G.L.; Woolgar, J.A.; Homer, J.; Liloglou, T.; Field, J.K.; et al. Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer. Br. J. Cancer 2009, 101, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Presneau, N.; Dewar, K.; Forgetta, V.; Provencher, D.; Mes-Masson, A.M.; Tonin, P.N. Loss of heterozygosity and transcriptome analyses of a 1.2 Mb candidate ovarian cancer tumor suppressor locus region at 17q25.1-q25.2. Mol. Carcinog. 2005, 43, 141–154. [Google Scholar] [CrossRef]
- Wojnarowicz, P.M.; Provencher, D.M.; Mes-Masson, A.M.; Tonin, P.N. Chromosome 17q25 genes, RHBDF2 and CYGB, in ovarian cancer. Int. J. Oncol. 2012, 40, 1865–1880. [Google Scholar] [PubMed] [Green Version]
- Hubers, A.J.; Heideman, D.A.; Burgers, S.A.; Herder, G.J.; Sterk, P.J.; Rhodius, R.J.; Smit, H.J.; Krouwels, F.; Welling, A.; Witte, B.I.; et al. DNA hypermethylation analysis in sputum for the diagnosis of lung cancer: Training validation set approach. Br. J. Cancer 2015, 112, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schörg, A.; Santambrogio, S.; Platt, J.L.; Schödel, J.; Lindenmeyer, M.T.; Cohen, C.D.; Schrödter, K.; Mole, D.R.; Wenger, R.H.; Hoogewijs, D. Destruction of a distal hypoxia response element abolishes trans-activation of the PAG1 gene mediated by HIF-independent chromatin looping. Nucleic Acids Res. 2015, 43, 5810–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koay, T.W.; Osterhof, C.; Orlando, I.M.C.; Keppner, A.; Andre, D.; Yousefian, S.; Suárez Alonso, M.; Correia, M.; Markworth, R.; Schödel, J.; et al. Androglobin gene expression patterns and FOXJ1-dependent regulation indicate its functional association with ciliogenesis. J. Biol. Chem. 2021, 296, 100291. [Google Scholar] [CrossRef]
- Storti, F.; Santambrogio, S.; Crowther, L.M.; Otto, T.; Abreu-Rodríguez, I.; Kaufmann, M.; Hu, C.J.; Dame, C.; Fandrey, J.; Wenger, R.H.; et al. A novel distal upstream hypoxia response element regulating oxygen-dependent erythropoietin gene expression. Haematologica 2014, 99, e45–e48. [Google Scholar] [CrossRef] [Green Version]
- Keppner, A.; Correia, M.; Santambrogio, S.; Koay, T.W.; Maric, D.; Osterhof, C.; Winter, D.V.; Clerc, A.; Stumpe, M.; Chalmel, F.; et al. Androglobin, a chimeric mammalian globin, is required for male fertility. eLife 2022, 11, e72374. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Korotkevich, G.; Sukhov, V.; Budin, N.; Shpak, B.; Artyomov, M.N.; and Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2021, 060012. [Google Scholar]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Huntley, R.P.; Sawford, T.; Mutowo-Meullenet, P.; Shypitsyna, A.; Bonilla, C.; Martin, M.J.; O’Donovan, C. The GOA database: Gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015, 43, D1057–D1063. [Google Scholar] [CrossRef]
- De Backer, J.; Lin, A.; Berghe, W.V.; Bogaerts, A.; Hoogewijs, D. Cytoglobin inhibits non-thermal plasma-induced apoptosis in melanoma cells through regulation of the NRF2-mediated Antioxid.ant response. Redox Biol. 2022, 55, 102399. [Google Scholar] [CrossRef]
- Yoshizato, K.; Thuy, L.T.T.; Shiota, G.; Kawada, N. Discovery of cytoglobin and its roles in physiology and pathology of hepatic stellate cells. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2016, 92, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Keppner, A.; Maric, D.; Correia, M.; Koay, T.W.; Orlando, I.M.C.; Vinogradov, S.N.; Hoogewijs, D. Lessons from the post-genomic era: Globin diversity beyond oxygen binding and transport. Redox Biol. 2020, 37, 101687. [Google Scholar] [CrossRef]
- Kamga, C.; Krishnamurthy, S.; Shiva, S. Myoglobin and mitochondria: A relationship bound by oxygen and nitric oxide. Nitric Oxide Biol. Chem. 2012, 26, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Rowland, L.K.; Campbell, P.S.; Mavingire, N.; Wooten, J.V.; McLean, L.; Zylstra, D.; Thorne, G.; Daly, D.; Boyle, K.; Whang, S.; et al. Putative tumor suppressor cytoglobin promotes aryl hydrocarbon receptor ligand-mediated triple negative breast cancer cell death. J. Cell Biochem. 2019, 120, 6004–6014. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; and Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Canseco, D.C.; Manda, S.M.; Shelton, J.M.; Chirumamilla, R.R.; Goetsch, S.C.; Ye, Q.; Gerard, R.D.; Schneider, J.W.; Richardson, J.A.; et al. Cytoglobin modulates myogenic progenitor cell viability and muscle regeneration. Proc. Natl. Acad. Sci. USA 2014, 111, E129–E138. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, X.; Jourd’heuil, F.L.; Qu, S.; Devejian, N.; Bennett, E.; Jourd’heuil, D.; Cai, C. Cytoglobin promotes cardiac progenitor cell survival against oxidative stress via the upregulation of the NFκB/iNOS Signal. pathway and nitric oxide production. Sci. Rep. 2017, 7, 10754. [Google Scholar] [CrossRef]
- Thorne, L.S.; Rochford, G.; Williams, T.D.; Southam, A.D.; Rodriguez-Blanco, G.; Dunn, W.B.; Hodges, N.J. Cytoglobin protects cancer cells from apoptosis by regulation of mitochondrial cardiolipin. Sci. Rep. 2021, 11, 985. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A dual role of heme oxygenase-1 in cancer cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme oxygenase 1 in the nervous system: Does it favor neuronal cell survival or induce neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Fan, F.Y.; Deng, R.; Yi, H.; Sun, H.P.; Zeng, Y.; He, G.C.; Su, Y. The inhibitory effect of MEG3/miR-214/AIFM2 axis on the growth of T-cell lymphoblastic lymphoma. Int. J. Oncol. 2017, 51, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhu, L.; Li, Y.; Zheng, Z.; Lin, X.; Yang, C. LncRNA MEG3 promotes melanoma growth, metastasis and formation through modulating miR-21/E-cadherin axis. Cancer Cell Int. 2020, 20, 12. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Barbacid, M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Richmond, A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006, 19, 112–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug. Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF Signal. transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starlets, D.; Gore, Y.; Binsky, I.; Haran, M.; Harpaz, N.; Shvidel, L.; Becker-Herman, S.; Berrebi, A.; Shachar, I. Cell-surface CD74 initiates a Signal.ing cascade leading to cell proliferation and survival. Blood 2006, 107, 4807–4816. [Google Scholar] [CrossRef] [PubMed]
- Lantner, F.; Starlets, D.; Gore, Y.; Flaishon, L.; Yamit-Hezi, A.; Dikstein, R.; Leng, L.; Bucala, R.; Machluf, Y.; Oren, M.; et al. CD74 induces TAp63 expression leading to B-cell survival. Blood 2007, 110, 4303–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, Y.; Starlets, D.; Maharshak, N.; Becker-Herman, S.; Kaneyuki, U.; Leng, L.; Bucala, R.; Shachar, I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex*. J. Biol. Chem. 2008, 283, 2784–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF Signaling. Cell. Signal. 2019, 57, 76–88. [Google Scholar] [CrossRef]
- Li, T.; Sun, H.; Li, Y.; Su, L.; Jiang, J.; Liu, Y.; Jiang, N.; Huang, R.; Zhang, J.; Peng, Z. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI. Cell Death Discov. 2022, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef]
- Wen, J.; Wu, Y.; Wei, W.; Li, Z.; Wang, P.; Zhu, S.; Dong, W. Protective effects of recombinant human cytoglobin against chronic alcohol-induced liver disease in vivo and in vitro. Sci. Rep. 2017, 7, 41647. [Google Scholar] [CrossRef] [Green Version]
- Yassin, M.; Kissow, H.; Vainer, B.; Joseph, P.D.; Hay-Schmidt, A.; Olsen, J.; Pedersen, A.E. Cytoglobin affects tumorigenesis and the expression of ulcerative colitis-associated genes under chemically induced colitis in mice. Sci. Rep. 2018, 8, 6905. [Google Scholar] [CrossRef] [Green Version]
- Gomes, B.R.B.; de Sousa, G.L.S.; Ott, D.; Murgott, J.; de Sousa, M.V.; de Souza, P.E.N.; Roth, J.; Veiga-Souza, F.H. Cytoglobin attenuates neuroinflammation in lipopolysaccharide-activated primary preoptic area cells via NF-κB pathway inhibition. Front. Mol. Neurosci. 2019, 12, 307. [Google Scholar] [CrossRef] [Green Version]
- Ou, L.; Li, X.; Chen, B.; Ge, Z.; Zhang, J.; Zhang, Y.; Cai, G.; Li, Z.; Wang, P.; Dong, W. Recombinant human cytoglobin prevents atherosclerosis by regulating lipid metabolism and oxidative stress. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeder, B.J.; Svistunenko, D.A.; Wilson, M.T. Lipid binding to cytoglobin leads to a change in haem co-ordination: A role for cytoglobin in lipid Signal.ling of oxidative stress. Biochem. J. 2011, 434, 483–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Guo, T.; Zhang, X. Pyroptosis in cancer: Friend or foe? Cancers 2021, 13, 3620. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qin, X.; Liang, J.; Ge, P. Induction of pyroptosis: A promising strategy for cancer treatment. Front. Oncol. 2021, 11, 635774. [Google Scholar] [CrossRef]
- Cao, Z.; Qin, H.; Huang, Y.; Zhao, Y.; Chen, Z.; Hu, J.; Gao, Q. Crosstalk of pyroptosis, ferroptosis, and mitochondrial aldehyde dehydrogenase 2-related mechanisms in sepsis-induced lung injury in a mouse model. Bioengineered 2022, 13, 4810–4820. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 2018, 24, 97–108.e4. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Zhang, J.-Y.; Liu, X.-S.; Chen, H.-A.; Ai, Y.-L.; Cheng, K.; Sun, R.-Y.; Zhou, D.; Han, J.; Wu, Q. Tom20 senses iron-activated ROS Signal.ing to promote melanoma cell pyroptosis. Cell Res. 2018, 28, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Backer, J.; Maric, D.; Zuhra, K.; Bogaerts, A.; Szabo, C.; Vanden Berghe, W.; Hoogewijs, D. Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response. Antioxidants 2022, 11, 1548. https://doi.org/10.3390/antiox11081548
De Backer J, Maric D, Zuhra K, Bogaerts A, Szabo C, Vanden Berghe W, Hoogewijs D. Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response. Antioxidants. 2022; 11(8):1548. https://doi.org/10.3390/antiox11081548
Chicago/Turabian StyleDe Backer, Joey, Darko Maric, Karim Zuhra, Annemie Bogaerts, Csaba Szabo, Wim Vanden Berghe, and David Hoogewijs. 2022. "Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response" Antioxidants 11, no. 8: 1548. https://doi.org/10.3390/antiox11081548
APA StyleDe Backer, J., Maric, D., Zuhra, K., Bogaerts, A., Szabo, C., Vanden Berghe, W., & Hoogewijs, D. (2022). Cytoglobin Silencing Promotes Melanoma Malignancy but Sensitizes for Ferroptosis and Pyroptosis Therapy Response. Antioxidants, 11(8), 1548. https://doi.org/10.3390/antiox11081548