
Mitochondrial Glrx2 Knockout Augments Acetaminophen-Induced Hepatotoxicity in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Mice construction and Genotyping
2.3. Animal Treatment
2.4. Liver Injury Evaluation
2.5. Total Glutathione Assay
2.6. TrxR Activity Assay
2.7. Protein Expression Analysis
2.8. Protein Redox Status Detection
2.9. GSH/GSSG Assay
2.10. AIF Nuclear Translocation Detection
2.11. MDA Level Detection
2.12. Statistical Analysis
3. Results
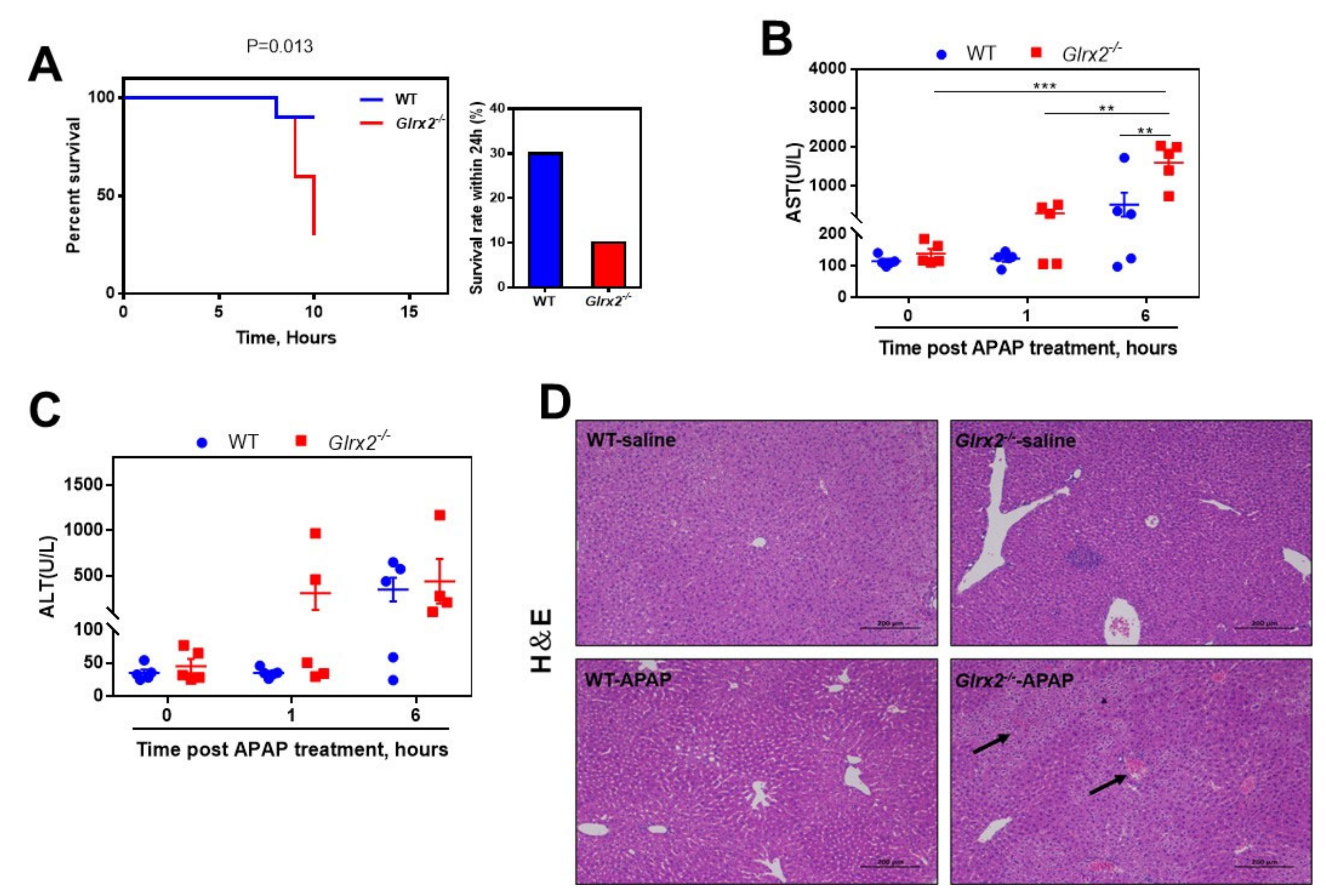
3.1. Glrx2 Deficiency Exacerbated APAP Caused Acute Hepatotoxicity
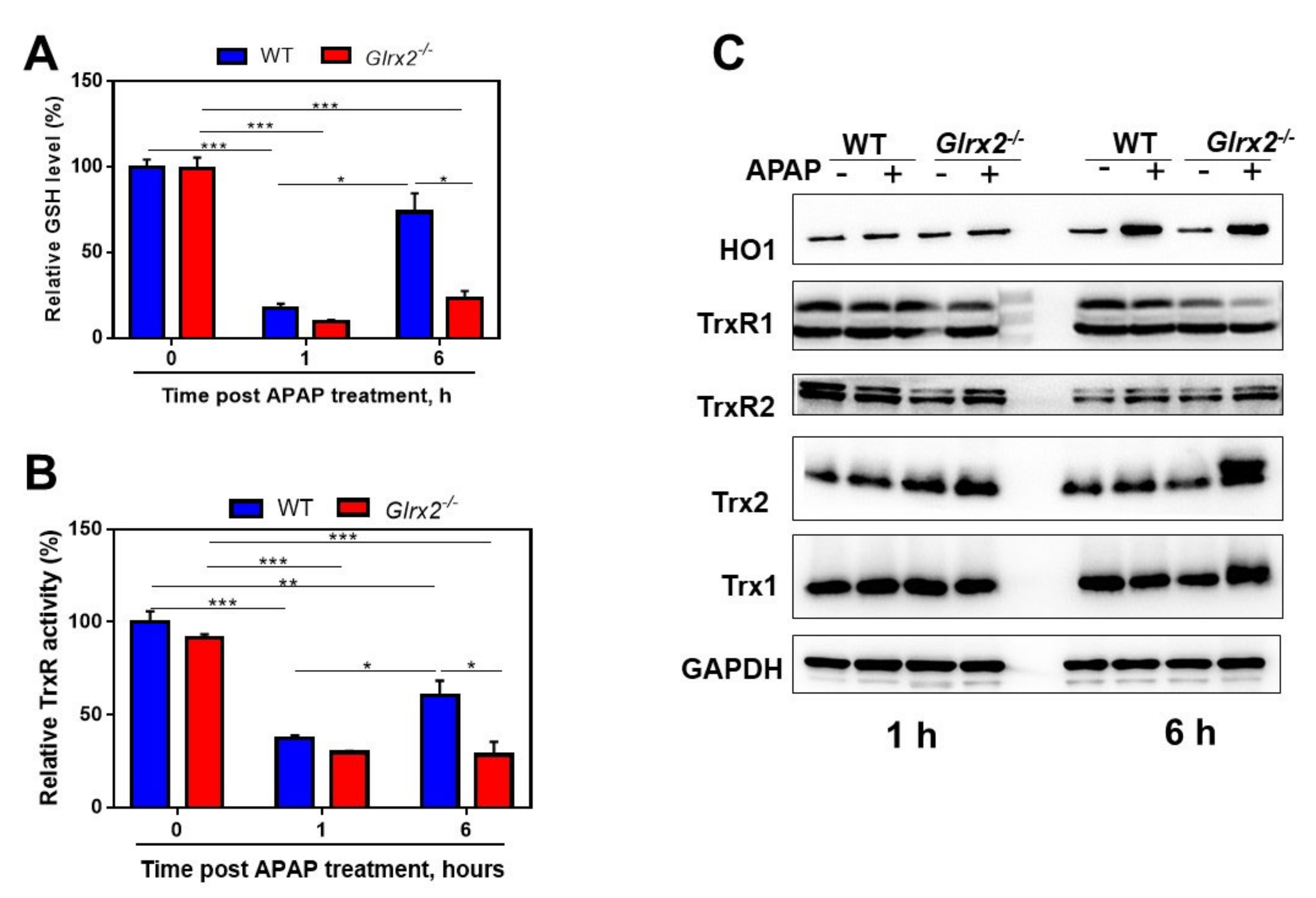
3.2. APAP Treatment Decreased Total Glutathione Content and TrxR Activity and Triggered Nrf2 Activation
3.3. Decreased GSH/GSSG Ratio in Glrx2−/− Mice after APAP Treatment Weaken Nrf2 Activation
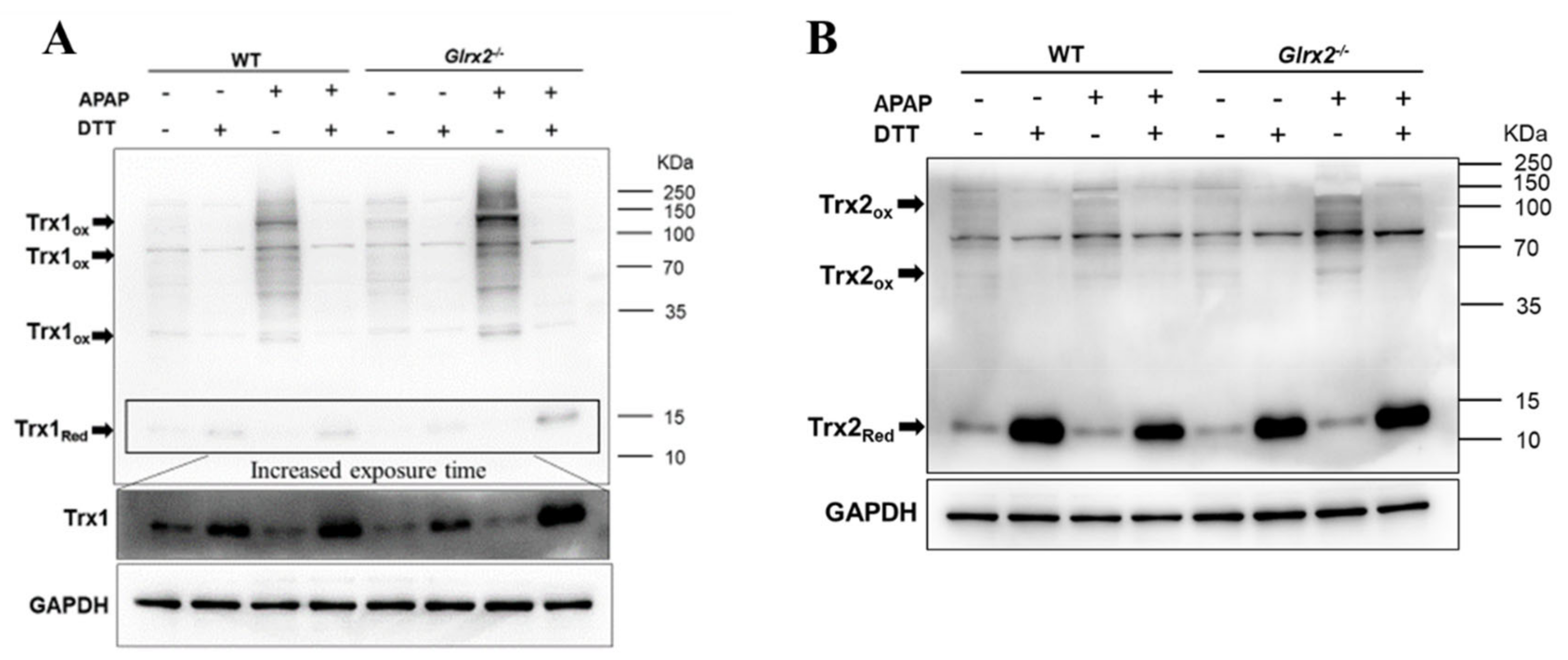
3.4. Elevated Trx Oxidation in Glrx2−/− Mice after APAP Treatment Weaken Nrf2 Activation
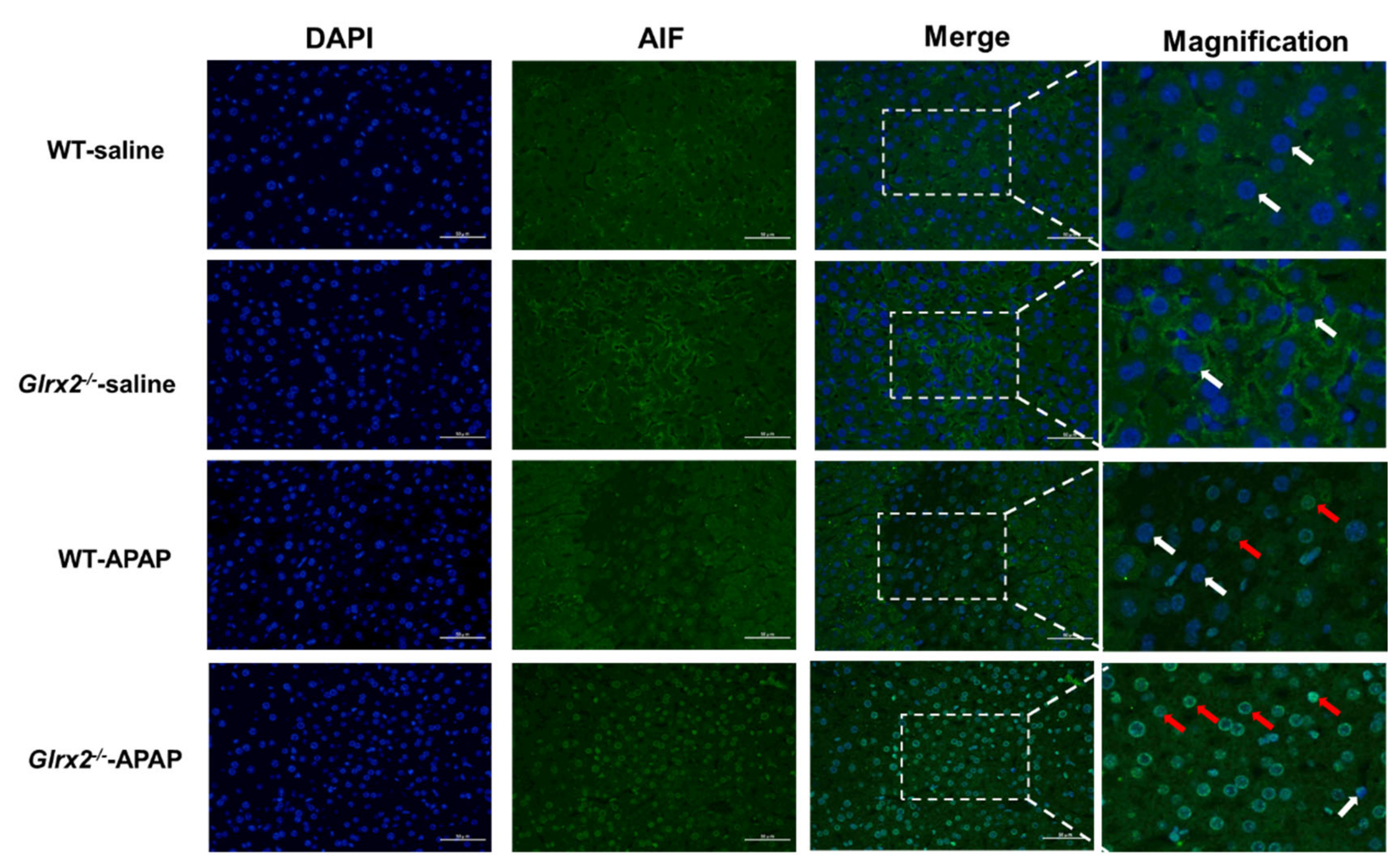
3.5. Glrx2 Deficiency Increased AIF Nuclear Translocation and MDA Level in Liver after APAP Administration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chalasani, N.P.; Maddur, H.; Russo, M.W.; Wong, R.J.; Reddy, K.R. ACG clinical guideline: Diagnosis and management of idiosyncratic drug-induced liver injury. Am. J. Gastroenterol. 2021, 116, 878–898. [Google Scholar] [CrossRef] [PubMed]
- Nithiyanandam, S.; Prince, S.E. Toxins mechanism in instigating hepatotoxicity. Toxin Rev. 2020, 40, 616–631. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ghosh, S.; Sil, P.C. Amelioration of aspirin induced oxidative impairment and apoptotic cell death by a novel antioxidant protein molecule isolated from the Herb Phyllanthus niruri. PLoS ONE 2014, 9, e89026. [Google Scholar] [CrossRef]
- Antherieu, S.; Bachour-El Azzi, P.; Dumont, J.; Abdel-Razzak, Z.; Guguen-Guillouzo, C.; Fromenty, B.; Robin, M.-A.; Guillouzo, A. Oxidative stress plays a major role in chlorpromazine-induced cholestasis in human HepaRG cells. Hepatology 2013, 57, 1518–1529. [Google Scholar] [CrossRef]
- Chan, K.M.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef]
- Larson, A.M.; Polson, J.; Fontana, R.J.; Davern, T.J.; Lalani, E.; Hynan, L.S.; Reisch, J.S.; Schiodt, F.V.; Ostapowicz, G.; Shakil, A.O.; et al. Acetaminophen-induced acute liver failure: Results of a United States multicenter, prospective study. Hepatology 2005, 42, 1364–1372. [Google Scholar] [CrossRef]
- Bunchorntavakul, C.; Reddy, K.R. Acetaminophen (APAP or N-Acetyl-p-Aminophenol) and Acute Liver Failure. Clin. Liver Dis. 2018, 22, 325–346. [Google Scholar] [CrossRef]
- Yan, M.Z.; Huo, Y.Z.; Yin, S.T.; Hu, H.B. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J. Clin. Transl. Res. 2017, 3, 157–169. [Google Scholar] [CrossRef]
- Iverson, S.V.; Eriksson, S.; Xu, J.; Prigge, J.R.; Talago, E.A.; Meade, T.A.; Meade, E.S.; Capecchi, M.R.; Arnér, E.S.; Schmidt, E.E. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic. Biol. Med. 2013, 63, 369–380. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; Mcgill, M.R.; Ramachandran, A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: Lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Liu, A.M.; Novoselov, S.V.; Krysan, K.; Sun, Q.A.; Kryukov, V.M.; Kryukov, G.V.; Lou, M.F. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J. Biol. Chem. 2001, 276, 30374–30380. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef] [PubMed]
- Stroher, E.; Millar, A.H. The biological roles of glutaredoxins. Biochem. J. 2012, 446, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins—Implications for mitochondrial redox regulation and antioxidant defense. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Vergnolle, O.; Lonn, M.E.; Hudemann, C.; Bill, E.; Holmgren, A. Characterization of human glutaredoxin 2 as iron-sulfur protein: A possible role as redox sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 8168–8173. [Google Scholar] [CrossRef]
- Ouyang, Y.F.; Peng, Y.; Li, J.; Holmgren, A.; Lu, J. Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems. Metallomics 2018, 10, 218–228. [Google Scholar] [CrossRef]
- Lee, D.W.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Andersen, J.K. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: Implications for Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 2083–2094. [Google Scholar] [CrossRef]
- Jung, E.U.; Yoon, J.H.; Lee, Y.J.; Lee, J.H.; Kim, B.H.; Yu, S.J.; Myung, S.J.; Kim, Y.J.; Lee, H.S. Hypoxia and retinoic acid-inducible NDRG1 expression is responsible for doxorubicin and retinoic acid resistance in hepatocellular carcinoma cells. Cancer Lett. 2010, 298, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Xu, W.M. Glutaredoxin 2 (GRX2) deficiency exacerbates high fat diet (HFD)-induced insulin resistance, inflammation and mitochondrial dysfunction in brain injury: A mechanism involving GSK-3 beta. Biomed. Pharmacother. 2019, 118, 108940. [Google Scholar] [CrossRef]
- Young, A.; Gardiner, D.; Kuksal, N.; Gill, R.; O’brien, M.; Mailloux, R.J. Deletion of the glutaredoxin-2 gene protects mice from diet-induced weight gain, which correlates with increased mitochondrial respiration and proton leaks in skeletal muscle. Antioxid. Redox Signal. 2019, 31, 1272–1288. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lin, L.; Giblin, F.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011, 51, 2108–2117. [Google Scholar] [CrossRef]
- Chalker, J.; Gardiner, D.; Kuksal, N.; Mailloux, R.J. Characterization of the impact of glutaredoxin-2 (GRX2) deficiency on superoxide/hydrogen peroxide release from cardiac and liver mitochondria. Redox Biol. 2018, 15, 216–227. [Google Scholar] [CrossRef]
- Scalcon, V.; Folda, A.; Lupo, M.G.; Tonolo, F.; Pei, N.; Battisti, I.; Ferri, N.; Arrigoni, G.; Bindoli, A.; Holmgren, A.; et al. Mitochondrial depletion of glutaredoxin 2 induces metabolic dysfunction-associated fatty liver disease in mice. Redox Biol. 2022, 51, 102277. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef]
- Arnaiz, S.L.; Llesuy, S.; Cutrin, J.C.; Boveris, A. Oxidative stress by acute acetaminophen administration in mouse-liver. Free. Radic. Biol. Med. 1995, 19, 303–310. [Google Scholar] [CrossRef]
- Li, J.; Cheng, P.; Li, S.F.; Zhao, P.F.; Han, B.; Ren, X.Y.; Zhong, J.L.; Lloyd, M.D.; Pourzand, C.; Holmgren, A.; et al. Selenium status in diet affects acetaminophen-induced hepatotoxicity via interruption of redox environment. Antioxid. Redox Signal. 2021, 34, 1355–1367. [Google Scholar] [CrossRef]
- Cheng, P.; Liu, H.; Li, Y.C.; Pi, P.L.; Jiang, Y.; Zang, S.Z.; Li, X.R.; Fu, A.L.; Ren, X.Y.; Xu, J.Q.; et al. Inhibition of thioredoxin reductase 1 correlates with platinum-based chemotherapeutic induced tissue injury. Biochem. Pharm. 2020, 175, 113873. [Google Scholar] [CrossRef]
- Adams, J.D.; Lauterburg, B.H.; Mitchell, J.R. Plasma glutathione and glutathione disulfide in the rat-Regulation and response to oxidative stress. J. Pharmacol. Exp. Ther. 1983, 227, 749–754. [Google Scholar] [PubMed]
- Motterlini, R.; Foresti, R.; Bassi, R.; Calabrese, V.; Clark, J.E.; Green, C.J. Endothelial heme oxygenase-1 induction by hypoxia—Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J. Biol. Chem. 2000, 275, 13613–13620. [Google Scholar] [CrossRef] [PubMed]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta-Gen. Subj. 2008, 1780, 1304–1317. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Wu, G.Y.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Watson, W.H.; Jones, D.P. Compartmentation of Nrf-2 redox control: Regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol. Sci. Off. J. Soc. Toxicol. 2004, 82, 308–317. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Thioredoxin system in cell death progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef] [PubMed]
- Shelar, S.B.; Kaminska, K.K.; Reddy, S.A.; Kumar, D.; Tan, C.T.; Yu, V.C.; Lu, J.; Holmgren, A.; Hagen, T.; Chew, E.H. Thioredoxin-dependent regulation of AIF-mediated DNA damage. Free Radic. Biol. Med. 2015, 87, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhang, X.; Wang, L.; Zhang, R.; Pu, X.; Wu, S.; Li, L.; Tong, P.; Wang, J.; Meng, Q.H.; et al. Inhibition of thioredoxin/thioredoxin reductase induces synthetic lethality in lung cancers with compromised glutathione homeostasis. Cancer Res. 2019, 79, 125–132. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Tang, X.; Wen, X.; Ren, X.; Zhang, H.; Du, Y.; Lu, J. Mitochondrial Glrx2 Knockout Augments Acetaminophen-Induced Hepatotoxicity in Mice. Antioxidants 2022, 11, 1643. https://doi.org/10.3390/antiox11091643
Li J, Tang X, Wen X, Ren X, Zhang H, Du Y, Lu J. Mitochondrial Glrx2 Knockout Augments Acetaminophen-Induced Hepatotoxicity in Mice. Antioxidants. 2022; 11(9):1643. https://doi.org/10.3390/antiox11091643
Chicago/Turabian StyleLi, Jing, Xuewen Tang, Xing Wen, Xiaoyuan Ren, Huihui Zhang, Yatao Du, and Jun Lu. 2022. "Mitochondrial Glrx2 Knockout Augments Acetaminophen-Induced Hepatotoxicity in Mice" Antioxidants 11, no. 9: 1643. https://doi.org/10.3390/antiox11091643