

Thermogenic Adipose Redox Mechanisms: Potential Targets for Metabolic Disease Therapies




Abstract
:1. Introduction
2. Adipose Tissue as a Modulator of Energy Homeostasis
2.1. Adipose Depots and Lipolysis
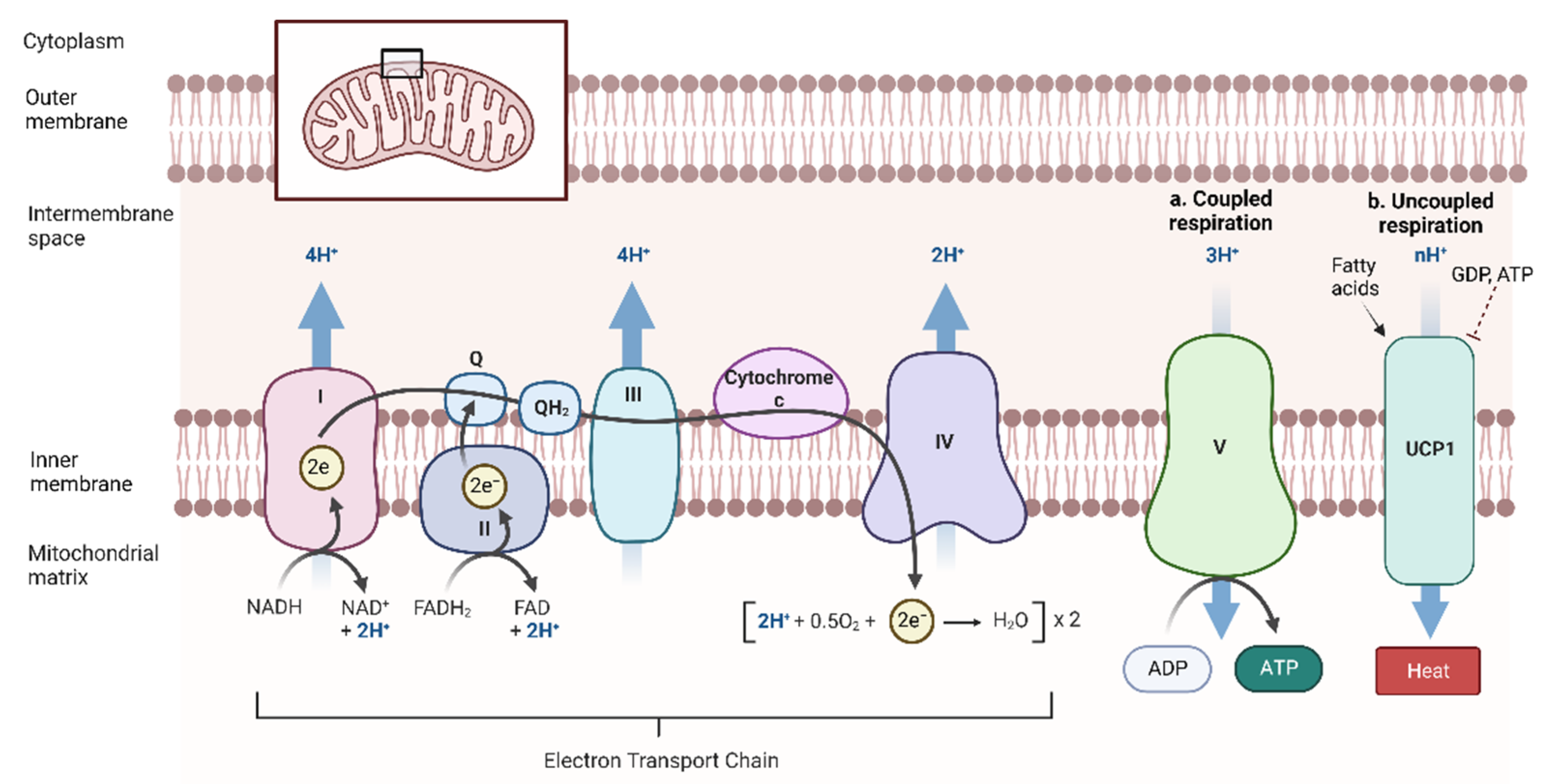
2.2. Beta-Oxidation and Coupled Respiration
2.3. Thermogenesis
3. Metabolic Redox Reactions in Thermogenic Adipose Tissue
3.1. Role of Reactive Metabolites
3.2. Antioxidant Defenses
3.2.1. Enzymatic Antioxidants
3.2.2. Non-enzymatic Antioxidants
3.3. Additional NST-Specific Redox Considerations
4. Manipulating Adipose Redox Mechanisms as Potential Metabolic Disease Therapies
4.1. Current Therapeutics Directed towards Thermogenesis
4.2. Current Use of Antioxidants to Combat Metabolic Disease
4.3. New Perspectives to Altering the Redox Balance in Thermogenic Adipose Tissue as Metabolic Disease Therapies
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nagib, A.M.; Elsayed Matter, Y.; Gheith, O.A.; Refaie, A.F.; Othman, N.F.; Al-Otaibi, T. Diabetic Nephropathy Following Posttransplant Diabetes Mellitus. Exp. Clin. Transpl. 2019, 17, 138–146. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Cohen, D.E.; Fisher, E.A. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 2013, 33, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.J.; Rochelau, S.K.; Nicholls, S.J. Managing Dyslipidemia in Type 2 Diabetes. Endocrinol. Metab. Clin. North Am. 2018, 47, 153–173. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. Mitochondrial reactive oxygen species and adipose tissue thermogenesis: Bridging physiology and mechanisms. J. Biol. Chem. 2017, 292, 16810–16816. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lu, W.; Shi, B.; Klein, S.; Su, X. Peroxisomal regulation of redox homeostasis and adipocyte metabolism. Redox Biol. 2019, 24, 101167. [Google Scholar] [CrossRef]
- Fransen, M.; Lismont, C. Peroxisomes and Cellular Oxidant/Antioxidant Balance: Protein Redox Modifications and Impact on Inter-organelle Communication. Subcell. Biochem. 2018, 89, 435–461. [Google Scholar] [CrossRef]
- Galinier, A.; Carriere, A.; Fernandez, Y.; Caspar-Bauguil, S.; Periquet, B.; Periquet, A.; Penicaud, L.; Casteilla, L. Site specific changes of redox metabolism in adipose tissue of obese Zucker rats. FEBS Lett. 2006, 580, 6391–6398. [Google Scholar] [CrossRef] [Green Version]
- Ro, S.H.; Nam, M.; Jang, I.; Park, H.W.; Park, H.; Semple, I.A.; Kim, M.; Kim, J.S.; Park, H.; Einat, P.; et al. Sestrin2 inhibits uncoupling protein 1 expression through suppressing reactive oxygen species. Proc. Natl. Acad. Sci. USA 2014, 111, 7849–7854. [Google Scholar] [CrossRef]
- Oo, S.M.; Oo, H.K.; Takayama, H.; Ishii, K.A.; Takeshita, Y.; Goto, H.; Nakano, Y.; Kohno, S.; Takahashi, C.; Nakamura, H.; et al. Selenoprotein P-mediated reductive stress impairs cold-induced thermogenesis in brown fat. Cell Rep. 2022, 38, 110566. [Google Scholar] [CrossRef] [PubMed]
- Cero, C.; Lea, H.J.; Zhu, K.Y.; Shamsi, F.; Tseng, Y.H.; Cypess, A.M. beta3-Adrenergic receptors regulate human brown/beige adipocyte lipolysis and thermogenesis. JCI Insight 2021, 6, e139160. [Google Scholar] [CrossRef] [PubMed]
- O’Mara, A.E.; Johnson, J.W.; Linderman, J.D.; Brychta, R.J.; McGehee, S.; Fletcher, L.A.; Fink, Y.A.; Kapuria, D.; Cassimatis, T.M.; Kelsey, N.; et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J. Clin. Investig. 2020, 130, 2209–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, T.; Palanisamy, S.; Kramer, D.J.; Eljalby, M.; Marx, S.J.; Wibmer, A.G.; Butler, S.D.; Jiang, C.S.; Vaughan, R.; Schoder, H.; et al. Brown adipose tissue is associated with cardiometabolic health. Nat. Med. 2021, 27, 58–65. [Google Scholar] [CrossRef]
- Pan, R.; Zhu, X.; Maretich, P.; Chen, Y. Combating Obesity With Thermogenic Fat: Current Challenges and Advancements. Front Endocrinol. (Lausanne) 2020, 11, 185. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.J.; Zhang, Z.S.; Onishi, K.; Ukai, T.; Sane, D.C.; Cheng, C.P. Upregulation of functional beta(3)-adrenergic receptor in the failing canine myocardium. Circ. Res. 2001, 89, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yun, K.; Mu, R. A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes. Lipids Health Dis. 2020, 19, 164. [Google Scholar] [CrossRef] [PubMed]
- Abou-Rjeileh, U.; Contreras, G.A. Redox Regulation of Lipid Mobilization in Adipose Tissues. Antioxidants 2021, 10, 1090. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [Green Version]
- Gavalda-Navarro, A.; Villarroya, J.; Cereijo, R.; Giralt, M.; Villarroya, F. The endocrine role of brown adipose tissue: An update on actors and actions. Rev. Endocr. Metab. Disord. 2022, 23, 31–41. [Google Scholar] [CrossRef]
- Kita, S.; Maeda, N.; Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Invest. 2019, 129, 4041–4049. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular mechanisms for lipid mobilization from fat stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Fenzl, A.; Kiefer, F.W. Brown adipose tissue and thermogenesis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 25–37. [Google Scholar] [CrossRef]
- Heaton, G.M.; Wagenvoord, R.J.; Kemp, A., Jr.; Nicholls, D.G. Brown-adipose-tissue mitochondria: Photoaffinity labelling of the regulatory site of energy dissipation. Eur. J. Biochem. 1978, 82, 515–521. [Google Scholar] [CrossRef]
- Cohen, P.; Kajimura, S. The cellular and functional complexity of thermogenic fat. Nat. Rev. Mol. Cell Biol. 2021, 22, 393–409. [Google Scholar] [CrossRef]
- Lee, Y.H.; Petkova, A.P.; Konkar, A.A.; Granneman, J.G. Cellular origins of cold-induced brown adipocytes in adult mice. FASEB J. 2015, 29, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Contreras, G.A.; Lee, Y.H.; Mottillo, E.P.; Granneman, J.G. Inducible brown adipocytes in subcutaneous inguinal white fat: The role of continuous sympathetic stimulation. Am. J. Physiol. -Endocrinol. Metab. 2014, 307, E793–E799. [Google Scholar] [CrossRef]
- Rockstroh, D.; Landgraf, K.; Wagner, I.V.; Gesing, J.; Tauscher, R.; Lakowa, N.; Kiess, W.; Buhligen, U.; Wojan, M.; Till, H.; et al. Direct evidence of brown adipocytes in different fat depots in children. PLoS ONE 2015, 10, e0117841. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [Green Version]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Atgie, C.; Faintrenie, G.; Carpene, C.; Bukowiecki, L.J.; Geloen, A. Effects of chronic treatment with noradrenaline or a specific beta3-adrenergic agonist, CL 316 243, on energy expenditure and epididymal adipocyte lipolytic activity in rat. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 1998, 119, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Collins, S. beta-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front Endocrinol. (Lausanne) 2011, 2, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruhbeck, G.; Mendez-Gimenez, L.; Fernandez-Formoso, J.A.; Fernandez, S.; Rodriguez, A. Regulation of adipocyte lipolysis. Nutr. Res. Rev. 2014, 27, 63–93. [Google Scholar] [CrossRef] [Green Version]
- Zu, L.; He, J.; Jiang, H.; Xu, C.; Pu, S.; Xu, G. Bacterial endotoxin stimulates adipose lipolysis via toll-like receptor 4 and extracellular signal-regulated kinase pathway. J. Biol. Chem. 2009, 284, 5915–5926. [Google Scholar] [CrossRef] [Green Version]
- Chirivi, M.; Rendon, C.J.; Myers, M.N.; Prom, C.M.; Roy, S.; Sen, A.; Lock, A.L.; Contreras, G.A. Lipopolysaccharide induces lipolysis and insulin resistance in adipose tissue from dairy cows. J. Dairy Sci. 2022, 105, 842–855. [Google Scholar] [CrossRef]
- Yang, A.; Mottillo, E.P. Adipocyte lipolysis: From molecular mechanisms of regulation to disease and therapeutics. Biochem. J. 2020, 477, 985–1008. [Google Scholar] [CrossRef]
- Haemmerle, G.; Zimmermann, R.; Strauss, J.G.; Kratky, D.; Riederer, M.; Knipping, G.; Zechner, R. Hormone-sensitive lipase deficiency in mice changes the plasma lipid profile by affecting the tissue-specific expression pattern of lipoprotein lipase in adipose tissue and muscle. J. Biol. Chem. 2002, 277, 12946–12952. [Google Scholar] [CrossRef]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Contreras, J.A.; Hellman, U.; Tornqvist, H.; Holm, C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997, 272, 27218–27223. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banke, N.H.; Wende, A.R.; Leone, T.C.; O’Donnell, J.M.; Abel, E.D.; Kelly, D.P.; Lewandowski, E.D. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circ. Res. 2010, 107, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernandez-Fernandez, C.; Mourino-Bayolo, D. Mitochondrial beta-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Spiegelman, B.M. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019, 29, 27–37. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Kozak, L.P. Brown fat and the myth of diet-induced thermogenesis. Cell Metab. 2010, 11, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.; Yu, H.; Gong, J.; Jiang, J.; Qiao, X.; Perkey, E.; Kim, D.I.; Emont, M.P.; Zestos, A.G.; Cho, J.S.; et al. An immune-beige adipocyte communication via nicotinic acetylcholine receptor signaling. Nat. Med. 2018, 24, 814–822. [Google Scholar] [CrossRef]
- Ricquier, D.; Kader, J.C. Mitochondrial protein alteration in active brown fat: A soidum dodecyl sulfate-polyacrylamide gel electrophoretic study. Biochem. Biophys. Res. Commun. 1976, 73, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Bast-Habersbrunner, A.; Fromme, T. Purine Nucleotides in the Regulation of Brown Adipose Tissue Activity. Front Endocrinol. (Lausanne) 2020, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.E. Thermogenic mechanisms and their hormonal regulation. Physiol. Rev. 2006, 86, 435–464. [Google Scholar] [CrossRef]
- Ukropec, J.; Anunciado, R.P.; Ravussin, Y.; Hulver, M.W.; Kozak, L.P. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1-/- mice. J. Biol. Chem. 2006, 281, 31894–31908. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, W.E.; Liu, X.; Bearden, C.M.; Harper, M.E.; Kozak, L.P. Effects of genetic background on thermoregulation and fatty acid-induced uncoupling of mitochondria in UCP1-deficient mice. J. Biol. Chem. 2001, 276, 12460–12465. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Rossmeisl, M.; McClaine, J.; Riachi, M.; Harper, M.E.; Kozak, L.P. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J. Clin. Invest. 2003, 111, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Mottillo, E.P.; Balasubramanian, P.; Lee, Y.H.; Weng, C.; Kershaw, E.E.; Granneman, J.G. Coupling of lipolysis and de novo lipogenesis in brown, beige, and white adipose tissues during chronic beta3-adrenergic receptor activation. J. Lipid Res. 2014, 55, 2276–2286. [Google Scholar] [CrossRef]
- Kazak, L.; Chouchani, E.T.; Lu, G.Z.; Jedrychowski, M.P.; Bare, C.J.; Mina, A.I.; Kumari, M.; Zhang, S.; Vuckovic, I.; Laznik-Bogoslavski, D.; et al. Genetic Depletion of Adipocyte Creatine Metabolism Inhibits Diet-Induced Thermogenesis and Drives Obesity. Cell Metab. 2017, 26, 660–671.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, M.J.; Enerback, S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat. Rev. Endocrinol. 2018, 14, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, M.; Kuroshima, A. Creatine metabolism in skeletal muscle of cold-acclimated rats. J. Appl. Physiol. Respir. Env. Exerc. Physiol. 1978, 44, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Nisoli, E. Nitric oxide, interorganelle communication, and energy flow: A novel route to slow aging. Front Cell Dev. Biol. 2015, 3, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, A.B.; Song, A.; Wang, Q.A. Brown Adipose Tissue Heterogeneity, Energy Metabolism, and Beyond. Front Endocrinol. (Lausanne) 2021, 12, 651763. [Google Scholar] [CrossRef]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Muller, S.; Balaz, M.; Stefanicka, P.; Varga, L.; Amri, E.Z.; Ukropec, J.; Wollscheid, B.; Wolfrum, C. Proteomic Analysis of Human Brown Adipose Tissue Reveals Utilization of Coupled and Uncoupled Energy Expenditure Pathways. Sci. Rep. 2016, 6, 30030. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Park, A.; Oh, K.J.; Lee, S.C.; Kim, W.K.; Bae, K.H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B.; et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Han, Y.H.; Buffolo, M.; Pires, K.M.; Pei, S.; Scherer, P.E.; Boudina, S. Adipocyte-Specific Deletion of Manganese Superoxide Dismutase Protects From Diet-Induced Obesity Through Increased Mitochondrial Uncoupling and Biogenesis. Diabetes 2016, 65, 2639–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossini, E.; Farruggio, S.; Raina, G.; Mary, D.; Deiro, G.; Gentilli, S. Effects of Genistein on Differentiation and Viability of Human Visceral Adipocytes. Nutrients 2018, 10, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Little, S.A.; de Haen, C. Effects of hydrogen peroxide on basal and hormone-stimulated lipolysis in perifused rat fat cells in relation to the mechanism of action of insulin. J. Biol. Chem. 1980, 255, 10888–10895. [Google Scholar] [CrossRef]
- Nagashima, T.; Ohinata, H.; Kuroshima, A. Involvement of nitric oxide in noradrenaline-induced increase in blood flow through brown adipose tissue. Life Sci. 1994, 54, 17–25. [Google Scholar] [CrossRef]
- Petrovic, V.; Korac, A.; Buzadzic, B.; Korac, B. The effects of L-arginine and L-NAME supplementation on redox-regulation and thermogenesis in interscapular brown adipose tissue. J. Exp. Biol. 2005, 208, 4263–4271. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vrbacky, M.; Pecinova, A.; Kalinovich, A.V.; Drahota, Z.; Houstek, J.; Mracek, T.; Cannon, B.; Nedergaard, J. ROS production in brown adipose tissue mitochondria: The question of UCP1-dependence. Biochim. Biophys. Acta 2014, 1837, 2017–2030. [Google Scholar] [CrossRef]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Semple, I.; Ho, A.; Park, H.W.; Lee, J.H. Sestrin2, a Regulator of Thermogenesis and Mitohormesis in Brown Adipose Tissue. Front Endocrinol (Lausanne) 2015, 6, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef] [PubMed]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Aoshima, H.; Saitoh, Y.; Miwa, N. Highly hydroxylated fullerene localizes at the cytoskeleton and inhibits oxidative stress in adipocytes and a subcutaneous adipose-tissue equivalent. Free. Radic. Biol. Med. 2011, 51, 1376–1389. [Google Scholar] [CrossRef]
- Haushalter, K.J.; Schilling, J.M.; Song, Y.; Sastri, M.; Perkins, G.A.; Strack, S.; Taylor, S.S.; Patel, H.H. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIalpha. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1231–H1242. [Google Scholar] [CrossRef]
- Galigniana, N.M.; Charo, N.L.; Uranga, R.; Cabanillas, A.M.; Piwien-Pilipuk, G. Oxidative stress induces transcription of telomeric repeat-containing RNA (TERRA) by engaging PKA signaling and cytoskeleton dynamics. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118643. [Google Scholar] [CrossRef]
- Morrow, J.D.; Minton, T.A.; Mukundan, C.R.; Campbell, M.D.; Zackert, W.E.; Daniel, V.C.; Badr, K.F.; Blair, I.A.; Roberts, L.J., 2nd. Free radical-induced generation of isoprostanes in vivo. Evidence for the formation of D-ring and E-ring isoprostanes. J. Biol. Chem. 1994, 269, 4317–4326. [Google Scholar] [CrossRef]
- Yamakura, F.; Taka, H.; Fujimura, T.; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 1998, 273, 14085–14089. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; Chen, B.; Wang, L.; Taghizadeh, K.; Demott, M.S.; Dedon, P.C. Quantification of the 2-deoxyribonolactone and nucleoside 5’-aldehyde products of 2-deoxyribose oxidation in DNA and cells by isotope-dilution gas chromatography mass spectrometry: Differential effects of gamma-radiation and Fe2+-EDTA. J. Am. Chem. Soc. 2010, 132, 6145–6153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir Aslani, B.; Ghobadi, S. Studies on oxidants and antioxidants with a brief glance at their relevance to the immune system. Life Sci. 2016, 146, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, V.; Buzadzic, B.; Korac, A.; Korac, B. Antioxidative defense and mitochondrial thermogenic response in brown adipose tissue. Genes Nutr. 2010, 5, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niakao, C.; Ookawara, T.; Kizaki, T.; Suzuki, K.; Haga, S.; Sato, Y.; Ohno, H. Effects of acute cold stress on mrna expression and immunoreactivity of three superoxide dismutase isoenzymes in genetically obese mice. Res. Commun. Mol. Pathol. Pharm. 1999, 106, 47–61. [Google Scholar]
- Dutta, R.K.; Lee, J.N.; Maharjan, Y.; Park, C.; Choe, S.K.; Ho, Y.S.; Park, R. Catalase deficiency facilitates the shuttling of free fatty acid to brown adipose tissue through lipolysis mediated by ROS during sustained fasting. Cell Biosci. 2021, 11, 201. [Google Scholar] [CrossRef]
- Spasic, M.B.; Saicic, Z.S.; Buzadzic, B.; Korac, B.; Blagojevic, D.; Petrovic, V.M. Effect of long-term exposure to cold on the antioxidant defense system in the rat. Free Radic. Biol. Med. 1993, 15, 291–299. [Google Scholar] [CrossRef]
- Lettieri Barbato, D.; Tatulli, G.; Maria Cannata, S.; Bernardini, S.; Aquilano, K.; Ciriolo, M.R. Glutathione Decrement Drives Thermogenic Program In Adipose Cells. Sci. Rep. 2015, 5, 13091. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.M.; Kazantzis, M.; Wang, J.; Venkatraman, S.; Goncalves, R.L.; Quinlan, C.L.; Ng, R.; Jastroch, M.; Benjamin, D.I.; Nie, B.; et al. Dependence of brown adipose tissue function on CD36-mediated coenzyme Q uptake. Cell Rep. 2015, 10, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Fu, X.; Liang, X.; Deavila, J.M.; Wang, Z.; Zhao, L.; Tian, Q.; Zhao, J.; Gomez, N.A.; Trombetta, S.C.; et al. Retinoic acid induces white adipose tissue browning by increasing adipose vascularity and inducing beige adipogenesis of PDGFRalpha(+) adipose progenitors. Cell Discov. 2017, 3, 17036. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.Q.; Pan, D.X.; Hu, C.Q.; Zhu, Y.L.; Liu, X.J. Vitamin D-vitamin D receptor system down-regulates expression of uncoupling proteins in brown adipocyte through interaction with Hairless protein. Biosci. Rep. 2020, 40, BSR20194294. [Google Scholar] [CrossRef] [PubMed]
- Tanaka-Yachi, R.; Takahashi-Muto, C.; Adachi, K.; Tanimura, Y.; Aoki, Y.; Koike, T.; Kiyose, C. Promoting Effect of alpha-Tocopherol on Beige Adipocyte Differentiation in 3T3-L1 Cells and Rat White Adipose Tissue. J. Oleo Sci. 2017, 66, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinkov, A.A.; Ajsuvakova, O.P.; Filippini, T.; Zhou, J.C.; Lei, X.G.; Gatiatulina, E.R.; Michalke, B.; Skalnaya, M.G.; Vinceti, M.; Aschner, M.; et al. Selenium and Selenoproteins in Adipose Tissue Physiology and Obesity. Biomolecules 2020, 10, 658. [Google Scholar] [CrossRef] [PubMed]
- Jedrychowski, M.P.; Lu, G.Z.; Szpyt, J.; Mariotti, M.; Garrity, R.; Paulo, J.A.; Schweppe, D.K.; Laznik-Bogoslavski, D.; Kazak, L.; Murphy, M.P.; et al. Facultative protein selenation regulates redox sensitivity, adipose tissue thermogenesis, and obesity. Proc. Natl. Acad. Sci. USA 2020, 117, 10789–10796. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, L.; Urbanowicz, I.; Kepinska, M.; Milnerowicz, H. Concentration/activity of superoxide dismutase isozymes and the pro-/antioxidative status, in context of type 2 diabetes and selected single nucleotide polymorphisms (genes: INS, SOD1, SOD2, SOD3)—Preliminary findings. Biomed. Pharm. 2021, 137, 111396. [Google Scholar] [CrossRef] [PubMed]
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435. [Google Scholar] [CrossRef]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Palace, V.P.; Khaper, N.; Qin, Q.; Singal, P.K. Antioxidant potentials of vitamin A and carotenoids and their relevance to heart disease. Free Radic. Biol. Med. 1999, 26, 746–761. [Google Scholar] [CrossRef]
- Wiseman, H. Vitamin D is a membrane antioxidant. Ability to inhibit iron-dependent lipid peroxidation in liposomes compared to cholesterol, ergosterol and tamoxifen and relevance to anticancer action. FEBS Lett. 1993, 326, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Stevens, S.L. Fat-Soluble Vitamins. Nurs. Clin. North Am. 2021, 56, 33–45. [Google Scholar] [CrossRef]
- Blaner, W.S.; Shmarakov, I.O.; Traber, M.G. Vitamin A and Vitamin E: Will the Real Antioxidant Please Stand Up? Annu. Rev. Nutr. 2021, 41, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71. [Google Scholar] [CrossRef] [PubMed]
- Abdali, D.; Samson, S.E.; Grover, A.K. How effective are antioxidant supplements in obesity and diabetes? Med. Princ. Pract. 2015, 24, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Wallert, M.; Ziegler, M.; Wang, X.; Maluenda, A.; Xu, X.; Yap, M.L.; Witt, R.; Giles, C.; Kluge, S.; Hortmann, M.; et al. alpha-Tocopherol preserves cardiac function by reducing oxidative stress and inflammation in ischemia/reperfusion injury. Redox Biol. 2019, 26, 101292. [Google Scholar] [CrossRef]
- Traber, M.G.; Head, B. Vitamin E: How much is enough, too much and why! Free Radic. Biol. Med. 2021, 177, 212–225. [Google Scholar] [CrossRef]
- Lu, X.; Solmonson, A.; Lodi, A.; Nowinski, S.M.; Sentandreu, E.; Riley, C.L.; Mills, E.M.; Tiziani, S. The early metabolomic response of adipose tissue during acute cold exposure in mice. Sci. Rep. 2017, 7, 3455. [Google Scholar] [CrossRef] [Green Version]
- Green, C.L.; Mitchell, S.E.; Derous, D.; Wang, Y.; Chen, L.; Han, J.J.; Promislow, D.E.L.; Lusseau, D.; Douglas, A.; Speakman, J.R. The Effects of Graded Levels of Calorie Restriction: XIV. Global Metabolomics Screen Reveals Brown Adipose Tissue Changes in Amino Acids, Catecholamines, and Antioxidants After Short-Term Restriction in C57BL/6 Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 218–229. [Google Scholar] [CrossRef]
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The Uncoupling Proteins: A Systematic Review on the Mechanism Used in the Prevention of Oxidative Stress. Antioxidants 2022, 11, 322. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial proton leaks and uncoupling proteins. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148428. [Google Scholar] [CrossRef]
- Srinivasan, S.; Avadhani, N.G. Cytochrome c oxidase dysfunction in oxidative stress. Free Radic. Biol. Med. 2012, 53, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Murdolo, G.; Piroddi, M.; Luchetti, F.; Tortoioli, C.; Canonico, B.; Zerbinati, C.; Galli, F.; Iuliano, L. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie 2013, 95, 585–594. [Google Scholar] [CrossRef]
- Sun, X.; Seidman, J.S.; Zhao, P.; Troutman, T.D.; Spann, N.J.; Que, X.; Zhou, F.; Liao, Z.; Pasillas, M.; Yang, X.; et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 189–206.e188. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, A.; Korac, A.; Buzadzic, B.; Otasevic, V.; Stancic, A.; Daiber, A.; Korac, B. Redox implications in adipose tissue (dys)function--A new look at old acquaintances. Redox Biol. 2015, 6, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2019, 224, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Schwarzler, J.; Mayr, L.; Radlinger, B.; Grabherr, F.; Philipp, M.; Texler, B.; Grander, C.; Ritsch, A.; Hunjadi, M.; Enrich, B.; et al. Adipocyte GPX4 protects against inflammation, hepatic insulin resistance and metabolic dysregulation. Int. J. Obes. (Lond) 2022, 46, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Matsushita, M.; Yoneshiro, T.; Okamatsu-Ogura, Y. Brown Adipose Tissue, Diet-Induced Thermogenesis, and Thermogenic Food Ingredients: From Mice to Men. Front Endocrinol. (Lausanne) 2020, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, M.; Claus, T.H.; Himms-Hagen, J. Hypertrophy of brown adipocytes in brown and white adipose tissues and reversal of diet-induced obesity in rats treated with a beta3-adrenoceptor agonist. Biochem. Pharm. 1997, 54, 121–131. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, C.; Ma, Y.; Liu, D. Cold Exposure Improves the Anti-diabetic Effect of T0901317 in Streptozotocin-Induced Diabetic Mice. AAPS J. 2015, 17, 700–710. [Google Scholar] [CrossRef] [Green Version]
- Yoneshiro, T.; Rodriguez-Rodriguez, R.; Betz, M.J.; Rensen, P.C.N. Editorial: Current Challenges for Targeting Brown Fat Thermogenesis to Combat Obesity. Front Endocrinol. (Lausanne) 2020, 11, 600341. [Google Scholar] [CrossRef]
- Vijgen, G.H.; Bouvy, N.D.; Teule, G.J.; Brans, B.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Brown adipose tissue in morbidly obese subjects. PLoS ONE 2011, 6, e17247. [Google Scholar] [CrossRef] [Green Version]
- Celi, F.S.; Le, T.N.; Ni, B. Physiology and relevance of human adaptive thermogenesis response. Trends Endocrinol. Metab. 2015, 26, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.H. Of mice and men: Novel insights regarding constitutive and recruitable brown adipocytes. Int. J. Obes. Suppl. 2015, 5, S15–S20. [Google Scholar] [CrossRef] [Green Version]
- Cannon, B.; de Jong, J.M.A.; Fischer, A.W.; Nedergaard, J.; Petrovic, N. Human brown adipose tissue: Classical brown rather than brite/beige? Exp. Physiol. 2020, 105, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Riis-Vestergaard, M.J.; Richelsen, B.; Bruun, J.M.; Li, W.; Hansen, J.B.; Pedersen, S.B. Beta-1 and Not Beta-3 Adrenergic Receptors May Be the Primary Regulator of Human Brown Adipocyte Metabolism. J. Clin. Endocrinol. Metab. 2020, 105, e994–e1005. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.K.; Saxena, A.K. Structural basis for the beta-adrenergic receptor subtype selectivity of the representative agonists and antagonists. J Chem Inf Model 2011, 51, 1405–1422. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Lin, L. Small molecules for fat combustion: Targeting obesity. Acta Pharm. Sin. B 2019, 9, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, T.; Wu, X.; Nice, E.C.; Huang, C.; Zhang, Y. Oxidative stress and diabetes: Antioxidative strategies. Front Med. 2020, 14, 583–600. [Google Scholar] [CrossRef]
- Choi, S.W.; Ho, C.K. Antioxidant properties of drugs used in Type 2 diabetes management: Could they contribute to, confound or conceal effects of antioxidant therapy? Redox Rep. 2018, 23, 1–24. [Google Scholar] [CrossRef]
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front Biosci. (Landmark Ed.) 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Kornhauser, C.; Garcia-Ramirez, J.R.; Wrobel, K.; Perez-Luque, E.L.; Garay-Sevilla, M.E.; Wrobel, K. Serum selenium and glutathione peroxidase concentrations in type 2 diabetes mellitus patients. Prim. Care Diabetes 2008, 2, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Loguercio, C.; De Simone, T.; D’Auria, M.V.; de Sio, I.; Federico, A.; Tuccillo, C.; Abbatecola, A.M.; Del Vecchio Blanco, C.; Italian, A.C.G. Non-alcoholic fatty liver disease: A multicentre clinical study by the Italian Association for the Study of the Liver. Dig. Liver Dis. 2004, 36, 398–405. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Pedram, P.; Shahidi, M.; Du, J.; Yi, Y.; Gulliver, W.; Zhang, H.; Sun, G. Significant Beneficial Association of High Dietary Selenium Intake with Reduced Body Fat in the CODING Study. Nutrients 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, W.C.; Keim, N.L. Dietary selenium intake modulates thyroid hormone and energy metabolism in men. J. Nutr. 2003, 133, 3443–3448. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.B.; Hayward, M.L.; Griffith, O.W. Analytical and preparative separation of the diastereomers of L-buthionine (SR)-sulfoximine, a potent inhibitor of glutathione biosynthesis. Anal. Biochem. 1991, 194, 268–277. [Google Scholar] [CrossRef]
- Bailey, H.H.; Ripple, G.; Tutsch, K.D.; Arzoomanian, R.Z.; Alberti, D.; Feierabend, C.; Mahvi, D.; Schink, J.; Pomplun, M.; Mulcahy, R.T.; et al. Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl. Cancer Inst. 1997, 89, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Nederveen, J.P.; Manta, K.; Bujak, A.L.; Simone, A.C.; Fuda, M.R.; Nilsson, M.I.; Hettinga, B.P.; Hughes, M.C.; Perry, C.G.R.; Tarnopolsky, M.A. A Novel Multi-Ingredient Supplement Activates a Browning Program in White Adipose Tissue and Mitigates Weight Gain in High-Fat Diet-Fed Mice. Nutrients 2021, 13, 3726. [Google Scholar] [CrossRef]
- Mills, E.L.; Harmon, C.; Jedrychowski, M.P.; Xiao, H.; Gruszczyk, A.V.; Bradshaw, G.A.; Tran, N.; Garrity, R.; Laznik-Bogoslavski, D.; Szpyt, J.; et al. Cysteine 253 of UCP1 regulates energy expenditure and sex-dependent adipose tissue inflammation. Cell Metab. 2022, 34, 140–157.e148. [Google Scholar] [CrossRef]

{kind=link}
| Reactive Metabolite | Association with Thermogenic Fat | Source |
|---|---|---|
| Superoxide (O2●−) | Increases in vivo after cold exposure | [71] |
| Enhances UCP1 1 proton transport | [72] | |
| Stimulates mitochondrial uncoupling and fatty acid oxidation | [73] | |
| Promotes beige phenotype in WAT 1 | [73] | |
| Hydrogen peroxide (H2O2) | Increases in vivo after cold exposure | [71] |
| Decreases mitochondrial membrane potential | [74] | |
| Inhibits lipolysis | [75] | |
| Nitric oxide (NO●) | Increases blood flow following catecholamine stimulation | [76] |
| Increases BAT 1 mass and UCP1 content | [77] |
| Antioxidant | Association with Thermogenic Fat | Source |
|---|---|---|
| Superoxide dismutase (SOD) | mRNA expression and antioxidant activity decrease after acute and chronic cold exposure | [94,95] |
| Glutathione peroxidase (GPx) | Upregulation of GPx4 by selenoprotein P promotes resistance to norepinephrine-induced NST 1 | [11] |
| Catalase | Deficiency leads to enhanced fatty acid shuttling to BAT 1, induces NST | [96] |
| Activity increases after prolonged cold exposure in BAT | [97] | |
| Glutathione reductase | Increases in BAT after chronic cold exposure | [97] |
| Selenoprotein P | mRNA downregulates in BAT after acute cold exposure; negatively correlates with human BAT activity | [11] |
| Sestrin2 | Decreases Ucp1 1 mRNA; decreases NST after cold exposure | [10] |
| Glutathione (GSH) | Increases in BAT after chronic cold exposure | [97] |
| Decreases during white-to-brown phenotype conversion after acute cold exposure and β3-adrenergic agonist stimulation; mRNA levels of Ucp1 upregulate in BAT and WAT 1 | [98] | |
| Ubiquinone (Q) | Electron carrier in the mitochondrial electron transport chain | [67] |
| Uptake into BAT required for normal NST | [99] | |
| Vitamin A | Retinoic acid promotes a brown adipocyte phenotype in WAT | [100] |
| Vitamin D | Decreases uncoupling protein expression in rat brown adipocytes | [101] |
| Vitamin E | Promotes a beige phenotype | [102] |
| Selenium | Cofactor for many antioxidants relevant to NST, including GPx | [103] |
| Improves UCP1 efficiency in BAT | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putman, A.K.; Contreras, G.A.; Mottillo, E.P. Thermogenic Adipose Redox Mechanisms: Potential Targets for Metabolic Disease Therapies. Antioxidants 2023, 12, 196. https://doi.org/10.3390/antiox12010196
Putman AK, Contreras GA, Mottillo EP. Thermogenic Adipose Redox Mechanisms: Potential Targets for Metabolic Disease Therapies. Antioxidants. 2023; 12(1):196. https://doi.org/10.3390/antiox12010196
Chicago/Turabian StylePutman, Ashley K., G. Andres Contreras, and Emilio P. Mottillo. 2023. "Thermogenic Adipose Redox Mechanisms: Potential Targets for Metabolic Disease Therapies" Antioxidants 12, no. 1: 196. https://doi.org/10.3390/antiox12010196
APA StylePutman, A. K., Contreras, G. A., & Mottillo, E. P. (2023). Thermogenic Adipose Redox Mechanisms: Potential Targets for Metabolic Disease Therapies. Antioxidants, 12(1), 196. https://doi.org/10.3390/antiox12010196