
Paraoxonase 2 Deficiency Causes Mitochondrial Dysfunction in Retinal Pigment Epithelial Cells and Retinal Degeneration in Mice

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Retinal Pigment Epithelium (RPE) Culture
2.2. Effect of Oxidative Stress on PON2 Expression and Subcellular Localization
2.3. siRNA-Mediated Knockdown of PON2 and Mitochondrial Functional Studies
2.4. NaIO3-Induced RPE Degeneration Mouse Model
2.5. Spectral Domain Optical Coherence Tomography (SD-OCT) and Fundus Imaging
2.6. Electroretinography
2.7. Retinal Histology
2.8. Terminal Deoxynucleotide Transferase dUTP Nick End Labeling (TUNEL) Staining
2.9. Immunofluorescence Staining
2.10. Data Analysis
3. Results
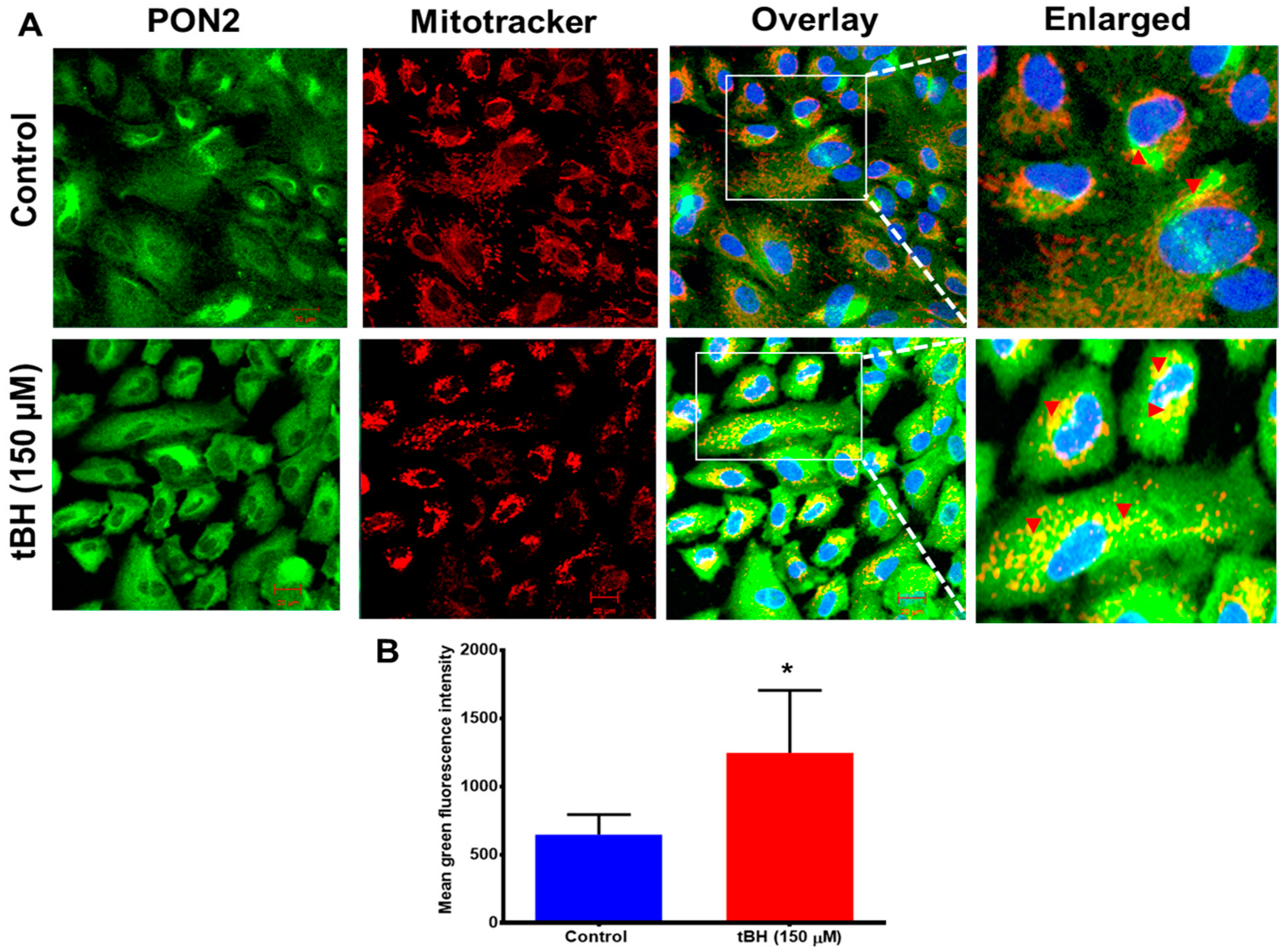
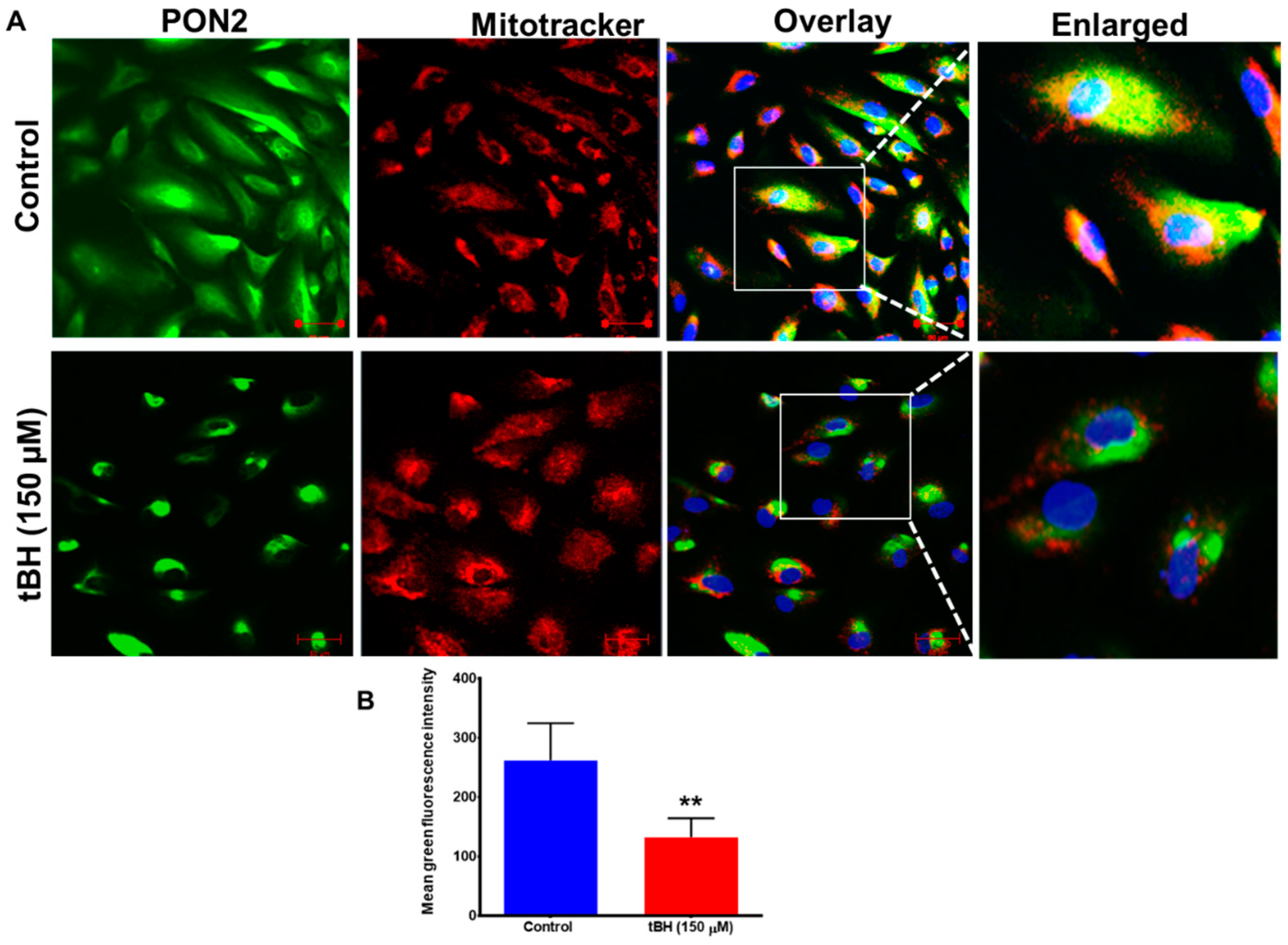
3.1. Effect of Oxidative Stress on PON2 Expression in Human RPE Cells, and Its Subcellular Localization
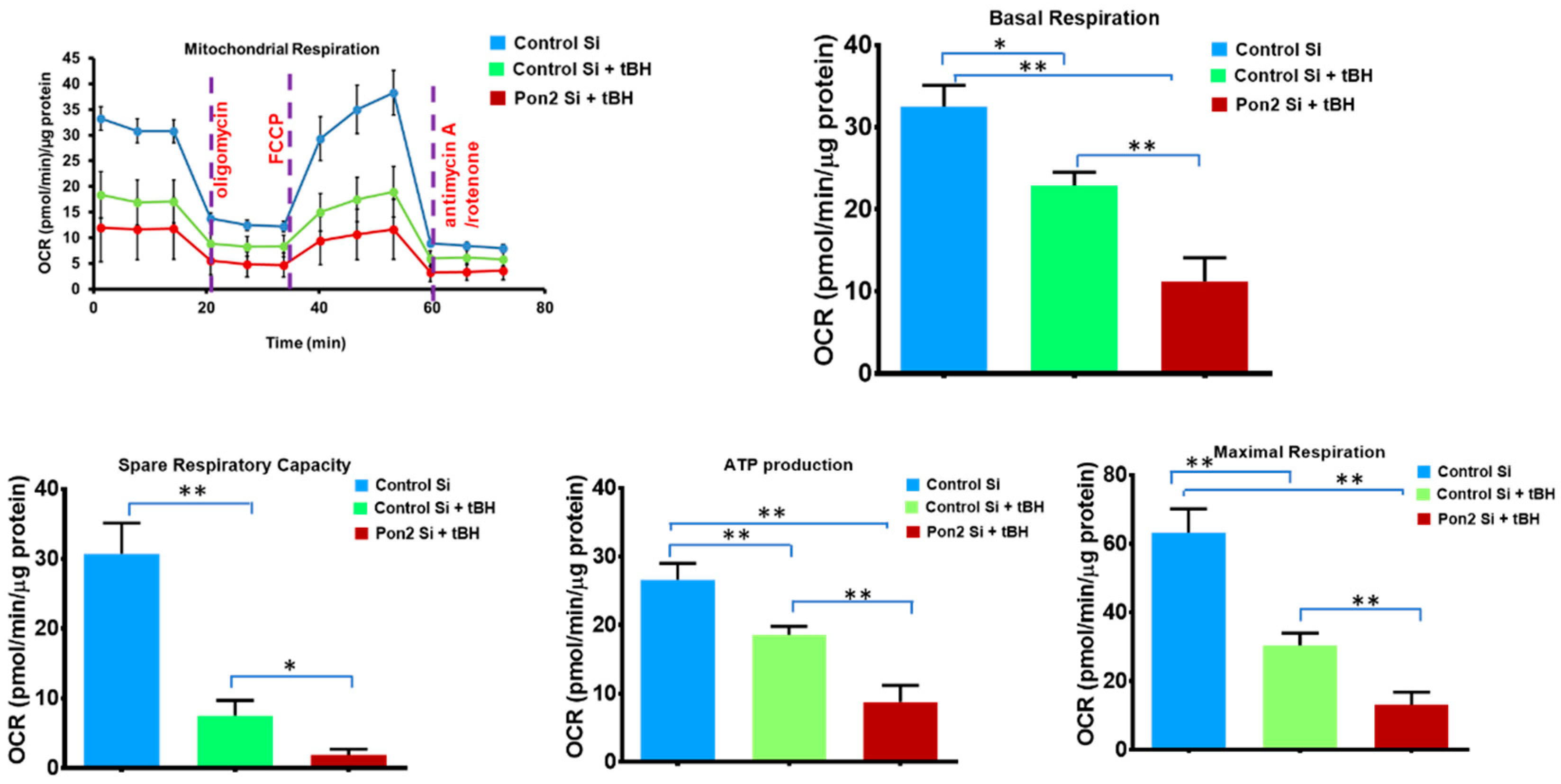
3.2. Mitochondrial Bioenergetics Is Altered in tBH Treated PON2 Knockdown RPE Cells
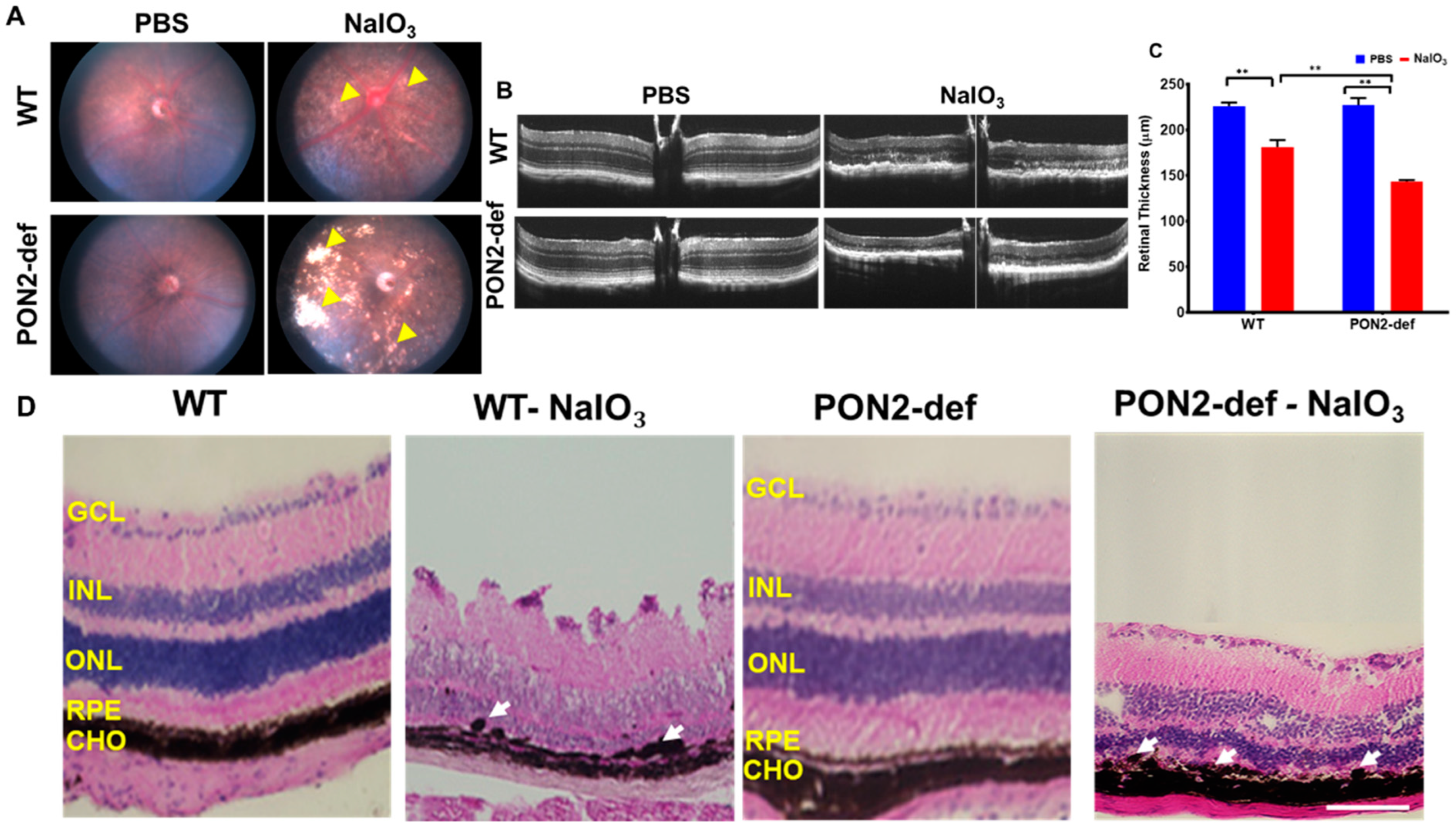
3.3. PON2-Deficiency Accelerates Retinal Degeneration Induced by NaIO3
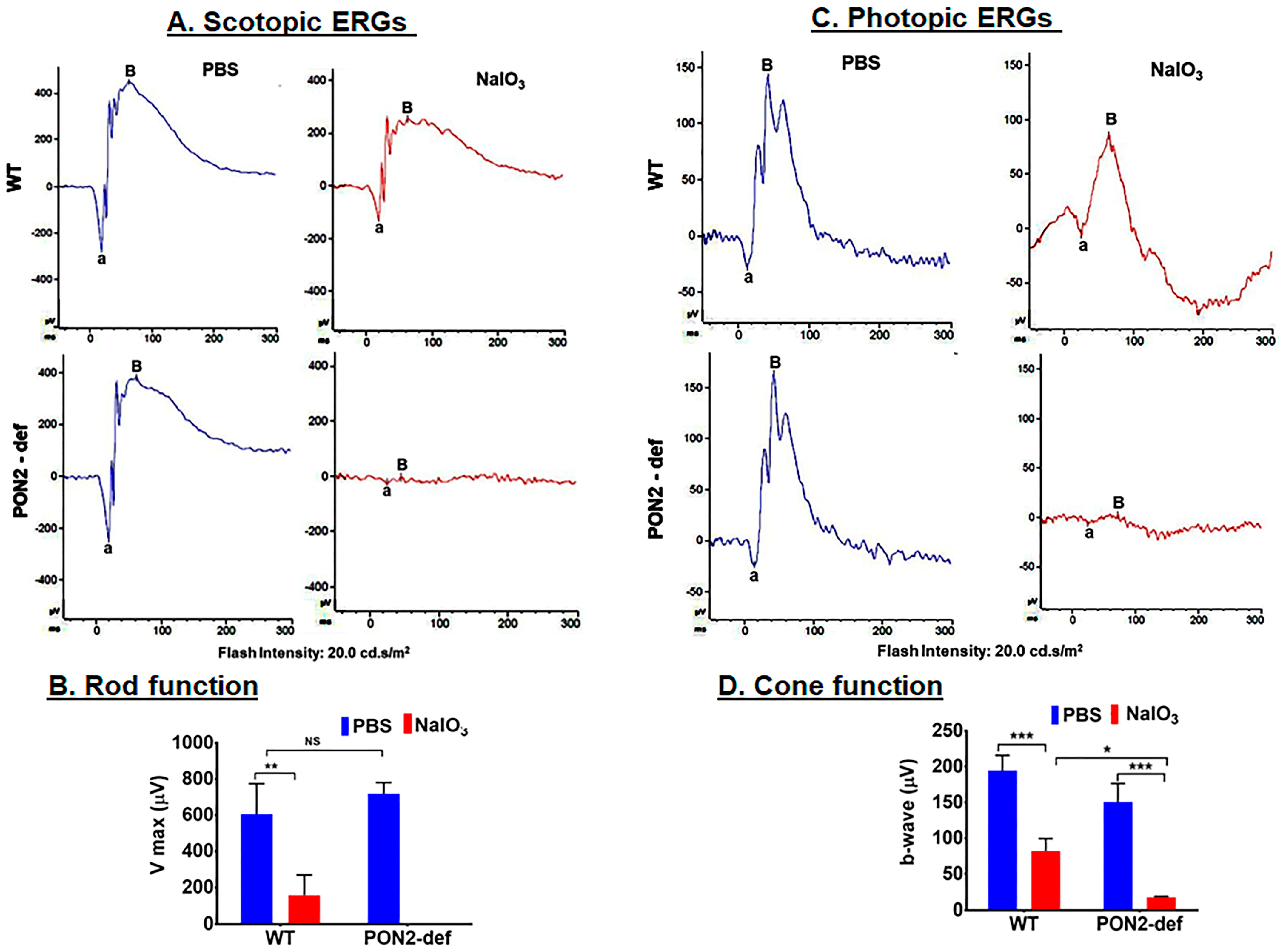
3.4. PON2-Deficient Mice Display Altered Scotopic and Photopic ERGs
3.5. PON2-Deficient Exacerbates Cell Death in Mouse Retina
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106-16. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lytvynchuk, L.; Ardan, T.; Studenovska, H.; Faura, G.; Eide, L.; Znaor, L.; Erceg, S.; Stieger, K.; Motlik, J.; et al. Retinal Pigment Epithelium Cell Development: Extrapolating Basic Biology to Stem Cell Research. Biomedicines 2023, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef] [PubMed]
- Karlov, V.D.; Pestov, N.B.; Shakhparonov, M.I.; Korneenko, T.V. Interactome of Paraoxonase PON2 Reveals New Pathways for Tumor Growth Regulation. Dokl. Biochem. Biophys. 2023, 508, 1–6. [Google Scholar] [CrossRef]
- Draganov, D.I.; La Du, B.N. Pharmacogenetics of paraoxonases: A brief review. Naunyn. Schmiedebergs Arch. Pharmacol. 2004, 369, 78–88. [Google Scholar] [CrossRef]
- Ng, C.J.; Wadleigh, D.J.; Gangopadhyay, A.; Hama, S.; Grijalva, V.R.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Paraoxonase-2 Is a Ubiquitously Expressed Protein with Antioxidant Properties and Is Capable of Preventing Cell-mediated Oxidative Modification of Low Density Lipoprotein. J. Biol. Chem. 2001, 276, 44444–44449. [Google Scholar] [CrossRef]
- Horke, S.; Witte, I.; Wilgenbus, P.; Krüger, M.; Strand, D.; Förstermann, U. Paraoxonase-2 Reduces Oxidative Stress in Vascular Cells and Decreases Endoplasmic Reticulum Stress–Induced Caspase Activation. Circulation 2007, 115, 2055–2064. [Google Scholar] [CrossRef]
- Horke, S.; Witte, I.; Wilgenbus, P.; Altenhöfer, S.; Krüger, M.; Li, H.; Förstermann, U. Protective effect of paraoxonase-2 against endoplasmic reticulum stress-induced apoptosis is lost upon disturbance of calcium homoeostasis. Biochem. J. 2008, 416, 395–405. [Google Scholar] [CrossRef]
- Taler-Vercic, A.; Golicnik, M.; Bavec, A. The Structure and Function of Paraoxonase-1 and Its Comparison to Paraoxonase-2 and -3. Molecules 2020, 25, 5980. [Google Scholar] [CrossRef]
- Witte, I.; Altenhöfer, S.; Wilgenbus, P.; Amort, J.; Clement, A.M.; Pautz, A.; Li, H.; Förstermann, U.; Horke, S. Beyond reduction of atherosclerosis: PON2 provides apoptosis resistance and stabilizes tumor cells. Cell Death Dis. 2011, 2, e112. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Nagaraj, N.R.; Rajesh, B.S. Effect of advanced glycation end product on paraoxonase 2 expression: Its impact on endoplasmic reticulum stress and inflammation in HUVECs. Life Sci. 2020, 246, 117397. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Rajesh, B.S. Paraoxonase 2 protects against the CML mediated mitochondrial dysfunction through modulating JNK pathway in human retinal cells. Biochim. Biophys. Acta (BBA) Gen. Subj. 2022, 1866, 130043. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Buck, T.M.; Nickerson, A.J.; Brodsky, J.L.; Kleyman, T.R. Paraoxonase 2 is an ER chaperone that regulates the epithelial Na+ channel. Am. J. Physiol. Physiol. 2022, 322, C111–C121. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M. Paraoxonases; oxidative stress, and macrophage foam cell formation during atherosclerosis development. Free Radic. Biol. Med. 2004, 37, 1304–1316. [Google Scholar] [CrossRef]
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 Deficiency Alters Mitochondrial Function and Exacerbates the Development of Atherosclerosis. Antioxid. Redox. Sig. 2011, 14, 341–351. [Google Scholar] [CrossRef]
- Javadzadeh, A.; Ghorbanihaghjo, A.; Bahreini, E.; Rashtchizadeh, N.; Argani, H.; Alizadeh, S. Serum paraoxonase phenotype distribution in exudative age-related macular degeneration and its relationship to homocysteine and oxidized low-density lipoprotein. Retina 2012, 32, 658–666. [Google Scholar] [CrossRef]
- Hashim, Z.; Ilyas, A.; Saleem, A.; Salim, A.; Zarina, S. Expression and activity of paraoxonase 1 in human cataractous lens tissue. Free Radic. Biol. Med. 2009, 46, 1089–1095. [Google Scholar] [CrossRef]
- Nowak, M.; Wielkoszynski, T.; Marek, B.; Kos-Kudla, B.; Swietochowska, E.; Sieminska, L.; Karpe, J.; Kajdaniuk, D.; Glogowska-Szelag, J.; Nowak, K. Antioxidant potential; paraoxonase, ceruloplasmin activity and C-reactive protein concentration in diabetic retinopathy. Clin. Exp. Med. 2010, 10, 185–192. [Google Scholar] [CrossRef]
- Sonoda, S.; Sreekumar, P.G.; Kase, S.; Spee, C.; Ryan, S.J.; Kannan, R.; Hinton, D.R. Attainment of polarity promotes growth factor secretion by retinal pigment epithelial cells: Relevance to age-related macular degeneration. Aging 2009, 2, 28–42. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Su, F.; Spee, C.; Araujo, E.; Nusinowitz, S.; Reddy, S.T.; Kannan, R. Oxidative Stress and Lipid Accumulation Augments Cell Death in LDLR-Deficient RPE Cells and Ldlr(-/-) Mice. Cells 2022, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.J.; Bourquard, N.; Grijalva, V.; Hama, S.; Shih, D.M.; Navab, M.; Fogelman, A.M.; Lusis, A.J.; Young, S.; Reddy, S.T. Paraoxonase-2 deficiency aggravates atherosclerosis in mice despite lower apolipoprotein-B-containing lipoproteins: Anti-atherogenic role for paraoxonase-2. J. Biol. Chem. 2006, 281, 29491–29500. [Google Scholar] [CrossRef] [PubMed]
- Stryke, D.; Kawamoto, M.; Huang, C.C.; Johns, S.J.; King, L.A.; Harper, C.A.; Meng, E.C.; Lee, R.E.; Yee, A.; L’Italien, L.; et al. BayGenomics: A resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003, 31, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Townley, D.J.; Avery, B.J.; Rosen, B.; Skarnes, W.C. Rapid sequence analysis of gene trap integrations to generate a resource of insertional mutations in mice. Genome Res. 1997, 7, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Spee, C.; Araujo, E.; Barron, E.; Wang, M.; Ghione, C.; Hinton, D.R.; Nusinowitz, S.; Kannan, R.; Reddy, S.T.; et al. A Novel HDL-Mimetic Peptide HM-10/10 Protects RPE and Photoreceptors in Murine Models of Retinal Degeneration. Int. J. Mol. Sci. 2019, 20, 4807. [Google Scholar] [CrossRef]
- Mochizuki, H.; Scherer, S.W.; Xi, T.; Nickle, D.C.; Majer, M.; Huizenga, J.J.; Tsui, L.-C.; Prochazka, M. Human PON2 gene at 7q21.3: Cloning, multiple mRNA forms, and missense polymorphisms in the coding sequence. Gene 1998, 213, 149–157. [Google Scholar] [CrossRef]
- Rosenblat, M.; Draganov, D.; Watson, C.E.; Bisgaier, C.L.; La Du, B.N.; Aviram, M. Mouse Macrophage Paraoxonase 2 Activity Is Increased Whereas Cellular Paraoxonase 3 Activity Is Decreased Under Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 468–474. [Google Scholar] [CrossRef]
- Levy, E.; Trudel, K.; Bendayan, M.; Seidman, E.; Delvin, E.; Elchebly, M.; Lavoie, J.-C.; Precourt, L.-P.; Amre, D.; Sinnett, D. Biological role, protein expression, subcellular localization, and oxidative stress response of paraoxonase 2 in the intestine of humans and rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G1252–G1261. [Google Scholar]
- Costa, L.G.; de Laat, R.; Dao, K.; Pellacani, C.; Cole, T.B.; Furlong, C.E. Paraoxonase-2 (PON2) in brain and its potential role in neuroprotection. NeuroToxicology 2014, 43, 3–9. [Google Scholar] [CrossRef]
- Bharathidevi, S.R.; Babu, K.A.; Jain, N.; Muthukumaran, S.; Umashankar, V.; Biswas, J.; Angayarkanni, N. Ocular distribution of antioxidant enzyme paraoxonase & its alteration in cataractous lens & diabetic retina. Indian J. Med Res. 2017, 145, 513–520. [Google Scholar]
- Précourt, L.-P.; Marcil, V.; Ntimbane, T.; Taha, R.; Lavoie, J.-C.; Delvin, E.; Seidman, E.G.; Beaulieu, J.-F.; Levy, E. Antioxidative properties of paraoxonase 2 in intestinal epithelial cells. Am. J. Physiol. Liver Physiol. 2012, 303, G623–G634. [Google Scholar] [CrossRef] [PubMed]
- Morresi, C.; Cianfruglia, L.; Sartini, D.; Cecati, M.; Fumarola, S.; Emanuelli, M.; Armeni, T.; Ferretti, G.; Bacchetti, T. Effect of High Glucose-Induced Oxidative Stress on Paraoxonase 2 Expression and Activity in Caco-2 Cells. Cells 2019, 8, 1616. [Google Scholar] [CrossRef]
- Parween, F.; Gupta, R.D. Insights into the role of paraoxonase 2 in human pathophysiology. J. Biosci. 2022, 47, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Carusone, T.M.; Cardiero, G.; Cerreta, M.; Mandrich, L.; Moran, O.; Porzio, E.; Catara, G.; Lacerra, G.; Manco, G. WTAP and BIRC3 are involved in the posttranscriptional mechanisms that impact on the expression and activity of the human lactonase PON2. Cell Death Dis. 2020, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Baig, A.; Rehman, A.U.; Zarina, S. Association of PON2 and PON3 polymorphism with risk of developing cataract. Saudi J. Ophthalmol. 2019, 33, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pinizzotto, M.; Castillo, E.; Fiaux, M.; Temler, E.; Gaillard, R.C.; Ruiz, J. Paraoxonase2 polymorphisms are associated with nephropathy in Type II diabetes. Diabetologia 2001, 44, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Janka, Z.; Juhasz, A.; Rimanoczy, A.A.; Boda, K.; Marki-Zay, J.; Kalman, J. Codon 311 (Cys --> Ser) polymorphism of paraoxonase-2 gene is associated with apolipoprotein E4 allele in both Alzheimer’s and vascular dementias. Mol. Psychiatry 2002, 7, 110–112. [Google Scholar]
- Nie, Y.; Luo, D.; Yang, M.; Wang, Y.; Xiong, L.; Gao, L.; Liu, Y.; Liu, H. A Meta-Analysis on the Relationship of the PON Genes and Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2017, 30, 303–310. [Google Scholar] [CrossRef]
- Yehuda, I.; Madar, Z.; Szuchman-Sapir, A.; Tamir, S. Glabridin, a phytoestrogen from licorice root, up-regulates manganese superoxide dismutase, catalase and paraoxonase 2 under glucose stress. Phytother. Res. 2011, 25, 659–667. [Google Scholar] [CrossRef]
- Yehuda, I.; Madar, Z.; Leikin-Frenkel, A.; Szuchman-Sapir, A.; Magzal, F.; Markman, G.; Tamir, S. Glabridin, an isoflavan from licorice root, upregulates paraoxonase 2 expression under hyperglycemia and protects it from oxidation. Mol. Nutr. Food Res. 2016, 60, 287–299. [Google Scholar] [CrossRef]
- Sulaiman, D.; Li, J.; Devarajan, A.; Cunningham, C.M.; Li, M.; Fishbein, G.A.; Fogelman, A.M.; Eghbali, M.; Reddy, S.T. Paraoxonase 2 protects against acute myocardial ischemia-reperfusion injury by modulating mitochondrial function and oxidative stress via the PI3K/Akt/GSK-3beta RISK pathway. J. Mol. Cell Cardiol. 2019, 129, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Shin, G.-C.; Lee, H.M.; Kim, N.; Yoo, S.-K.; Park, H.S.; Choi, L.S.; Kim, K.P.; Lee, A.-R.; Seo, S.-U.; Kim, K.-H. Paraoxonase-2 contributes to promoting lipid metabolism and mitochondrial function via autophagy activation. Sci. Rep. 2022, 12, 21483. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Reiter, R.J. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Witte, I.; Teiber, J.F.; Wilgenbus, P.; Pautz, A.; Li, H.; Daiber, A.; Witan, H.; Clement, A.M.; Forstermann, U.; et al. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J. Biol. Chem. 2010, 285, 24398–24403. [Google Scholar] [CrossRef]
- Jasna, J.M.; Anandbabu, K.; Bharathi, S.R.; Angayarkanni, N. Paraoxonase Enzyme Protects Retinal Pigment Epithelium from Chlorpyrifos Insult. PLoS ONE 2014, 9, e101380. [Google Scholar] [CrossRef]
- Shih, D.M.; Meng, Y.; Sallam, T.; Vergnes, L.; Shu, M.L.; Reue, K.; Tontonoz, P.; Fogelman, A.M.; Lusis, A.J.; Reddy, S.T. PON2 Deficiency Leads to Increased Susceptibility to Diet-Induced Obesity. Antioxidants 2019, 8, 19. [Google Scholar] [CrossRef]
- Koster, C.; Hurk, K.T.v.D.; Brink, J.B.T.; Lewallen, C.F.; Stanzel, B.V.; Bharti, K.; Bergen, A.A. Sodium-Iodate Injection Can Replicate Retinal Degenerative Disease Stages in Pigmented Mice and Rats: Non-Invasive Follow-Up Using OCT and ERG. Int. J. Mol. Sci. 2022, 23, 2918. [Google Scholar] [CrossRef]
- Mizota, A.; Adachi-Usami, E. Functional recovery of retina after sodium iodate injection in mice. Vis. Res. 1997, 37, 1859–1865. [Google Scholar] [CrossRef]
- Enzmann, V.; Row, B.W.; Yamauchi, Y.; Kheirandish, L.; Gozal, D.; Kaplan, H.J.; McCall, M.A. Behavioral and anatomical abnormalities in a sodium iodate-induced model of retinal pigment epithelium degeneration. Exp. Eye Res. 2006, 82, 441–448. [Google Scholar] [CrossRef]
- He, H.; Wei, D.; Liu, H.; Zhu, C.; Lu, Y.; Ke, Z.; Jiang, S.; Huang, J. Glycyrrhizin protects against sodium iodate-induced RPE and retinal injury though activation of AKT and Nrf2/HO-1 pathway. J. Cell Mol. Med. 2019, 23, 3495–3504. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.E.G.; Knave, B.; Persson, H.E. Changes in ultrastructure and function of the sheep pigment epithelium and retina induced by sodium iodate1. Acta Ophthalmol. 1977, 55, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Mones, J.; Leiva, M.; Pena, T.; Martinez, G.; Biarnes, M.; Garcia, M.; Serrano, A.; Fernandez, E. A Swine Model of Selective Geographic Atrophy of Outer Retinal Layers Mimicking Atrophic AMD: A Phase I Escalating Dose of Subretinal Sodium Iodate. Invest. Ophthalmol. Vis. Sci. 2016, 57, 3974–3983. [Google Scholar] [CrossRef] [PubMed]
- Obata, R.; Yanagi, Y.; Tamaki, Y.; Hozumi, K.; Mutoh, M.; Tanaka, Y. Retinal degeneration is delayed by tissue factor pathway inhibitor-2 in RCS rats and a sodium-iodate-induced model in rabbits. Eye 2005, 19, 464–468. [Google Scholar] [CrossRef]
- Ou, Q.; Zhu, T.; Li, P.; Li, Z.; Wang, L.; Lian, C.; Xu, H.; Jin, C.; Gao, F.; Xu, J.-Y.; et al. Establishment of Retinal Degeneration Model in Rat and Monkey by Intravitreal Injection of Sodium Iodate. Curr. Mol. Med. 2018, 18, 352–364. [Google Scholar] [CrossRef]
- Aung, K.H.; Liu, H.; Ke, Z.; Jiang, S.; Huang, J. Glabridin Attenuates the Retinal Degeneration Induced by Sodium Iodate In Vitro and In Vivo. Front. Pharmacol. 2020, 11, 566699. [Google Scholar] [CrossRef]
- Li, D.; Wei, T.-T.; Cai, J.; Xie, T.-H.; Yao, Y.; Zhu, L. Smurf1: A possible therapeutic target in dry age-related macular degeneration. Exp. Eye Res. 2023, 233, 109549. [Google Scholar] [CrossRef]
- Pan, T.; Shen, H.; Yuan, S.; Lu, G.; Zhang, Y.; Wang, H.; Zhao, Y.; Sun, X.; Liu, Q. Combined Transplantation with Human Mesenchymal Stem Cells Improves Retinal Rescue Effect of Human Fetal RPE Cells in Retinal Degeneration Mouse Model. Investig. Opthalmol. Vis. Sci. 2020, 61, 9. [Google Scholar] [CrossRef]
- Moriguchi, M.; Nakamura, S.; Inoue, Y.; Nishinaka, A.; Nakamura, M.; Shimazawa, M.; Hara, H. Irreversible Photoreceptors and RPE Cells Damage by Intravenous Sodium Iodate in Mice Is Related to Macrophage Accumulation. Investig. Opthalmol. Vis. Sci. 2018, 59, 3476–3487. [Google Scholar] [CrossRef]
- Enzbrenner, A.; Zulliger, R.; Biber, J.; Pousa, A.M.Q.; Schäfer, N.; Stucki, C.; Giroud, N.; Berrera, M.; Kortvely, E.; Schmucki, R.; et al. Sodium Iodate-Induced Degeneration Results in Local Complement Changes and Inflammatory Processes in Murine Retina. Int. J. Mol. Sci. 2021, 22, 9218. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Demirci, F.Y.; Dressen, A.S.; Kao, A.H.; Rhew, E.Y.; Ramsey-Goldman, R.; Manzi, S.; Kammerer, C.M.; Kamboh, M.I. Association analysis of PON2 genetic variants with serum paraoxonase activity and systemic lupus erythematosus. BMC Med. Genet. 2011, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Agbaga, M.-P.; Merriman, D.K.; Brush, R.S.; Lydic, T.A.; Conley, S.M.; Naash, M.I.; Jackson, S.; Woods, A.S.; Reid, G.E.; Busik, J.V.; et al. Differential composition of DHA and very-long-chain PUFAs in rod and cone photoreceptors. J. Lipid Res. 2018, 59, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, T.; Liu, W.; Wang, Y.; Xu, R.; Zeng, S.; Zhang, R.; Zhu, S.; Gillies, M.C.; Zhu, L.; et al. Metabolic Features of Mouse and Human Retinas: Rods versus Cones, Macula versus Periphery, Retina versus RPE. iScience 2020, 23, 101672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-Related Retinopathy in NRF2-Deficient Mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef]
- Ren, X.; Li, C.; Liu, J.; Zhang, C.; Fu, Y.; Wang, N.; Ma, H.; Lu, H.; Kong, H.; Kong, L. Thioredoxin plays a key role in retinal neuropathy prior to endothelial damage in diabetic mice. Oncotarget 2017, 8, 61350–61364. [Google Scholar] [CrossRef]
- Garrick, J.M.; Cole, T.B.; Bammler, T.K.; MacDonald, J.W.; Marsillach, J.; Furlong, C.E.; Costa, L.G. Paraoxonase 2 deficiency in mice alters motor behavior and causes region-specific transcript changes in the brain. Neurotoxicol. Teratol. 2021, 87, 107010. [Google Scholar] [CrossRef]
- Li, W.; Kennedy, D.; Shao, Z.; Wang, X.; Kamdar, A.K.; Weber, M.; Mislick, K.; Kiefer, K.; Morales, R.; Agatisa-Boyle, B.; et al. Paraoxonase 2 prevents the development of heart failure. Free. Radic. Biol. Med. 2018, 121, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Qujeq, D.; Mahrooz, A.; Alizadeh, A.; Boorank, R. Paraoxonase-2 variants potentially influence insulin resistance, beta-cell function, and their interrelationships with alanine aminotransferase in type 2 diabetes. J. Res. Med. Sci. 2018, 23, 107. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sreekumar, P.G.; Su, F.; Spee, C.; Hong, E.; Komirisetty, R.; Araujo, E.; Nusinowitz, S.; Reddy, S.T.; Kannan, R. Paraoxonase 2 Deficiency Causes Mitochondrial Dysfunction in Retinal Pigment Epithelial Cells and Retinal Degeneration in Mice. Antioxidants 2023, 12, 1820. https://doi.org/10.3390/antiox12101820
Sreekumar PG, Su F, Spee C, Hong E, Komirisetty R, Araujo E, Nusinowitz S, Reddy ST, Kannan R. Paraoxonase 2 Deficiency Causes Mitochondrial Dysfunction in Retinal Pigment Epithelial Cells and Retinal Degeneration in Mice. Antioxidants. 2023; 12(10):1820. https://doi.org/10.3390/antiox12101820
Chicago/Turabian StyleSreekumar, Parameswaran Gangadharan, Feng Su, Christine Spee, Elise Hong, Ravikiran Komirisetty, Eduardo Araujo, Steven Nusinowitz, Srinivasa T. Reddy, and Ram Kannan. 2023. "Paraoxonase 2 Deficiency Causes Mitochondrial Dysfunction in Retinal Pigment Epithelial Cells and Retinal Degeneration in Mice" Antioxidants 12, no. 10: 1820. https://doi.org/10.3390/antiox12101820