
NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. AF Pathophysiology
1.1. Introduction
1.2. AF Classification
1.3. Arrhythmogenic Mechanisms in Atrial Fibrillation
2. Sources of Cardiac ROS
3. Oxidative Stress in the Natural History of AF
3.1. Role of NADPH Oxidases and Oxidative Stress in the Onset of Paroxysmal AF
3.2. Role of NADPH Oxidases and Oxidative Stress in Permanent AF
3.3. ROS in the Transition from Paroxysmal to Permanent AF
4. Oxidative Stress and Arrhythmogenic Mechanisms in AF
4.1. Oxidative Stress Is Associated with AF-Triggered Activity
4.2. ROS Alter the Intracellular Ca2+ Handling
4.3. Oxidative Stress Is Associated with Functional Reentry
4.4. ROS Promote Anatomical Reentry Mechanisms
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| Ang II | angiotensin II |
| AF | atrial fibrillation |
| AP | action potential |
| APD | action potential duration |
| CaMKII | Calcium-calmodulin (CaM)-dependent protein kinase II |
| CD44 | cluster of differentiation 44 |
| CM | cardiomyocyte |
| CXCL1 | C-X-C motif chemokine ligand 1 |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| Cx40/Cx43 | connexin 40/43 |
| DAD | delayed afterdepolarization |
| DD | diastolic dysfunction |
| EAD | early afterdepolarization |
| FOXO | forkhead box O |
| HA | hyaluronan |
| HAS | Hyaluronan synthase |
| ICaL | L-type Ca2 current |
| IK1 | inwardly rectifying potassium current |
| IKur | ultra-rapid delayed-rectifier K+ current |
| INa | sodium current |
| INaL | late sodium current |
| Iti | inward sodium current |
| Ito | outward potassium current |
| Kir | inwardly rectifying potassium channel |
| MitoOS | mitochondrial oxidative stress |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NaV1.5/SCN5A | pore-forming α-subunit of the voltage-dependent cardiac Na+ channel |
| NOS | nitric oxide synthase |
| nNOS | neuronal nitric oxide synthase |
| NOX | NADPH oxidase |
| NOX4 | NADPH oxidase 4 |
| PAF | paroxysmal atrial fibrillation |
| PKCε | protein kinase C epsilon |
| POAF | postoperative atrial fibrillation |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| ROS | reactive oxygen species |
| RyR2 | ryanodine receptor 2 |
| SERCA | sarco-endoplasmic reticulum calcium ATPase |
| SOICR | store overload induced Ca2+ release |
| SR | sarcoplasmic reticulum |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF-β1 | transforming growth factor-β1 |
References
- Benjamin, E.J.; Wolf, P.A.; D’Agostino, R.B.; Silbershatz, H.; Kannel, W.B.; Levy, D. Impact of atrial fibrillation on the risk of death: The Framingham Heart Study. Circulation 1998, 98, 946–952. [Google Scholar] [CrossRef]
- Hijazi, Z.; Oldgren, J.; Siegbahn, A.; Wallentin, L. Application of biomarkers for risk stratification in patients with atrial fibrillation. Clin. Chem. 2017, 63, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Colilla, S.; Crow, A.; Petkun, W.; Singer, D.E.; Simon, T.; Liu, X. Estimates of current and future incidence and prevalence of atrial fibrillation in the U.S. adult population. Am. J. Cardiol. 2013, 112, 1142–1147. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Levy, D.; Vaziri, S.M.; D’Agostino, R.B.; Belanger, A.J.; Wolf, P.A. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. JAMA 1994, 271, 840–844. [Google Scholar] [CrossRef]
- Kannel, W.B.; Wolf, P.A.; Benjamin, E.J.; Levy, D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: Population-based estimates. Am. J. Cardiol. 1998, 82, 2N–9N. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, S.A.; Yin, X.; Fontes, J.D.; Magnani, J.W.; Rienstra, M.; Pai, M.; Villalon, M.L.; Vasan, R.S.; Pencina, M.J.; Levy, D.; et al. Association between familial atrial fibrillation and risk of new-onset atrial fibrillation. JAMA 2010, 304, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2014, 64, e1–e76. [Google Scholar] [PubMed]
- Haïssaguerre, M.; Jaïs, P.; Shah, D.C.; Takahashi, A.; Hocini, M.; Quiniou, G.; Garrigue, S.; Le Mouroux, A.; Le Métayer, P.; Clémenty, J. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 1998, 339, 659–666. [Google Scholar] [CrossRef]
- Wit, A.L.; Boyden, P.A. Triggered activity and atrial fibrillation. Heart Rhythm 2007, 4, S17–S23. [Google Scholar] [CrossRef]
- Smeets, J.L.; Allessie, M.A.; Lammers, W.J.; Bonke, F.I.; Hollen, J. The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. The role of heart rate, autonomic transmitters, temperature, and potassium. Circ. Res. 1986, 58, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Papadatos, G.A.; Wallerstein, P.M.; Head, C.E.; Ratcliff, R.; Brady, P.A.; Benndorf, K.; Saumarez, R.C.; Trezise, A.E.; Huang, C.L.; Vandenberg, J.I.; et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. USA 2002, 99, 6210–6215. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, S.; Nelles, E.; Hagendorff, A.; Krüger, O.; Traub, O.; Willecke, K. Reduced cardiac conduction velocity and predisposition to arrhythmias in connexin40-deficient mice. Curr. Biol. 1998, 8, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.A.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Vranka, I.; Penz, P.; Dukát, A. Atrial conduction delay and its association with left atrial dimension, left atrial pressure and left ventricular diastolic dysfunction in patients at risk of atrial fibrillation. Exp. Clin. Cardiol. 2007, 12, 197–201. [Google Scholar]
- Eijsbouts, S.C.; Majidi, M.; van Zandvoort, M.; Allessie, M.A. Effects of acute atrial dilation on heterogeneity in conduction in the isolated rabbit heart. J. Cardiovasc. Electrophysiol. 2003, 14, 269–278. [Google Scholar] [CrossRef]
- Krul, S.P.J.; Berger, W.R.; Veldkamp, M.W.; Driessen, A.H.G.; Wilde, A.A.M.; Deneke, T.; de Bakker, J.M.T.; Coronel, R.; de Groot, J.R. Treatment of atrial and ventricular arrhythmias through autonomic modulation. JACC Clin. Electrophysiol. 2015, 1, 496–508. [Google Scholar] [CrossRef]
- Verheule, S.; Sato, T.; Everett, T., 4th; Engle, S.K.; Otten, D.; Rubart-von der Lohe, M.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef]
- Li, N.; Chiang, D.Y.; Wang, S.; Wang, Q.; Sun, L.; Voigt, N.; Respress, J.L.; Ather, S.; Skapura, D.G.; Jordan, V.K.; et al. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 2014, 129, 1276–1285. [Google Scholar] [CrossRef]
- Bukowska, A.; Lendeckel, U.; Hirte, D.; Wolke, C.; Striggow, F.; Röhnert, P.; Huth, C.; Klein, H.U.; Goette, A. Activation of the calcineurin signaling pathway induces atrial hypertrophy during atrial fibrillation. Cell. Mol. Life Sci. 2006, 63, 333–342. [Google Scholar] [CrossRef]
- Kanthan, A.; Fahmy, P.; Rao, R.; Pouliopoulos, J.; Alexander, I.E.; Thomas, S.P.; Kizana, E. Human connexin40 mutations slow conduction and increase propensity for atrial fibrillation. Heart Lung Circ. 2018, 27, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, J.M.; Tyml, K.; Jones, D.L. Atrial tachycardia/fibrillation in the connexin 43 G60S mutant (Oculodentodigital dysplasia) mouse. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1402–H1411. [Google Scholar] [CrossRef]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH oxidases in heart failure: Poachers or gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Duranteau, J.; Chandel, N.S.; Kulisz, A.; Shao, Z.; Schumacker, P.T. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998, 273, 11619–11624. [Google Scholar] [CrossRef] [PubMed]
- Oudot, A.; Vergely, C.; Ecarnot-Laubriet, A.; Rochette, L. Angiotensin II activates NADPH oxidase in isolated rat hearts subjected to ischaemia-reperfusion. Eur. J. Pharmacol. 2003, 462, 145–154. [Google Scholar] [CrossRef]
- Rude, M.K.; Duhaney, T.A.; Kuster, G.M.; Judge, S.; Heo, J.; Colucci, W.S.; Siwik, D.A.; Sam, F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension 2005, 46, 555–561. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Xiao, L.; Pimentel, D.R.; Wang, J.; Singh, K.; Colucci, W.S.; Sawyer, D.B. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. Cell Physiol. 2002, 282, C926–C934. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef]
- Dutta, S.; Rittinger, K. Regulation of NOXO1 activity through reversible interactions with p22 and NOXA1. PLoS ONE 2010, 5, e10478. [Google Scholar] [CrossRef] [PubMed]
- Honjo, T.; Otsui, K.; Shiraki, R.; Kawashima, S.; Sawamura, T.; Yokoyama, M.; Inoue, N. Essential role of NOXA1 in generation of reactive oxygen species induced by oxidized low-density lipoprotein in human vascular endothelial cells. Endothelium 2008, 15, 137–141. [Google Scholar] [CrossRef]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Schreiber, J.G.; Janiszewski, M.; Busse, R.; Brandes, R.P. Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic. Biol. Med. 2006, 41, 193–201. [Google Scholar] [CrossRef]
- Niu, X.L.; Madamanchi, N.R.; Vendrov, A.E.; Tchivilev, I.; Rojas, M.; Madamanchi, C.; Brandes, R.P.; Krause, K.H.; Humphries, J.; Smith, A.; et al. Nox activator 1: A potential target for modulation of vascular reactive oxygen species in atherosclerotic arteries. Circulation 2010, 121, 549–559. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Stevenson, M.D.; Lozhkin, A.; Hayami, T.; Holland, N.A.; Yang, X.; Moss, N.; Pan, H.; Wickline, S.A.; Stockand, J.D.; et al. Renal NOXA1/NOX1 signaling regulates epithelial sodium channel and sodium retention in Angiotensin II-induced hypertension. Antioxid. Redox Signal. 2022, 36, 550–566. [Google Scholar] [CrossRef]
- Stevenson, M.D.; Vendrov, A.E.; Yang, X.; Chen, Y.; Navarro, H.A.; Moss, N.; Runge, M.S.; Arendshorst, W.J.; Madamanchi, N.R. Reactivity of renal and mesenteric resistance vessels to angiotensin II is mediated by NOXA1/NOX1 and superoxide signaling. Am. J. Physiol. Renal Physiol. 2023, 324, F335–F352. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xu, N.; Zhang, Z.; Wang, F.; Xiao, J.; Ji, X. Setanaxib (GKT137831) Ameliorates doxorubicin-induced cardiotoxicity by inhibiting the NOX1/NOX4/reactive oxygen species/MAPK pathway. Front. Pharmacol. 2022, 13, 823975. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Cong, T.; Xu, G.; Hao, Z.; Liao, J.; Xie, Y.; Lin, Y.; Yang, X.; Li, Q.; Liu, Y.; et al. Anthracycline-induced atrial structural and electrical remodeling characterizes early cardiotoxicity and contributes to atrial conductive instability and dysfunction. Antioxid. Redox Signal. 2022, 37, 19–39. [Google Scholar] [CrossRef]
- Zduniak, A.; Lévêque, E.; Perdrix, A.; Etancelin, P.; Ménard, A.L.; Lenain, P.; Contentin, N.; Pépin, L.F.; Leprêtre, S.; Lemasle, E.; et al. Cardiovascular outcomes of patients treated for non-Hodgkin lymphoma with first-line doxorubicin-based chemotherapy. Leuk. Lymphoma. 2022, 63, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.S.; Barnes, M.E.; Gersh, B.J.; Bailey, K.R.; Seward, J.B. Risks for atrial fibrillation and congestive heart failure in patients >/=65 years of age with abnormal left ventricular diastolic relaxation. Am. J. Cardiol. 2004, 93, 54–58. [Google Scholar] [CrossRef]
- Tsang, T.S.; Gersh, B.J.; Appleton, C.P.; Tajik, A.J.; Barnes, M.E.; Bailey, K.R.; Oh, J.K.; Leibson, C.; Montgomery, S.C.; Seward, J.B. Left ventricular diastolic dysfunction as a predictor of the first diagnosed nonvalvular atrial fibrillation in 840 elderly men and women. J. Am. Coll. Cardiol. 2002, 40, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Schirmer, H.; Jacobsen, B.K.; Hopstock, L.A.; Nyrnes, A.; Heggelund, G.; Njølstad, I.; Mathiesen, E.B.; Løchen, M.L. Association between diastolic dysfunction and future atrial fibrillation in the Tromsø Study from 1994 to 2010. Heart 2015, 101, 1302–1308. [Google Scholar] [CrossRef]
- Bonapace, S.; Rossi, A.; Cicoira, M.; Targher, G.; Marino, P.; Benfari, G.; Mugnai, G.; Arcaro, G.; Vassanelli, C. Echocardiographically derived pulse wave velocity and diastolic dysfunction are associated with an Increased Incidence of atrial fibrillation in patients with systolic heart failure. Echocardiography 2016, 33, 1024–1031. [Google Scholar] [CrossRef]
- Rosenberg, M.A.; Gottdiener, J.S.; Heckbert, S.R.; Mukamal, K.J. Echocardiographic diastolic parameters and risk of atrial fibrillation: The Cardiovascular Health Study. Eur. Heart J. 2012, 33, 904–912. [Google Scholar] [CrossRef]
- Kosiuk, J.; Breithardt, O.A.; Bode, K.; Kornej, J.; Arya, A.; Piorkowski, C.; Gaspar, T.; Sommer, P.; Husser, D.; Hindricks, G.; et al. The predictive value of echocardiographic parameters associated with left ventricular diastolic dysfunction on short- and long-term outcomes of catheter ablation of atrial fibrillation. Europace 2014, 16, 1168–1174. [Google Scholar] [CrossRef]
- Li, D.L.; Quispe, R.; Madan, N.; Zhang, L.; Taub, C.C. A risk score for predicting atrial fibrillation in individuals with preclinical diastolic dysfunction: A retrospective study in a single large urban center in the United States. BMC Cardiovasc. Disord. 2019, 19, 47. [Google Scholar] [CrossRef]
- Watanabe, H.; Tanabe, N.; Watanabe, T.; Darbar, D.; Roden, D.M.; Sasaki, S.; Aizawa, Y. Metabolic syndrome and risk of development of atrial fibrillation: The Niigata preventive medicine study. Circulation 2008, 117, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Balzarolo, M.; Robinson, E.L.; Lorenz, V.; Verde, G.D.; Joray, L.; Mochizuki, M.; Kaufmann, B.A.; Valstar, G.; de Jager, S.C.A.; et al. NOX1 mediates metabolic heart disease in mice and is upregulated in monocytes of humans with diastolic dysfunction. Cardiovasc. Res. 2022, 118, 2973–2984. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z.G. NADPH oxidases are essential for macrophage differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef]
- Barton, M.; Meyer, M.R.; Prossnitz, E.R. Nox1 down regulators: A new class of therapeutics. Steroids 2019, 152, 108494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Teng, F.; Han, X.; Li, P.B.; Yan, X.; Guo, S.B.; Li, H.H. Selective blocking of CXCR2 prevents and reverses atrial fibrillation in spontaneously hypertensive rats. J. Cell. Mol. Med. 2020, 24, 11272–11282. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, Z.; Goette, A.; Mera, F.; Honeycutt, C.; Feterik, K.; Wilcox, J.N.; Dudley, S.C., Jr.; Harrison, D.G.; Langberg, J.J. Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: Potential mechanisms for atrial thrombosis and stroke. Circulation 2002, 106, 2854–2858. [Google Scholar] [CrossRef] [PubMed]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Nabeebaccus, A.A.; Reumiller, C.M.; Shen, J.; Zoccarato, A.; Santos, C.X.C.; Shah, A.M. The regulation of cardiac intermediary metabolism by NADPH oxidases. Cardiovasc. Res. 2023, 118, 3305–3319. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, J.; Khaidakov, M.; Mitra, S.; Ding, Z.; Raina, S.; Goyal, T.; Mehta, J.L. Aspirin suppresses cardiac fibroblast proliferation and collagen formation through downregulation of angiotensin type 1 receptor transcription. Toxicol. Appl. Pharmacol. 2012, 259, 346–354. [Google Scholar] [CrossRef]
- Krijnen, P.A.; Meischl, C.; Hack, C.E.; Meijer, C.J.; Visser, C.A.; Roos, D.; Niessen, H.W. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J. Clin. Pathol. 2003, 56, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Nicolas-Avila, J.A.; Li, J.L.; Garcia-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; González, N.A.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 2008, 51, 68–74. [Google Scholar] [CrossRef]
- Cangemi, R.; Celestini, A.; Calvieri, C.; Carnevale, R.; Pastori, D.; Nocella, C.; Vicario, T.; Pignatelli, P.; Violi, F. Different behaviour of NOX2 activation in patients with paroxysmal/persistent or permanent atrial fibrillation. Heart 2012, 98, 1063–1066. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Kuo, C.T.; Chan, T.H.; Chang, G.J.; Qi, X.Y.; Tsai, F.; Nattel, S.; Chen, W.J. Transforming growth factor-β and oxidative stress mediate tachycardia-induced cellular remodeling in cultured atrial-derived myocytes. Cardiovasc. Res. 2011, 91, 62–70. [Google Scholar] [CrossRef]
- Violi, F.; Carnevale, R.; Calvieri, C.; Nocella, C.; Falcone, M.; Farcomeni, A.; Taliani, G.; Cangemi, R.; SIXTUS study group. Nox2 up-regulation is associated with an enhanced risk of atrial fibrillation in patients with pneumonia. Thorax 2015, 70, 961–966. [Google Scholar] [CrossRef]
- Somers, V.K.; White, D.P.; Amin, R.; Abraham, W.T.; Costa, F.; Culebras, A.; Daniels, S.; Floras, J.S.; Hunt, C.E.; Olson, L.J.; et al. Sleep apnea and cardiovascular disease: An American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. J. Am. Coll. Cardiol. 2008, 52, 686–717. [Google Scholar]
- Linz, D.; Linz, B.; Hohl, M.; Böhm, M. Atrial arrhythmogenesis in obstructive sleep apnea: Therapeutic implications. Sleep Med. Rev. 2016, 26, 87–94. [Google Scholar] [CrossRef]
- Gemel, J.; Su, Z.; Gileles-Hillel, A.; Khalyfa, A.; Gozal, D.; Beyer, E.C. Intermittent hypoxia causes NOX2-dependent remodeling of atrial connexins. BMC Cell Biol. 2017, 18 (Suppl. S1), 7. [Google Scholar] [CrossRef]
- Reilly, S.N.; Jayaram, R.; Nahar, K.; Antoniades, C.; Verheule, S.; Channon, K.M.; Alp, N.J.; Schotten, U.; Casadei, B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Mighiu, A.S.; Recalde, A.; Ziberna, K.; Carnicer, R.; Tomek, J.; Bub, G.; Brewer, A.C.; Verheule, S.; Shah, A.M.; Simon, J.N.; et al. Inducibility, but not stability, of atrial fibrillation is increased by NOX2 overexpression in mice. Cardiovasc. Res. 2021, 117, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Pfenniger, A.; Hoffman, J.; Zhang, W.; Ng, J.; Burrell, A.; Johnson, D.A.; Gussak, G.; Waugh, T.; Bull, S.; et al. Attenuation of oxidative injury with targeted expression of NADPH Oxidase 2 short hairpin RNA prevents onset and maintenance of electrical remodeling in the canine atrium: A novel gene therapy approach to atrial fibrillation. Circulation 2020, 142, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Voigt, N.; Friedrich, A.; Bock, M.; Wettwer, E.; Christ, T.; Knaut, M.; Strasser, R.H.; Ravens, U.; Dobrev, D. Differential phosphorylation-dependent regulation of constitutively active and muscarinic receptor-activated IK, ACh channels in patients with chronic atrial fibrillation. Cardiovasc. Res. 2007, 74, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S. New ideas about atrial fibrillation 50 years on. Nature 2002, 415, 219–226. [Google Scholar] [CrossRef]
- Healey, J.S.; Baranchuk, A.; Crystal, E.; Morillo, C.A.; Garfinkle, M.; Yusuf, S.; Connolly, S.J. Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: A meta-analysis. J. Am. Coll. Cardiol 2005, 45, 1832–1839. [Google Scholar]
- Ehrlich, J.R.; Hohnloser, S.H.; Nattel, S. Role of angiotensin system and effects of its inhibition in atrial fibrillation clinical and experimental evidence. Eur. Heart J. 2006, 27, 512–518. [Google Scholar] [CrossRef]
- Xiao, H.D.; Fuchs, S.; Campbell, D.J.; Lewis, W.; Dudley, S.C., Jr.; Kasi, V.S.; Hoit, B.D.; Keshelava, G.; Zhao, H.; Capecchi, M.R.; et al. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am. J. Pathol. 2004, 165, 1019–1032. [Google Scholar] [CrossRef]
- Zhao, Z.; Fefelova, N.; Shanmugam, M.; Bishara, P.; Babu, G.J.; Xie, L.H. Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell. Cardiol. 2011, 50, 128–136. [Google Scholar] [CrossRef]
- Swaminathan, P.D.; Purohit, A.; Soni, S.; Voigt, N.; Singh, M.V.; Glukhov, A.V.; Gao, Z.; He, B.J.; Luczak, E.D.; Joiner, M.L.; et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J. Clin. Investig. 2011, 121, 3277–3288. [Google Scholar] [CrossRef]
- Zhang, Y.; Qi, Y.; Li, J.J.; He, W.J.; Gao, X.H.; Zhang, Y.; Sun, X.; Tong, J.; Zhang, J.; Deng, X.L.; et al. Stretch-induced sarcoplasmic reticulum calcium leak is causatively associated with atrial fibrillation in pressure-overloaded hearts. Cardiovasc. Res. 2021, 117, 1091–1102. [Google Scholar] [CrossRef]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Scho¨Ndube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Herting, J.; Mason, F.E.; Hartmann, N.; Watanabe, S.; Nikolaev, V.O.; Sprenger, J.U.; Fan, P.; Yao, L.; Popov, A.F.; et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc. Res. 2015, 107, 184–196. [Google Scholar] [CrossRef]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Muller, F.U.; Schmitz, W.; et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Investig. 2009, 119, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- DeSantiago, J.; Bare, D.J.; Varma, D.; Solaro, R.J.; Arora, R.; Banach, K. Loss of p21-activated kinase 1 (Pak1) promotes atrial arrhythmic activity. Heart Rhythm 2018, 15, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef]
- Desai, L.P.; Zhou, Y.; Estrada, A.V.; Ding, Q.; Cheng, G.; Collawn, J.F.; Thannickal, V.J. Negative regulation of NADPH oxidase 4 by hydrogen peroxide-inducible clone 5 (Hic-5) protein. J. Biol. Chem. 2014, 289, 18270–18278. [Google Scholar] [CrossRef]
- Prior, K.K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Dürr, P.; Shah, A.M.; Brandes, R.P. The endoplasmic reticulum chaperone calnexin is a NADPH oxidase NOX4 interacting protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid. Redox Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef]
- Lozhkin, A.; Vendrov, A.E.; Pan, H.; Wickline, S.A.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates vascular inflammation in aging and atherosclerosis. J. Mol. Cell. Cardiol. 2017, 102, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chamseddine, A.H.; Carrell, S.; Miller, F.J., Jr. Nox4 NADPH oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biol. 2014, 2, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Stevenson, M.D.; Vendrov, A.E.; Hayami, T.; Robidoux, J.; Xiao, H.; Zhang, Y.Y.; Eitzman, D.T.; Runge, M.S.; Madamanchi, N.R. Increased mitochondrial NADPH oxidase 4 (NOX4) expression in aging is a causative factor in aortic stiffening. Redox Biol. 2019, 26, 101288. [Google Scholar] [CrossRef]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef]
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 dual inhibitor GKT137831 or Nox4 Knockdown inhibits Angiotensin-II-induced adult mouse cardiac fibroblast proliferation and migration. AT1 physically associates with Nox4. J. Cell Physiol. 2016, 231, 1130–1141. [Google Scholar] [CrossRef]
- Yeh, Y.H.; Kuo, C.T.; Chang, G.J.; Qi, X.Y.; Nattel, S.; Chen, W.J. Nicotinamide adenine dinucleotide phosphate oxidase 4 mediates the differential responsiveness of atrial versus ventricular fibroblasts to transforming growth factor-β. Circ. Arrhythm. Electrophysiol. 2013, 6, 790–798. [Google Scholar] [CrossRef]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef]
- Lozhkin, A.; Vendrov, A.E.; Ramos-Mondragón, R.; Canugovi, C.; Stevenson, M.D.; Herron, T.J.; Hummel, S.L.; Figueroa, C.A.; Bowles, D.E.; Isom, L.L.; et al. Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biol. 2022, 57, 102474. [Google Scholar] [CrossRef] [PubMed]
- Rolski, F.; Czepiel, M.; Tkacz, K.; Fryt, K.; Siedlar, M.; Kania, G.; Błyszczuk, P. T Lymphocyte-derived exosomes transport MEK1/2 and ERK1/2 and induce NOX4-dependent oxidative stress in cardiac microvascular endothelial cells. Oxid. Med. Cell. Longev. 2022, 2022, 2457687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Wang, Z.J.; Miao, L.; Wang, Y.; Chang, L.L.; Guo, W.; Zhu, Y.Z. Neuroprotective effect of SCM-198 through stabilizing endothelial cell function. Oxid. Med. Cell. Longev. 2019, 2019, 7850154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Brewer, A.C.; Schröder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef]
- Stevenson, M.D.; Canugovi, C.; Vendrov, A.E.; Hayami, T.; Bowles, D.E.; Krause, K.H.; Madamanchi, N.R.; Runge, M.S. NADPH oxidase 4 regulates inflammation in ischemic heart failure, role of soluble epoxide hydrolase. Antioxid. Redox Signal. 2019, 31, 39–58. [Google Scholar] [CrossRef]
- Varga, Z.V.; Pipicz, M.; Baán, J.A.; Baranyai, T.; Koncsos, G.; Leszek, P.; Kuśmierczyk, M.; Sánchez-Cabo, F.; García-Pavía, P.; Brenner, G.J.; et al. Alternative splicing of NOX4 in the failing human heart. Front. Physiol. 2017, 8, 935. [Google Scholar] [CrossRef]
- Zhang, J.; Youn, J.Y.; Kim, A.Y.; Ramirez, R.J.; Gao, L.; Ngo, D.; Chen, P.; Scovotti, J.; Mahajan, A.; Cai, H. NOX4-dependent hydrogen peroxide overproduction in human atrial fibrillation and HL-1 atrial cells, Relationship to hypertension. Front. Physiol. 2012, 3, 140. [Google Scholar] [CrossRef]
- Zhang, Y.; Shimizu, H.; Siu, K.L.; Mahajan, A.; Chen, J.N.; Cai, H. NADPH oxidase 4 induces cardiac arrhythmic phenotype in zebrafish. J. Biol. Chem. 2014, 289, 23200–23208. [Google Scholar] [CrossRef]
- Burstein, B.; Nattel, S. Atrial structural remodeling as an antiarrhythmic target. J. Cardiovasc. Pharmacol. 2008, 52, 4–10. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Bellocci, F.; Morgante, E.; Russo, M.A.; Maseri, A. Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 1997, 96, 1180–1184. [Google Scholar] [CrossRef]
- Kostin, S.; Klein, G.; Szalay, Z.; Hein, S.; Bauer, E.P.; Schaper, J. Structural correlate of atrial fibrillation in human patients. Cardiovasc. Res. 2002, 54, 361–379. [Google Scholar] [CrossRef]
- Hayashi, H.; Wang, C.; Miyauchi, Y.; Omichi, C.; Pak, H.N.; Zhou, S.; Ohara, T.; Mandel, W.J.; Lin, S.F.; Fishbein, M.C.; et al. Aging-related increase to inducible atrial fibrillation in the rat model. J. Cardiovasc. Electrophysiol. 2002, 13, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Anyukhovsky, E.P.; Sosunov, E.A.; Plotnikov, A.; Gainullin, R.Z.; Jhang, J.S.; Marboe, C.C.; Rosen, M.R. Cellular electrophysiologic properties of old canine atria provide a substrate for arrhythmogenesis. Cardiovasc. Res. 2002, 54, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Wilson, E.; Everett, T., 4th; Shanbhag, S.; Golden, C.; Olgin, J. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation 2003, 107, 2615–2622. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fareh, S.; Leung, T.K.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs, atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Nakajima, H.O.; Salcher, O.; Dittiè, A.S.; Dembowsky, K.; Jing, S.; Field, L.J. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ. Res. 2000, 86, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shao, Q.; Korantzopoulos, P.; Liu, E.; Xu, G.; Li, G. Serum levels of nicotinamide adenine dinucleotide phosphate oxidase 4 are associated with non-valvular atrial fibrillation. Biomed Rep. 2015, 3, 864–868. [Google Scholar] [CrossRef]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation, a translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Heijman, J.; Voigt, N.; Nattel, S.; Dobrev, D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ. Res. 2014, 114, 1483–1499. [Google Scholar] [CrossRef]
- Nattel, S.; Dobrev, D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat. Rev. Cardiol. 2016, 13, 575–590. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, W.J. Calcium sparks. Physiol. Rev. 2008, 88, 1491–1545. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca2+/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef]
- Chang, S.H.; Yeh, Y.H.; Lee, J.L.; Hsu, Y.J.; Kuo, C.T.; Chen, W.J. Transforming growth factor-β-mediated CD44/STAT3 signaling contributes to the development of atrial fibrosis and fibrillation. Basic Res. Cardiol. 2017, 112, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Chang, S.H.; Chan, Y.H.; Lee, J.L.; Lai, Y.J.; Chang, G.J.; Tsai, F.C.; Yeh, Y.H. Tachycardia-induced CD44/NOX4 signaling is involved in the development of atrial remodeling. J. Mol. Cell. Cardiol. 2019, 135, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Zhou, R.H.; Vendrov, A.E.; Tchivilev, I.; Niu, X.L.; Molnar, K.C.; Rojas, M.; Carter, J.D.; Tong, H.; Stouffer, G.A.; Madamanchi, N.R.; et al. Mitochondrial oxidative stress in aortic stiffening with age, the role of smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 745–755. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell. 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Valli, H.; Ahmad, S.; Chadda, K.R.; Al-Hadithi, A.B.A.K.; Grace, A.A.; Jeevaratnam, K.; Huang, C.L. Age-dependent atrial arrhythmic phenotype secondary to mitochondrial dysfunction in Pgc-1β deficient murine hearts. Mech. Ageing Dev. 2017, 167, 30–45. [Google Scholar] [CrossRef]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef]
- Schaper, J.; Meiser, E.; Stämmler, G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ. Res. 1985, 56, 377–391. [Google Scholar] [CrossRef]
- Morillo, C.A.; Klein, G.J.; Jones, D.L.; Guiraudon, C.M. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995, 91, 1588–1595. [Google Scholar]
- Ausma, J.; Wijffels, M.; Thoné, F.; Wouters, L.; Allessie, M.; Borgers, M. Structural changes of atrial myocardium due to sustained atrial fibrillation in the goat. Circulation 1997, 96, 3157–3163. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.M.S.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Lin, P.H.; Lee, S.H.; Su, C.P.; Wei, Y.H. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic. Biol. Med. 2003, 35, 1310–1318. [Google Scholar] [CrossRef]
- Tsuboi, M.; Hisatome, I.; Morisaki, T.; Tanaka, M.; Tomikura, Y.; Takeda, S.; Shimoyama, M.; Ohtahara, A.; Ogino, K.; Igawa, O.; et al. Mitochondrial DNA deletion associated with the reduction of adenine nucleotides in human atrium and atrial fibrillation. Eur. J. Clin. Investig. 2001, 31, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Pool, L.; Knops, P.; Manintveld, O.C.; Brugts, J.J.; Theuns, D.A.M.J.; Brundel, B.J.J.M.; de Groot, N.M.S. The HF-AF ENERGY Trial, Nicotinamide riboside for the treatment of atrial fibrillation in heart failure patients. Cardiovasc. Drugs Ther. 2002. [Google Scholar] [CrossRef]
- Bukowska, A.; Schild, L.; Keilhoff, G.; Hirte, D.; Neumann, M.; Gardemann, A.; Neumann, K.H.; Röhl, F.W.; Huth, C.; Goette, A.; et al. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp. Biol. Med. 2008, 233, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Muszyński, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation-Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Ad, N.; Schneider, A.; Khaliulin, I.; Borman, J.B.; Schwalb, H. Impaired mitochondrial response to simulated ischemic injury as a predictor of the development of atrial fibrillation after cardiac surgery, in vitro study in human myocardium. J. Thorac. Cardiovasc. Surg. 2005, 129, 41–45. [Google Scholar] [CrossRef]
- Pool, L.; Wijdeveld, L.F.J.M.; de Groot, N.M.S.; Brundel, B.J.J.M. The Role of mitochondrial dysfunction in atrial fibrillation, Translation to druggable target and biomarker discovery. Int. J. Mol. Sci. 2021, 22, 8463. [Google Scholar] [CrossRef]
- Manning, A.S.; Coltart, D.J.; Hearse, D.J. Ischemia and reperfusion-induced arrhythmias in the rat. Effects of xanthine oxidase inhibition with allopurinol. Circ. Res. 1984, 55, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Fosset, M.; De Weille, J.R.; Green, R.D.; Schmid-Antomarchi, H.; Lazdunski, M. Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. J. Biol. Chem. 1988, 263, 7933–7936. [Google Scholar] [CrossRef] [PubMed]
- Faivre, J.F.; Findlay, I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim. Biophys. Acta 1990, 1029, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional role of mitochondria in arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar]
- Brown, D.A.; Aon, M.A.; Frasier, C.R.; Sloan, R.C.; Maloney, A.H.; Anderson, E.J.; O’Rourke, B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J. Mol. Cell. Cardiol. 2010, 48, 673–679. [Google Scholar] [CrossRef]
- Chen, F.; De Diego, C.; Xie, L.H.; Yang, J.H.; Klitzner, T.S.; Weiss, J.N. Effects of metabolic inhibition on conduction, Ca transients, and arrhythmia vulnerability in embryonic mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2472–H2478. [Google Scholar] [CrossRef]
- Kurokawa, S.; Niwano, S.; Niwano, H.; Ishikawa, S.; Kishihara, J.; Aoyama, Y.; Kosukegawa, T.; Masaki, Y.; Izumi, T. Progression of ventricular remodeling and arrhythmia in the primary hyperoxidative state of glutathione-depleted rats. Circ. J. 2011, 75, 1386–1393. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Williams, G.R.; Rubinstein, J.D.; Hogue, T.S.; Horwitz, L.D.; Reiter, M.J. Hydrogen peroxide decreases effective refractory period in the isolated heart. Free Radic. Biol. Med. 1991, 11, 529–535. [Google Scholar] [CrossRef]
- King, J.H.; Huang, C.L.; Fraser, J.A. Determinants of myocardial conduction velocity, implications for arrhythmogenesis. Front. Physiol. 2013, 4, 154. [Google Scholar] [CrossRef]
- Gaspo, R.; Bosch, R.F.; Bou-Abboud, E.; Nattel, S. Tachycardia-induced changes in Na+ current in a chronic dog model of atrial fibrillation. Circ. Res. 1997, 81, 1045–1052. [Google Scholar] [CrossRef]
- Liu, M.; Liu, H.; Dudley, S.C., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Jin, D.; Hata, M.; Takai, S.; Yamanishi, K.; Shen, W.; El-Darawish, Y.; Yamanishi, H.; Okamura, H. Dysfunction of mitochondria and deformed gap junctions in the heart of IL-18-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H313–H325. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Rutledge, C.A.; Jeong, E.M.; Dolmatova, E.; Arasu, D.; Liu, H.; Vahdani, N.; Gu, L.; Zandieh, S.; Xiao, L.; et al. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ. Arrhythm. Electrophysiol. 2013, 6, 623–631. [Google Scholar] [CrossRef]
- Joshi, K.K.; Tiru, M.; Chin, T.; Fox, M.T.; Stefan, M.S. Postoperative atrial fibrillation in patients undergoing non-cardiac non-thoracic surgery, A practical approach for the hospitalist. Hosp. Pract. 2015, 43, 235–244. [Google Scholar] [CrossRef]
- Youn, J.Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation, an emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Marchioli, R.; Silletta, M.G.; Masson, S.; Sellke, F.W.; Libby, P.; Milne, G.L.; Brown, N.J.; Lombardi, F.; Damiano, R.J., Jr.; et al. Oxidative Stress biomarkers and incidence of postoperative atrial fibrillation in the Omega-3 fatty acids for prevention of postoperative atrial fibrillation (OPERA) trial. J. Am. Heart Assoc. 2015, 4, e001886. [Google Scholar] [CrossRef]
- Chang, J.P.; Chen, M.C.; Liu, W.H.; Yang, C.H.; Chen, C.J.; Chen, Y.L.; Pan, K.L.; Tsai, T.H.; Chang, H.W. Atrial myocardial NOX2 containing NADPH oxidase activity contribution to oxidative stress in mitral regurgitation, potential mechanism for atrial remodeling. Cardiovasc. Pathol. 2011, 20, 99–106. [Google Scholar] [CrossRef]
- Antoniades, C.; Demosthenous, M.; Reilly, S.; Margaritis, M.; Zhang, M.H.; Antonopoulos, A.; Marinou, K.; Nahar, K.; Jayaram, R.; Tousoulis, D.; et al. Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. J. Am. Coll. Cardiol. 2012, 59, 60–70. [Google Scholar] [CrossRef]
- Adam, O.; Frost, G.; Custodis, F.; Sussman, M.A.; Schafers, H.J.; Bohm, M.; Laufs, U. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 359–367. [Google Scholar] [CrossRef]
- Lavall, D.; Schuster, P.; Jacobs, N.; Kazakov, A.; Böhm, M.; Laufs, U. Rac1 GTPase regulates 11β hydroxysteroid dehydrogenase type 2 and fibrotic remodeling. J. Biol. Chem. 2017, 292, 7542–7553. [Google Scholar] [CrossRef]
- Sahoo, S.; Meijles, D.N.; Pagano, P.J. NADPH oxidases, key modulators in aging and age-related cardiovascular diseases? Clin. Sci. 2016, 130, 317–335. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.D.; Hong, L.; Sridhar, A.; Menon, A.; Perike, S.; Zhang, M.; da Silva, I.B.; Yan, J.; Bonini, M.G.; Ai, X.; et al. Ion channel and structural remodeling in obesity-mediated atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2020, 13, e008296. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Lefebvre, P.; Modine, T.; Fayad, G.; Dehondt, H.; Hurt, C.; Coisne, A.; Koussa, M.; Remy-Jouet, I.; et al. Mitochondrial dysfunction as an arrhythmogenic substrate, a translational proof-of-concept study in patients with metabolic syndrome in whom post-operative atrial fibrillation develops. J. Am. Coll. Cardiol. 2013, 62, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, J.; Saraf, R.; Mahmood, F.; Pal, A.; Bhasin, M.K.; Huang, T.; Mittel, A.; Knio, Z.; Simons, R.; Khabbaz, K.; et al. Mitochondrial dysfunction in atrial tissue of patients developing postoperative atrial fibrillation. Ann. Thorac. Surg. 2017, 104, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Mihm, M.J.; Yu, F.; Carnes, C.A.; Reiser, P.J.; McCarthy, P.M.; Van Wagoner, D.R.; Bauer, J.A. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 2001, 104, 174–180. [Google Scholar] [CrossRef]
- Lamirault, G.; Gaborit, N.; Le Meur, N.; Chevalier, C.; Lande, G.; Demolombe, S.; Escande, D.; Nattel, S.; Léger, J.J.; Steenman, M. Gene expression profile associated with chronic atrial fibrillation and underlying valvular heart disease in man. J. Mol. Cell. Cardiol. 2006, 40, 173–184. [Google Scholar] [CrossRef]
- Brown, D.A.; O’Rourke, B. Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 2010, 88, 241–249. [Google Scholar] [CrossRef]
- Jeong, E.M.; Liu, M.; Sturdy, M.; Gao, G.; Varghese, S.T.; Sovari, A.A.; Dudley, S.C., Jr. Metabolic stress, reactive oxygen species, and arrhythmia. J. Mol. Cell. Cardiol. 2012, 52, 454–463. [Google Scholar] [CrossRef]
- Kanaan, G.N.; Patten, D.A.; Redpath, C.J.; Harper, M.E. Atrial fibrillation is associated with impaired atrial mitochondrial energetics and supercomplex formation in adults with type 2 diabetes. Can. J. Diabetes 2019, 43, 67–75.e1. [Google Scholar] [CrossRef]
- Lenaerts, I.; Holemans, P.; Pokreisz, P.; Sipido, K.R.; Janssens, S.; Heidbüchel, H.; Willems, R. Nitric oxide delays atrial tachycardia-induced electrical remodelling in a sheep model. Europace 2011, 13, 747–754. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, Z.; Santulli, G.; Gu, L.; Yang, Z.G.; Yuan, Z.Q.; Zhao, Y.T.; Xin, H.B.; Deng, K.Y.; Wang, S.Q.; et al. Functional role of Calstabin2 in age-related cardiac alterations. Sci. Rep. 2014, 4, 7425. [Google Scholar] [CrossRef] [PubMed]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L., Jr.; Roden, D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.N.; Tester, D.J.; Perry, J.; Salisbury, B.A.; Reed, C.R.; Ackerman, M.J. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 2008, 5, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.D.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef] [PubMed]
- Avula, U.M.R.; Dridi, H.; Chen, B.X.; Yuan, Q.; Katchman, A.N.; Reiken, S.R.; Desai, A.D.; Parsons, S.; Baksh, H.; Ma, E.; et al. Attenuating persistent sodium current-induced atrial myopathy and fibrillation by preventing mitochondrial oxidative stress. JCI Insight 2021, 6, e147371. [Google Scholar] [CrossRef]
- Kim, Y.H.; Lim, D.S.; Lee, J.H.; Shim, W.J.; Ro, Y.M.; Park, G.H.; Becker, K.G.; Cho-Chung, Y.S.; Kim, M.K. Gene expression profiling of oxidative stress on atrial fibrillation in humans. Exp. Mol. Med. 2003, 35, 336–349. [Google Scholar] [CrossRef]
- Neuman, R.B.; Bloom, H.L.; Shukrullah, I.; Darrow, L.A.; Kleinbaum, D.; Jones, D.P.; Dudley, S.C., Jr. Oxidative stress markers are associated with persistent atrial fibrillation. Clin. Chem. 2007, 53, 1652–1657. [Google Scholar] [CrossRef]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef]
- Martins, R.P.; Kaur, K.; Hwang, E.; Ramirez, R.J.; Willis, B.C.; Filgueiras-Rama, D.; Ennis, S.R.; Takemoto, Y.; Ponce-Balbuena, D.; Zarzoso, M.; et al. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 2014, 129, 1472–1482. [Google Scholar] [CrossRef]
- Alvarez-Franco, A.; Rouco, R.; Ramirez, R.J.; Guerrero-Serna, G.; Tiana, M.; Cogliati, S.; Kaur, K.; Saeed, M.; Magni, R.; Enriquez, J.A.; et al. Transcriptome and proteome mapping in the sheep atria reveal molecular features of atrial fibrillation progression. Cardiovasc. Res. 2021, 117, 1760–1775. [Google Scholar] [CrossRef]
- Goette, A.; Honeycutt, C.; Langberg, J.J. Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 1996, 94, 2968–2974. [Google Scholar]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef]
- Van Wagoner, D.R.; Pond, A.L.; Lamorgese, M.; Rossie, S.S.; McCarthy, P.M.; Nerbonne, J.M. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 1999, 85, 428–436. [Google Scholar] [CrossRef]
- Voigt, N.; Trausch, A.; Knaut, M.; Matschke, K.; Varró, A.; Van Wagoner, D.R.; Nattel, S.; Ravens, U.; Dobrev, D. Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2010, 3, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Caballero, R.; de la Fuente, M.G.; Gómez, R.; Barana, A.; Amorós, I.; Dolz-Gaitón, P.; Osuna, L.; Almendral, J.; Atienza, F.; Fernández-Avilés, F.; et al. In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J. Am. Coll. Cardiol. 2010, 55, 2346–2354. [Google Scholar] [CrossRef]
- Reilly, S.N.; Liu, X.; Carnicer, R.; Recalde, A.; Muszkiewicz, A.; Jayaram, R.; Carena, M.C.; Wijesurendra, R.; Stefanini, M.; Surdo, N.C.; et al. Up-regulation of miR-31 in human atrial fibrillation begets the arrhythmia by depleting dystrophin and neuronal nitric oxide synthase. Sci. Transl. Med. 2016, 8, 340ra74. [Google Scholar] [PubMed]
- Williams, J.C.; Armesilla, A.L.; Mohamed, T.M.; Hagarty, C.L.; McIntyre, F.H.; Schomburg, S.; Zaki, A.O.; Oceandy, D.; Cartwright, E.J.; Buch, M.H.; et al. The sarcolemmal calcium pump, alpha-1 syntrophin, and neuronal nitric-oxide synthase are parts of a macromolecular protein complex. J. Biol. Chem. 2006, 281, 23341–23348. [Google Scholar] [CrossRef]
- Tamargo, J.; Caballero, R.; Gómez, R.; Delpón, E. Cardiac electrophysiological effects of nitric oxide. Cardiovasc. Res. 2010, 87, 593–600. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Casadei, B. Sub-cellular targeting of constitutive NOS in health and disease. J. Mol. Cell. Cardiol. 2012, 52, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Sears, C.E.; Bryant, S.M.; Watkins, H.C.; Casadei, B. Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation 2002, 105, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Jian, Z.; Han, H.; Zhang, T.; Puglisi, J.; Izu, L.T.; Shaw, J.A.; Onofiok, E.; Erickson, J.R.; Chen, Y.J.; Horvath, B.; et al. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci. Signal. 2014, 7, ra27. [Google Scholar] [CrossRef] [PubMed]
- Hatem, S.N. Revealing the molecular history of the transition from paroxysmal to permanent atrial fibrillation. Cardiovasc. Res. 2021, 117, 1612–1613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-Te Bulte, L.; Wiersma, M.; Malik, N.U.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; et al. DNA damage induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1307. [Google Scholar]
- Lin, Y.K.; Lin, F.Z.; Chen, Y.C.; Cheng, C.C.; Lin, C.I.; Chen, Y.J.; Chen, S.A. Oxidative stress on pulmonary vein and left atrium arrhythmogenesis. Circ. J. 2010, 74, 1547–1556. [Google Scholar] [CrossRef]
- Pezhouman, A.; Cao, H.; Fishbein, M.C.; Belardinelli, L.; Weiss, J.N.; Karagueuzian, H.S. Atrial fibrillation initiated by early afterdepolarization-mediated triggered activity during acute oxidative stress, efficacy of late sodium current blockade. J. Heart Health 2018, 4, 10. [Google Scholar]
- Huang, M.; Fan, X.; Yang, Z.; Cyganek, L.; Li, X.; Yuecel, G.; Lan, H.; Li, Y.; Wendel, A.; Lang, S.; et al. Alpha 1-adrenoceptor signalling contributes to toxic effects of catecholamine on electrical properties in cardiomyocytes. Europace 2021, 23, 1137–1148. [Google Scholar] [CrossRef]
- Huang, M.; Yang, Z.; Li, Y.; Lan, H.; Cyganek, L.; Yuecel, G.; Lang, S.; Bieback, K.; El-Battrawy, I.; Zhou, X.; et al. Dopamine D1/D5 Receptor Signaling Is Involved in Arrhythmogenesis in the Setting of Takotsubo Cardiomyopathy. Front. Cardiovasc. Med. 2022, 8, 777463. [Google Scholar]
- Van Wagoner, D.R.; Pond, A.L.; McCarthy, P.M.; Trimmer, J.S.; Nerbonne, J.M. Outward K+ current densities and Kv1.5 expression are reduced in chronic human atrial fibrillation. Circ. Res. 1997, 80, 772–781. [Google Scholar] [CrossRef]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef]
- Núñez, L.; Vaquero, M.; Gómez, R.; Caballero, R.; Mateos-Cáceres, P.; Macaya, C.; Iriepa, I.; Gálvez, E.; López-Farré, A.; Tamargo, J.; et al. Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc. Res. 2006, 72, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef]
- Bers, D.M. Excitation-Contraction Coupling and Cardiac Contractile Force; Kluwer Academic Publishers Group: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Blatter, L.A.; Kockskämper, J.; Sheehan, K.A.; Zima, A.V.; Hüser, J.; Lipsius, S.L. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J. Physiol. 2003, 546, 19–31. [Google Scholar] [CrossRef]
- Sheehan, K.A.; Zima, A.V.; Blatter, L.A. Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J. Physiol. 2006, 572, 799–809. [Google Scholar] [CrossRef]
- Freestone, N.S.; Ribaric, S.; Scheuermann, M.; Mauser, U.; Paul, M.; Vetter, R. Differential lusitropic responsiveness to beta-adrenergic stimulation in rat atrial and ventricular cardiac myocytes. Pflugers Arch. 2000, 441, 78–87. [Google Scholar] [CrossRef]
- Walden, A.P.; Dibb, K.M.; Trafford, A.W. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 463–473. [Google Scholar] [CrossRef]
- Venetucci, L.A.; Trafford, A.W.; O’Neill, S.C.; Eisner, D.A. The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 2008, 77, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak, basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef]
- Bootman, M.D.; Smyrnias, I.; Thul, R.; Coombes, S.; Roderick, H.L. Atrial cardiomyocyte calcium signalling. Biochim. Biophys. Acta 2011, 1813, 922–934. [Google Scholar]
- Cherednichenko, G.; Zima, A.V.; Feng, W.; Schaefer, S.; Blatter, L.A.; Pessah, I.N. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ. Res. 2004, 94, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, G.; Pedrozo, Z.; Domenech, R.J.; Hidalgo, C.; Donoso, P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell. Cardiol. 2005, 39, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Nikolaienko, R.; Bovo, E.; Zima, A.V. Redox dependent modifications of ryanodine receptor, Basic mechanisms and implications in heart diseases. Front. Physiol. 2018, 9, 1775. [Google Scholar] [CrossRef] [PubMed]
- Anzai, K.; Ogawa, K.; Kuniyasu, A.; Ozawa, T.; Yamamoto, H.; Nakayama, H. Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem. Biophys. Res. Commun. 1998, 249, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Boraso, A.; Williams, A.J. Modification of the gating of the cardiac sarcoplasmic reticulum Ca(2+)-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 1994, 267, H1010–H1016. [Google Scholar] [CrossRef]
- Terentyev, D.; Györke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef]
- Sánchez, G.; Escobar, M.; Pedrozo, Z.; Macho, P.; Domenech, R.; Härtel, S.; Hidalgo, C.; Donoso, P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity, possible role in cardioprotection. Cardiovasc. Res. 2008, 77, 380–386. [Google Scholar] [CrossRef]
- Cotton, J.M.; Kearney, M.T.; Shah, A.M. Nitric oxide and myocardial function in heart failure, friend or foe? Heart 2002, 88, 564–566. [Google Scholar] [CrossRef]
- Gonzalez, D.R.; Beigi, F.; Treuer, A.V.; Hare, J.M. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 20612–20617. [Google Scholar] [CrossRef]
- Waddell, H.M.M.; Zhang, J.Z.; Hoeksema, K.J.; McLachlan, J.J.; McLay, J.C.; Jones, P.P. Oxidation of RyR2 has a biphasic effect on the threshold for store overload-induced calcium release. Biophys. J. 2016, 110, 2386–2396. [Google Scholar] [CrossRef]
- Vest, J.A.; Wehrens, X.H.; Reiken, S.R.; Lehnart, S.E.; Dobrev, D.; Chandra, P.; Danilo, P.; Ravens, U.; Rosen, M.R.; Marks, A.R. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005, 111, 2025–2032. [Google Scholar] [CrossRef]
- Sood, S.; Chelu, M.G.; van Oort, R.J.; Skapura, D.; Santonastasi, M.; Dobrev, D.; Wehrens, X.H. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 2008, 5, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Roussel, J.; Thireau, J.; Brenner, C.; Saint, N.; Scheuermann, V.; Lacampagne, A.; Le Guennec, J.Y.; Fauconnier, J. Palmitoyl-carnitine increases RyR2 oxidation and sarcoplasmic reticulum Ca2+ leak in cardiomyocytes, Role of adenine nucleotide translocase. Biochim. Biophys. Acta. 2015, 1852, 749–758. [Google Scholar] [CrossRef]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef]
- Hove-Madsen, L.; Llach, A.; Bayes-Genís, A.; Roura, S.; Rodriguez Font, E.; Arís, A.; Cinca, J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004, 110, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- El-Armouche, A.; Boknik, P.; Eschenhagen, T.; Carrier, L.; Knaut, M.; Ravens, U.; Dobrev, D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 2006, 114, 670–680. [Google Scholar] [CrossRef]
- Burashnikov, A.; Antzelevitch, C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation 2003, 107, 2355–2360. [Google Scholar] [CrossRef]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Dudley, S.C., Jr. Oxidative stress and atrial fibrillation, finding a missing piece to the puzzle. Circulation 2013, 128, 1724–1726. [Google Scholar] [CrossRef]
- Mesubi, O.O.; Rokita, A.G.; Abrol, N.; Wu, Y.; Chen, B.; Wang, Q.; Granger, J.M.; Tucker-Bartley, A.; Luczak, E.D.; Murphy, K.R.; et al. Oxidized CaMKII and O-GlcNAcylation cause increased atrial fibrillation in diabetic mice by distinct mechanisms. J. Clin. Investig. 2021, 131, e95747. [Google Scholar] [CrossRef]
- Yang, X.; An, N.; Zhong, C.; Guan, M.; Jiang, Y.; Li, X.; Zhang, H.; Wang, L.; Ruan, Y.; Gao, Y.; et al. Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib-induced atrial fibrillation. Redox Biol. 2020, 30, 101432. [Google Scholar] [CrossRef]
- Jiang, L.; Li, L.; Ruan, Y.; Zuo, S.; Wu, X.; Zhao, Q.; Xing, Y.; Zhao, X.; Xia, S.; Bai, R.; et al. Ibrutinib promotes atrial fibrillation by inducing structural remodeling and calcium dysregulation in the atrium. Heart Rhythm 2019, 16, 1374–1382. [Google Scholar] [CrossRef]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.M.; Hajjar, R.J. Sarcoplasmic reticulum Ca(2+) ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Ramírez, F.; Ramos-Mondragón, R.; García-Rivas, G. Mitochondrial and sarcoplasmic reticulum interconnection in cardiac arrhythmia. Front. Cell Dev. Biol. 2021, 8, 623381. [Google Scholar] [CrossRef]
- Qin, F.; Siwik, D.A.; Lancel, S.; Zhang, J.; Kuster, G.M.; Luptak, I.; Wang, L.; Tong, X.; Kang, Y.J.; Cohen, R.A.; et al. Hydrogen peroxide-mediated SERCA cysteine 674 oxidation contributes to impaired cardiac myocyte relaxation in senescent mouse heart. J. Am. Heart Assoc. 2013, 2, e000184. [Google Scholar] [CrossRef]
- Hobai, I.A.; Buys, E.S.; Morse, J.C.; Edgecomb, J.; Weiss, E.H.; Armoundas, A.A.; Hou, X.; Khandelwal, A.R.; Siwik, D.A.; Brouckaert, P.; et al. SERCA Cys674 sulphonylation and inhibition of L-type Ca2+ influx contribute to cardiac dysfunction in endotoxemic mice, independent of cGMP synthesis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1189–H1200. [Google Scholar] [CrossRef] [PubMed]
- Kuster, G.M.; Lancel, S.; Zhang, J.; Communal, C.; Trucillo, M.P.; Lim, C.C.; Pfister, O.; Weinberg, E.O.; Cohen, R.A.; Liao, R.; et al. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free Radic. Biol. Med. 2010, 48, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Herraiz-Martínez, A.; Álvarez-García, J.; Llach, A.; Molina, C.E.; Fernandes, J.; Ferrero-Gregori, A.; Rodríguez, C.; Vallmitjana, A.; Benítez, R.; Padró, J.M.; et al. Ageing is associated with deterioration of calcium homeostasis in isolated human right atrial myocytes. Cardiovasc. Res. 2015, 106, 76–86. [Google Scholar] [CrossRef]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.H.; Casadei, B.; Watkins, H.; Redwood, C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem. 2018, 293, 10487–10499. [Google Scholar] [CrossRef]
- Beuckelmann, D.J.; Erdmann, E. Ca(2+)-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res. Cardiol. 1992, 87 (Suppl. 1), 235–243. [Google Scholar]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.B.; Qin, F.; Morgan, R.J.; Chambers, J.M.; Croteau, D.; Siwik, D.A.; Hobai, I.; Panagia, M.; Luptak, I.; Bachschmid, M.; et al. Redox-resistant SERCA [Sarco(endo)plasmic Reticulum Calcium ATPase] attenuates oxidant-stimulated mitochondrial calcium and apoptosis in cardiac myocytes and pressure overload-induced myocardial failure in mice. Circulation 2020, 142, 2459–2469. [Google Scholar] [CrossRef] [PubMed]
- Cheniti, G.; Vlachos, K.; Pambrun, T.; Hooks, D.; Frontera, A.; Takigawa, M.; Bourier, F.; Kitamura, T.; Lam, A.; Martin, C.; et al. Atrial fibrillation mechanisms and implications for catheter ablation. Front. Physiol. 2018, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Waks, J.W.; Josephson, M.E. Mechanisms of atrial fibrillation—Reentry, rotors and reality. Arrhythm. Electrophysiol. Rev. 2014, 3, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.Y.; Yeh, Y.H.; Xiao, L.; Burstein, B.; Maguy, A.; Chartier, D.; Villeneuve, L.R.; Brundel, B.J.; Dobrev, D.; Nattel, S. Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circ. Res. 2008, 103, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Carnes, C.A.; Janssen, P.M.; Ruehr, M.L.; Nakayama, H.; Nakayama, T.; Haase, H.; Bauer, J.A.; Chung, M.K.; Fearon, I.M.; Gillinov, A.M.; et al. Atrial glutathione content, calcium current, and contractility. J. Biol. Chem. 2007, 282, 28063–28073. [Google Scholar] [CrossRef]
- Huang, S.Y.; Lu, Y.Y.; Chen, Y.C.; Chen, W.T.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Hydrogen peroxide modulates electrophysiological characteristics of left atrial myocytes. Acta Cardiol. Sin. 2014, 30, 38–45. [Google Scholar]
- Liu, M.; Sanyal, S.; Gao, G.; Gurung, I.S.; Zhu, X.; Gaconnet, G.; Kerchner, L.J.; Shang, L.L.; Huang, C.L.; Grace, A.; et al. Cardiac Na+ current regulation by pyridine nucleotides. Circ. Res. 2009, 105, 737–745. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Zhang, H. The effect of nicotinamide adenine dinucleotide (NADH) on electrical remodeling in atrial fibrillation. Int. J. Clin. Exp. Med. 2016, 9, 23513–23518. [Google Scholar]
- Shang, L.L.; Sanyal, S.; Pfahnl, A.E.; Jiao, Z.; Allen, J.; Liu, H.; Dudley, S.C., Jr. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am. J. Physiol. Cell Physiol. 2008, 294, C372–C379. [Google Scholar] [CrossRef]
- Carter, M.E.; Brunet, A. FOXO transcription factors. Curr. Biol. 2007, 17, R113–R114. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; You, T.; Ye, B.; Li, X.; Dong, H.H.; Hill, J.A.; Li, F.; Xu, H. Reactive oxygen species suppress cardiac NaV1.5 expression through Foxo1. PLoS ONE 2012, 7, e32738. [Google Scholar] [CrossRef] [PubMed]
- Tristani-Firouzi, M.; Chen, J.; Mitcheson, J.S.; Sanguinetti, M.C. Molecular biology of K(+) channels and their role in cardiac arrhythmias. Am. J. Med. 2001, 110, 50–59. [Google Scholar] [CrossRef]
- Dhamoon, A.S.; Jalife, J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005, 2, 316–324. [Google Scholar] [CrossRef]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef]
- Pandit, S.V.; Berenfeld, O.; Anumonwo, J.M.; Zaritski, R.M.; Kneller, J.; Nattel, S.; Jalife, J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys. J. 2005, 88, 3806–3821. [Google Scholar] [CrossRef]
- van Veen, A.A.; van Rijen, H.V.; Opthof, T. Cardiac gap junction channels, modulation of expression and channel properties. Cardiovasc. Res. 2001, 51, 217–229. [Google Scholar] [CrossRef]
- Rohr, S. Role of gap junctions in the propagation of the cardiac action potential. Cardiovasc. Res. 2004, 62, 309–322. [Google Scholar] [CrossRef]
- Wakili, R.; Voigt, N.; Kääb, S.; Dobrev, D.; Nattel, S. Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Investig. 2011, 121, 2955–2968. [Google Scholar] [CrossRef]
- Kato, T.; Iwasaki, Y.K.; Nattel, S. Connexins and atrial fibrillation, filling in the gaps. Circulation 2012, 125, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.N.; Jan, L.Y. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef]
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.S.; Mihm, M.J.; Cook, A.C.; Schanbacher, B.L.; Bauer, J.A. Alterations in connexin 43 during diabetic cardiomyopathy, competition of tyrosine nitration versus phosphorylation. J. Diabetes 2015, 7, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Krul, S.P.; Berger, W.R.; Smit, N.W.; van Amersfoorth, S.C.; Driessen, A.H.; van Boven, W.J.; Fiolet, J.W.; van Ginneken, A.C.; van der Wal, A.C.; de Bakker, J.M.; et al. Atrial fibrosis and conduction slowing in the left atrial appendage of patients undergoing thoracoscopic surgical pulmonary vein isolation for atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2015, 8, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Grieve, D.J.; Byrne, J.A.; Siva, A.; Layland, J.; Johar, S.; Cave, A.C.; Shah, A.M. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J. Am. Coll. Cardiol. 2006, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhu, H.; Shen, E.; Wan, L.; Arnold, J.M.; Peng, T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010, 59, 2033–2042. [Google Scholar] [CrossRef]
- Nakano, Y.; Niida, S.; Dote, K.; Takenaka, S.; Hirao, H.; Miura, F.; Ishida, M.; Shingu, T.; Sueda, T.; Yoshizumi, M.; et al. Matrix metalloproteinase-9 contributes to human atrial remodeling during atrial fibrillation. J. Am. Coll. Cardiol. 2004, 43, 818–825. [Google Scholar] [CrossRef]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; van der Vliet, A.; Maeda, H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J. Biol. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell. Physiol. 2001, 280, C53–C60. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.; Petrov, V.; van Pelt, J.; Fagard, R. Inhibition of superoxide dismutase induces collagen production in cardiac fibroblasts. Am. J. Hypertens. 2008, 21, 1129–1136. [Google Scholar] [CrossRef]
- Moe, G.W.; Laurent, G.; Doumanovskaia, L.; Konig, A.; Hu, X.; Dorian, P. Matrix metalloproteinase inhibition attenuates atrial remodeling and vulnerability to atrial fibrillation in a canine model of heart failure. J. Card. Fail. 2008, 14, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Ke, L.; Qi, X.Y.; Dijkhuis, A.J.; Chartier, D.; Nattel, S.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J. Mol. Cell. Cardiol. 2008, 45, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; Kampinga, H.H.; Henning, R.H. Calpain inhibition prevents pacing-induced cellular remodeling in a HL-1 myocyte model for atrial fibrillation. Cardiovasc. Res. 2004, 62, 521–528. [Google Scholar] [CrossRef]
- Rennison, J.H.; Li, L.; Lin, C.R.; Lovano, B.S.; Castel, L.; Wass, S.Y.; Cantlay, C.C.; McHale, M.; Gillinov, A.M.; Mehra, R.; et al. Atrial fibrillation rhythm is associated with marked changes in metabolic and myofibrillar protein expression in left atrial appendage. Pflugers Arch. 2021, 473, 461–475. [Google Scholar] [PubMed]
- Andersen, J.S.; Egeblad, H.; Abildgaard, U.; Aldershvile, J.; Godtfredsen, J. Atrial fibrillation and left atrial enlargement, cause or effect? J. Intern. Med. 1991, 229, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Conen, D.; Glynn, R.J.; Sandhu, R.K.; Tedrow, U.B.; Albert, C.M. Risk factors for incident atrial fibrillation with and without left atrial enlargement in women. Int. J. Cardiol. 2013, 168, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Neuberger, H.R.; Allessie, M.A. The role of atrial dilatation in the domestication of atrial fibrillation. Prog. Biophys. Mol. Biol. 2003, 82, 151–162. [Google Scholar] [CrossRef]
- Tousoulis, D.; Zisimos, K.; Antoniades, C.; Stefanadi, E.; Siasos, G.; Tsioufis, C.; Papageorgiou, N.; Vavouranakis, E.; Vlachopoulos, C.; Stefanadis, C. Oxidative stress and inflammatory process in patients with atrial fibrillation, the role of left atrium distension. Int. J. Cardiol. 2009, 136, 258–262. [Google Scholar] [CrossRef]
- Chen, M.C.; Chang, J.P.; Liu, W.H.; Yang, C.H.; Chen, C.J.; Fang, C.Y.; Hsieh, Y.K.; Wang, Y.H.; Chang, H.W. Increased serum oxidative stress in patients with severe mitral regurgitation, a new finding and potential mechanism for atrial enlargement. Clin. Biochem. 2009, 42, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; He, Y.; Ke, H.H.; Jin, Y.; Jiang, Z.Y.; Zhong, G.Q. Plasma oxidative stress and inflammatory biomarkers are associated with the sizes of the left atrium and pulmonary vein in atrial fibrillation patients. Clin. Cardiol. 2017, 40, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Elnakish, M.T.; Ahmed, A.A.; Mohler, P.J.; Janssen, P.M. Role of oxidative stress in thyroid hormone-induced cardiomyocyte hypertrophy and associated cardiac dysfunction, an undisclosed story. Oxid. Med. Cell. Longev. 2015, 2015, 854265. [Google Scholar] [CrossRef]
- Moens, A.L.; Takimoto, E.; Tocchetti, C.G.; Chakir, K.; Bedja, D.; Cormaci, G.; Ketner, E.A.; Majmudar, M.; Gabrielson, K.; Halushka, M.K.; et al. Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin, efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 2008, 117, 2626–2636. [Google Scholar] [CrossRef]
- Li, J.M.; Gall, N.P.; Grieve, D.J.; Chen, M.; Shah, A.M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension 2002, 40, 477–484. [Google Scholar] [CrossRef]
- Murdoch, C.E.; Zhang, M.; Cave, A.C.; Shah, A.M. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodeling and failure. Cardiovasc. Res. 2006, 71, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Patten, M.; Pecha, S.; Aydin, A. Atrial Fibrillation in Hypertrophic Cardiomyopathy, Diagnosis and Considerations for Management. J. Atr. Fibrillation 2018, 10, 1556. [Google Scholar]
- De Marchi, S.F.; Allemann, Y.; Seiler, C. Relaxation in hypertrophic cardiomyopathy and hypertensive heart disease, relations between hypertrophy and diastolic function. Heart 2000, 83, 678–684. [Google Scholar] [CrossRef]
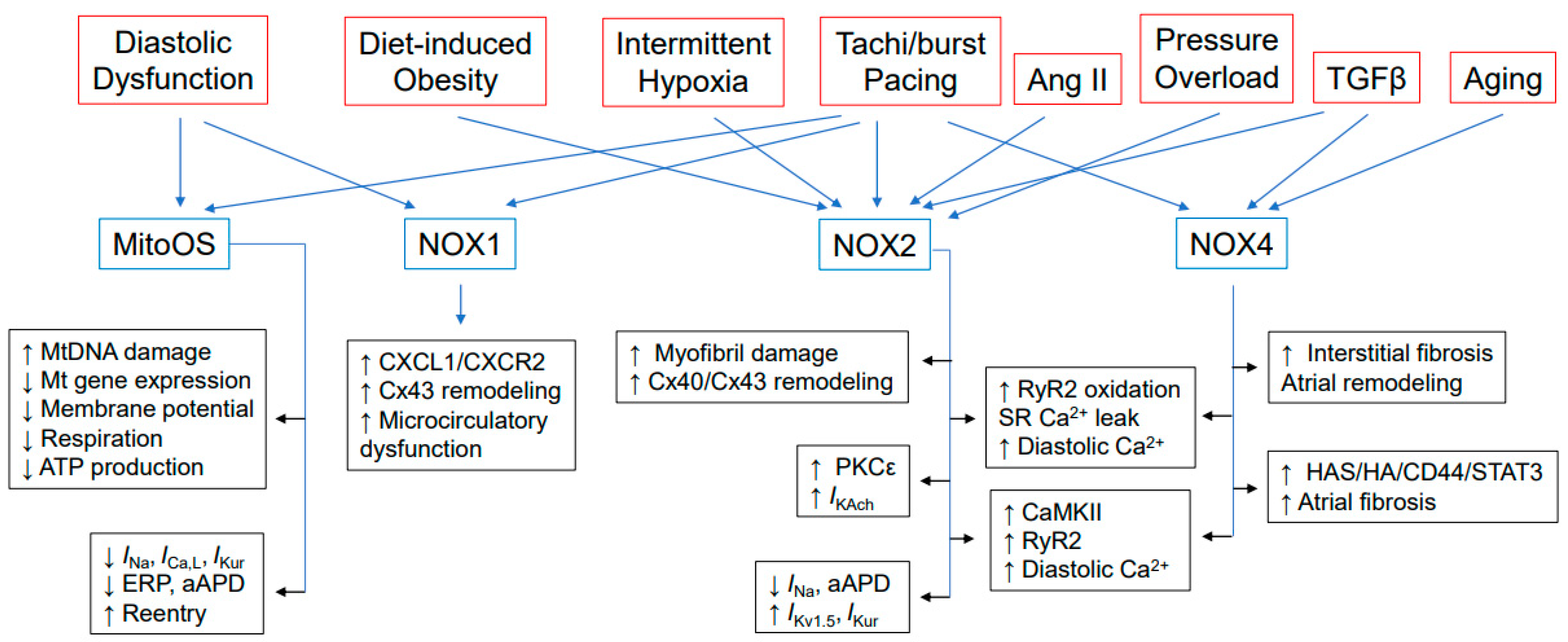
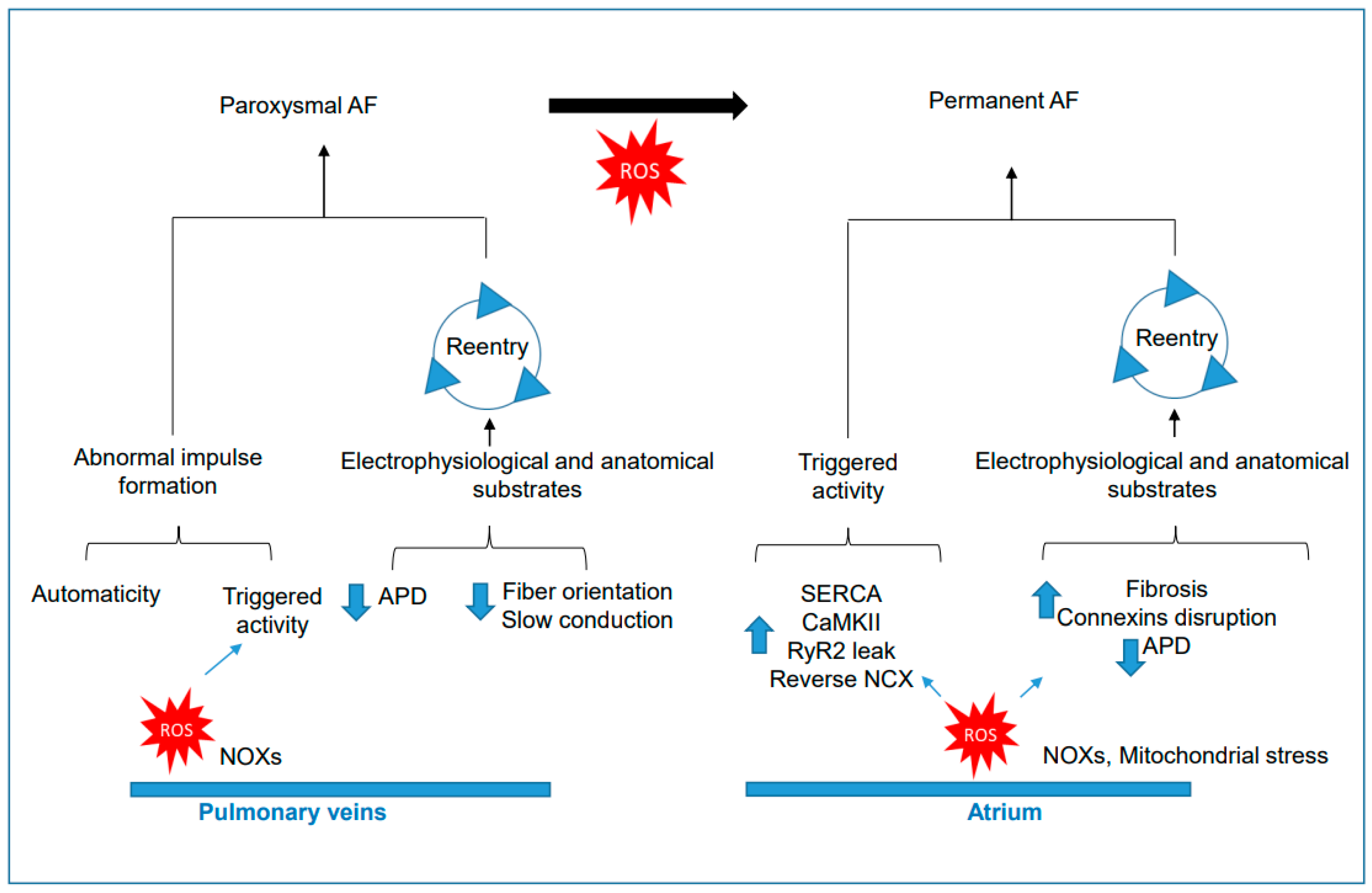
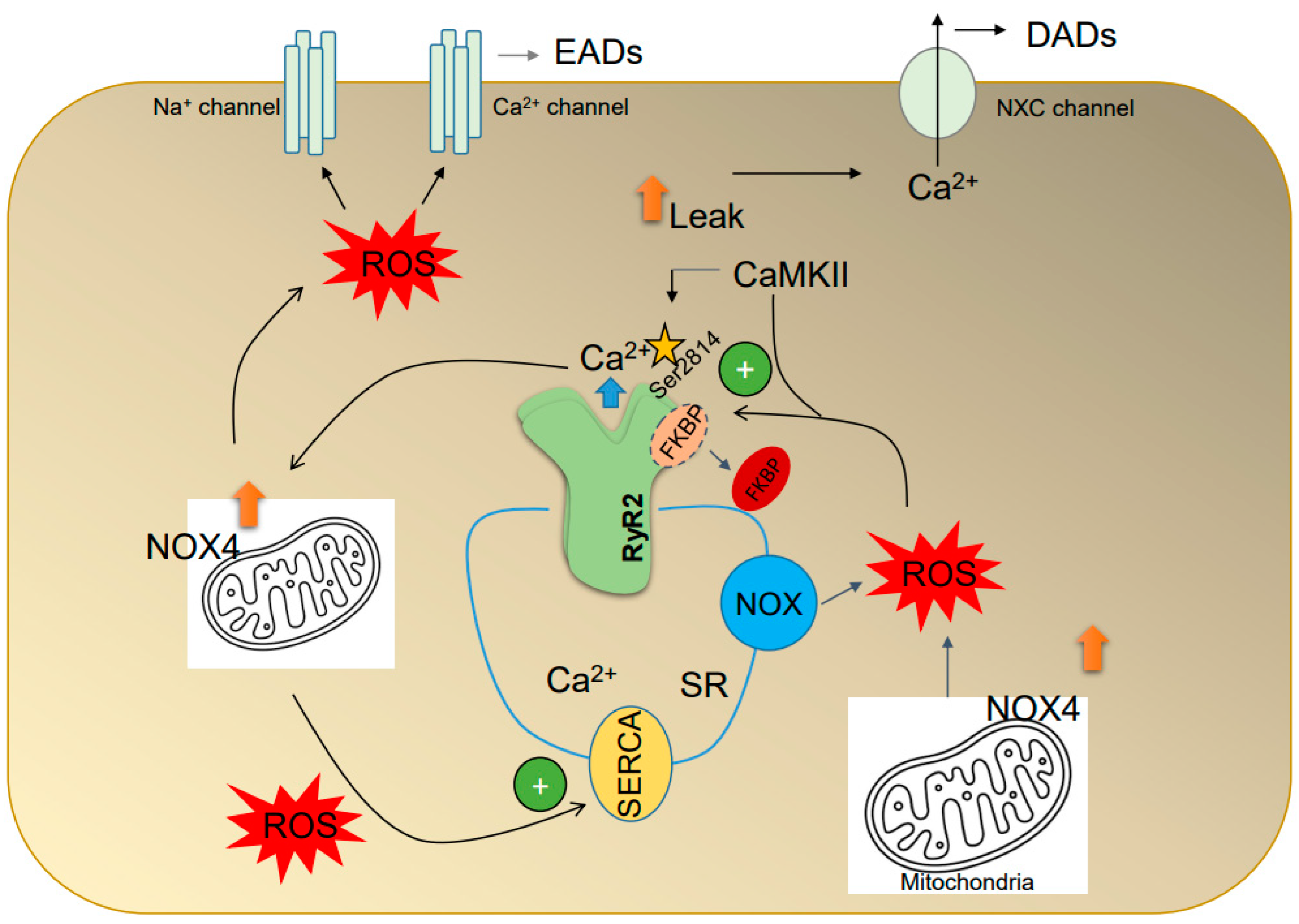
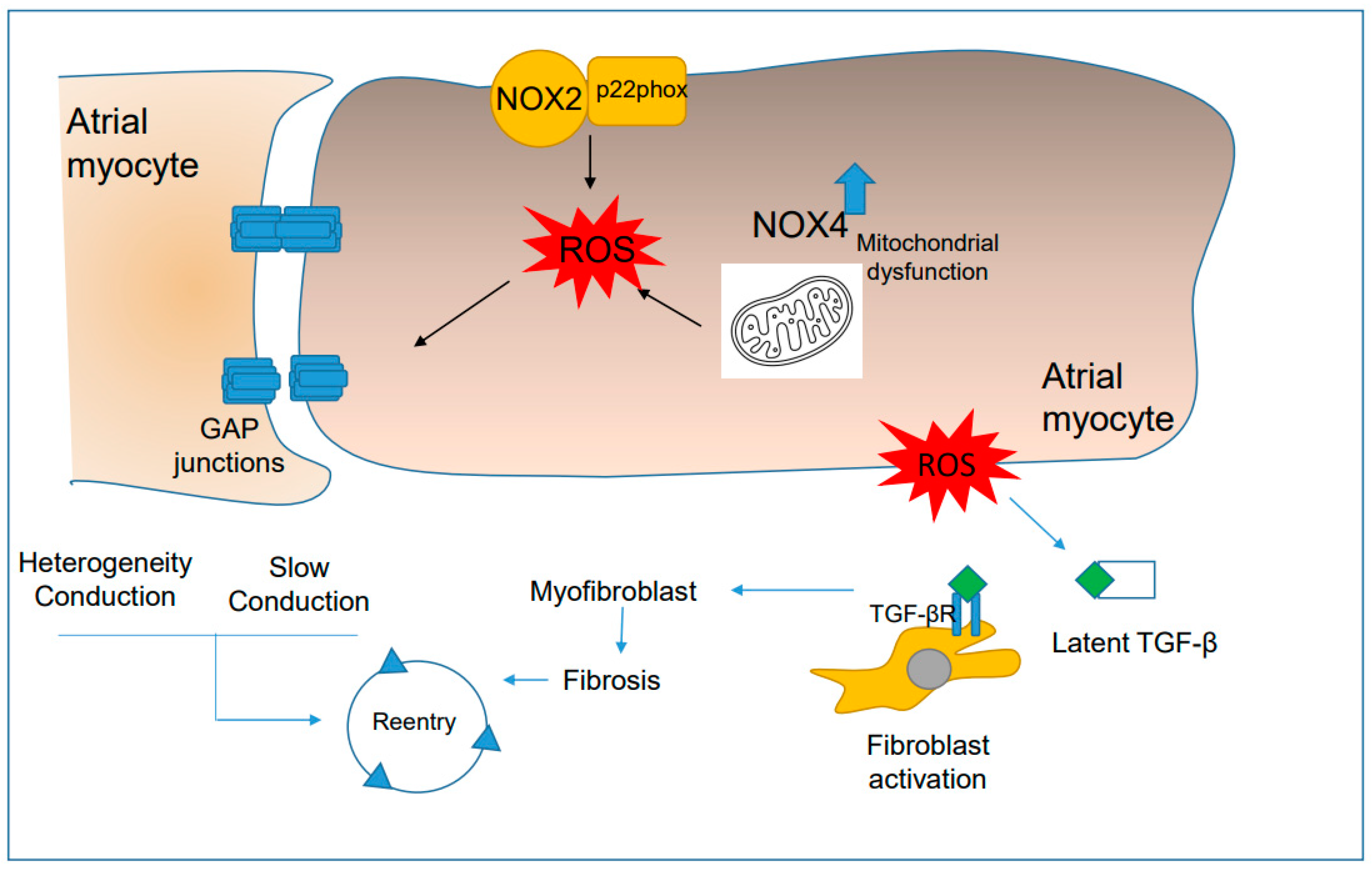
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos-Mondragón, R.; Lozhkin, A.; Vendrov, A.E.; Runge, M.S.; Isom, L.L.; Madamanchi, N.R. NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation. Antioxidants 2023, 12, 1833. https://doi.org/10.3390/antiox12101833
Ramos-Mondragón R, Lozhkin A, Vendrov AE, Runge MS, Isom LL, Madamanchi NR. NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation. Antioxidants. 2023; 12(10):1833. https://doi.org/10.3390/antiox12101833
Chicago/Turabian StyleRamos-Mondragón, Roberto, Andrey Lozhkin, Aleksandr E. Vendrov, Marschall S. Runge, Lori L. Isom, and Nageswara R. Madamanchi. 2023. "NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation" Antioxidants 12, no. 10: 1833. https://doi.org/10.3390/antiox12101833
APA StyleRamos-Mondragón, R., Lozhkin, A., Vendrov, A. E., Runge, M. S., Isom, L. L., & Madamanchi, N. R. (2023). NADPH Oxidases and Oxidative Stress in the Pathogenesis of Atrial Fibrillation. Antioxidants, 12(10), 1833. https://doi.org/10.3390/antiox12101833