

Hypoxia Aggravates Neuron Ferroptosis in Early Brain Injury Following Subarachnoid Hemorrhage via NCOA4-Meditated Ferritinophagy
 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animal and In Vivo SAH Models
2.3. Cell Culture and SAH In Vitro Model
2.4. Research Design and Drug Management
2.4.1. Experiment 1
2.4.2. Experiment 2
2.4.3. Experiment 3
2.4.4. Experiment 4
2.4.5. Experiment 5
2.5. Knockout NCOA4 with siRNA In Vitro
2.6. Knockout NCOA4 with Lentivirus In Vivo
2.7. Western Blot Analysis (WB)
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Measurement of Iron Content In Vivo
2.10. Measurement of Glutathione and Malondialdehyde (MDA) In Vivo
2.11. Transmission Electron Microscope (TEM)
2.12. Immunofluorescence (IF)
2.13. TUNEL Assay
2.14. Brain Water Content Evaluation
2.15. Neurological Function Evaluation
2.16. Analysis of Fe2+ In Vitro
2.17. Analysis of ROS and Lipid Peroxides In Vitro
2.18. Staining with Hematoxylin-Eosin (H&E, and Cresol Violet (Nissl))
2.19. Statistical Analysis
3. Results
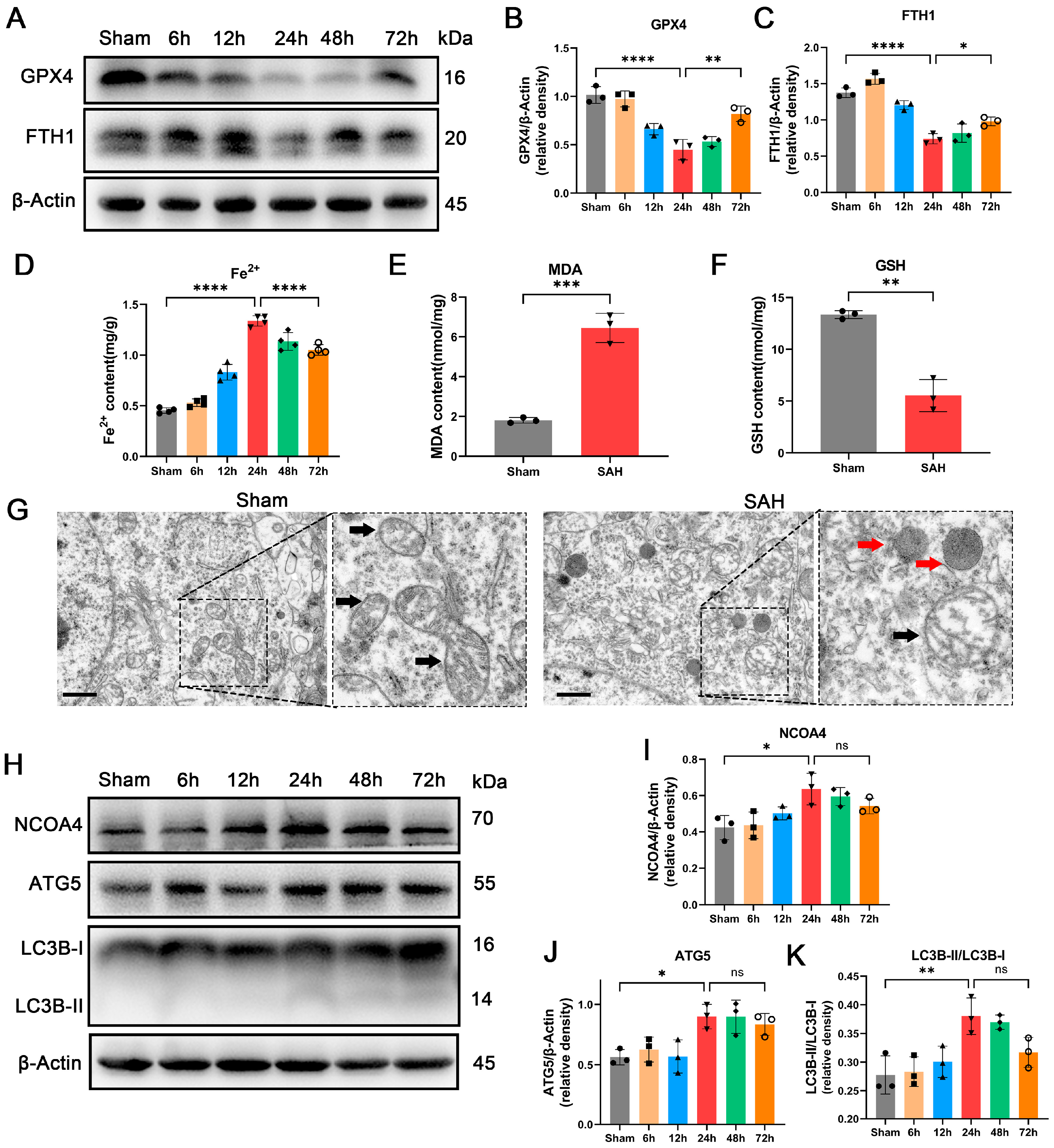
3.1. Occurrence of Ferroptosis Was Observed during EBI In Vivo
3.2. Enhancement of Ferritinophagy Was Observed during EBI In Vivo
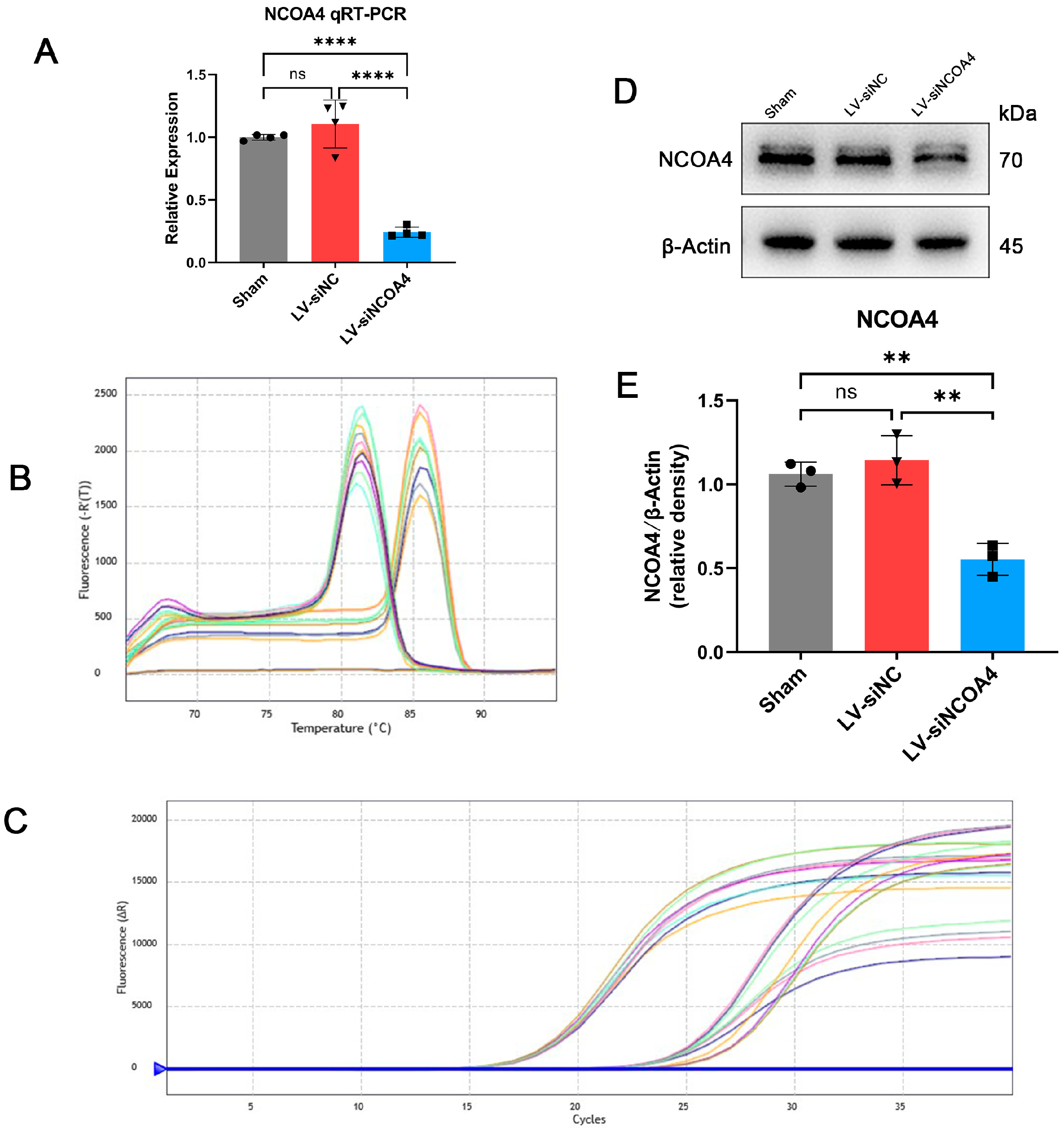
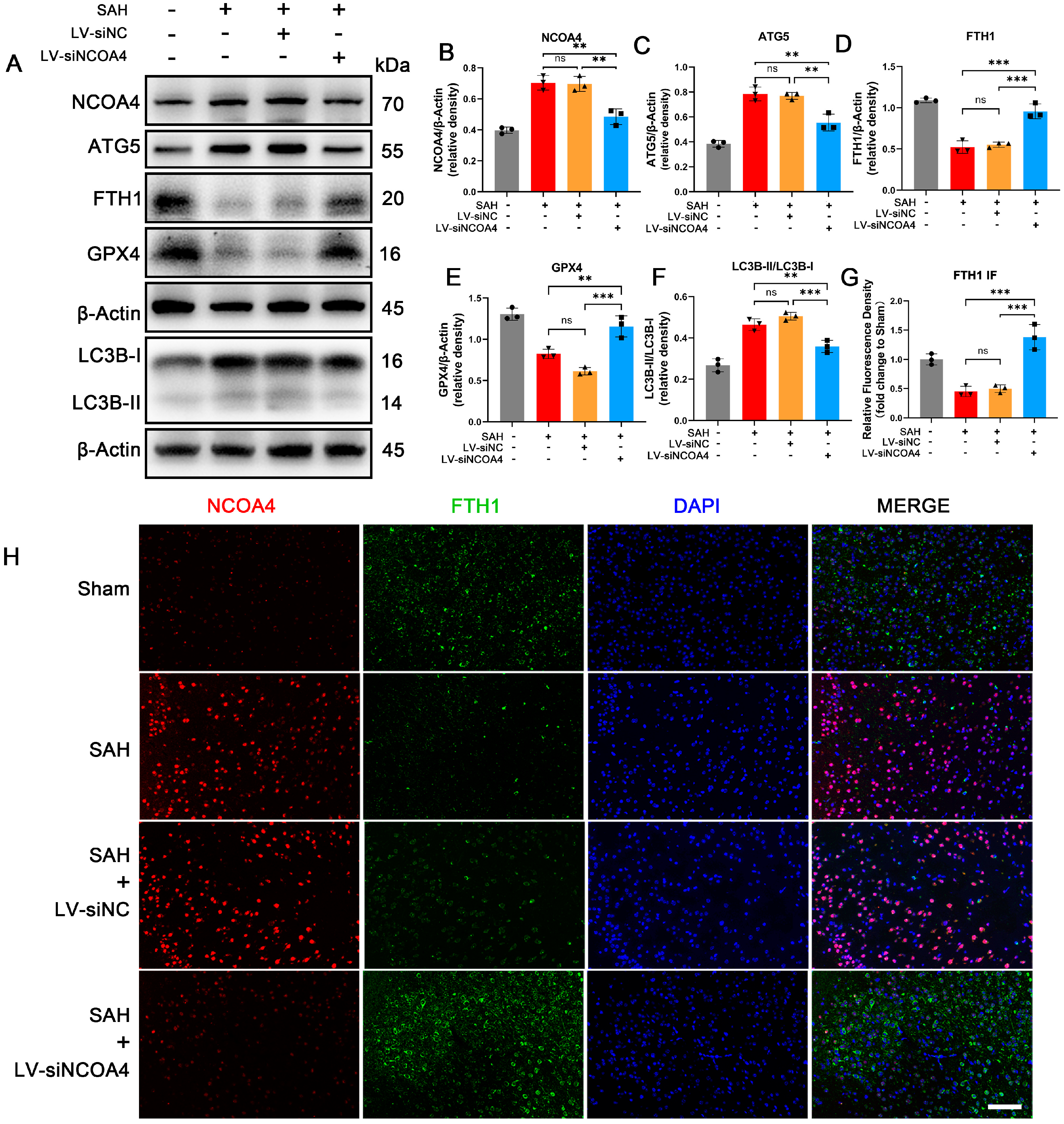
3.3. Knockdown of NCOA4 Effectively Suppressed Ferritinophagy during EBI In Vivo
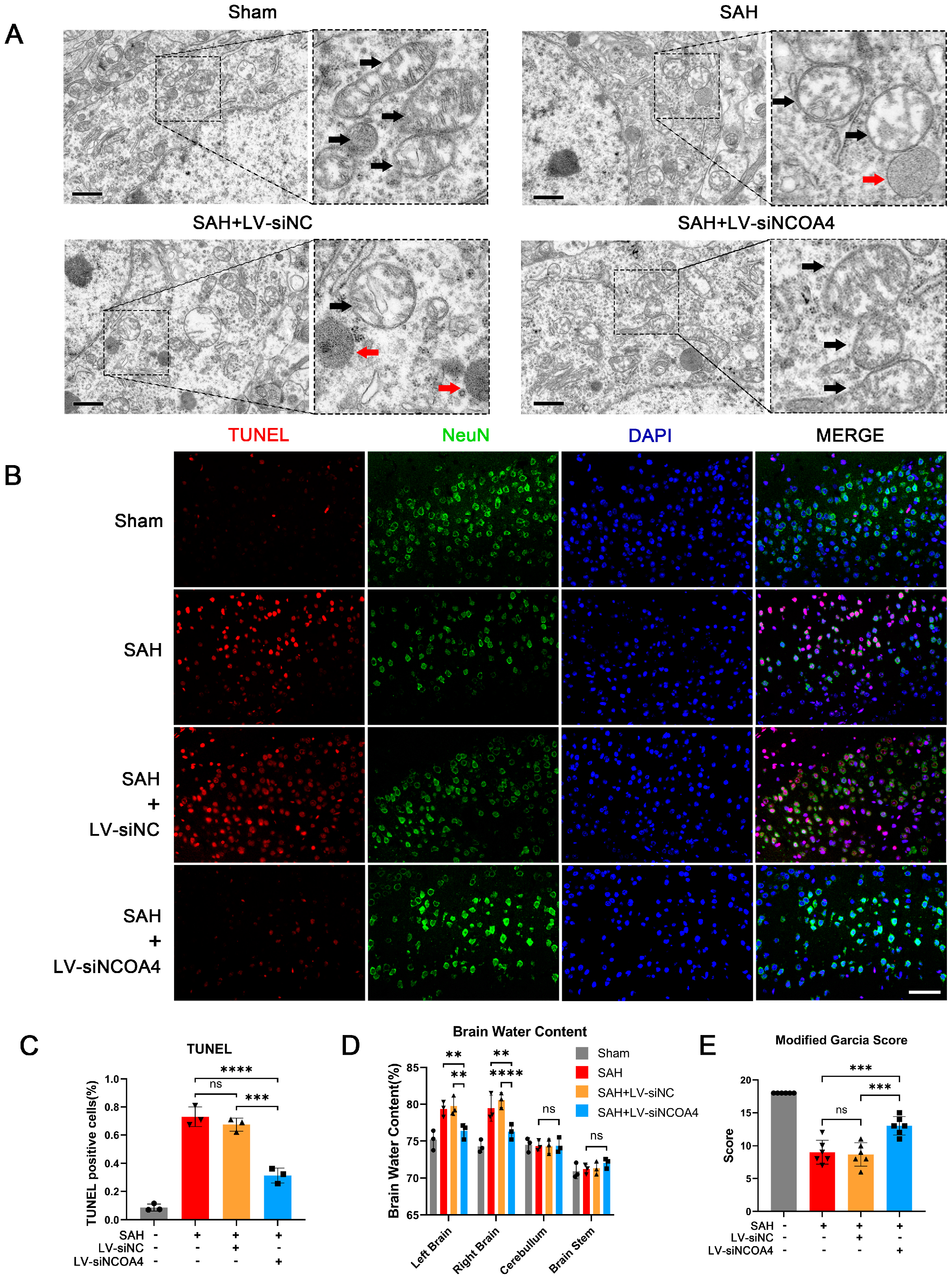
3.4. Knockdown of NCOA4 Effectively Suppressed Neuronal Ferroptosis and Enhanced the Prognosis In Vivo
3.5. Hypoxia Induced Ferritinophagy In Vitro
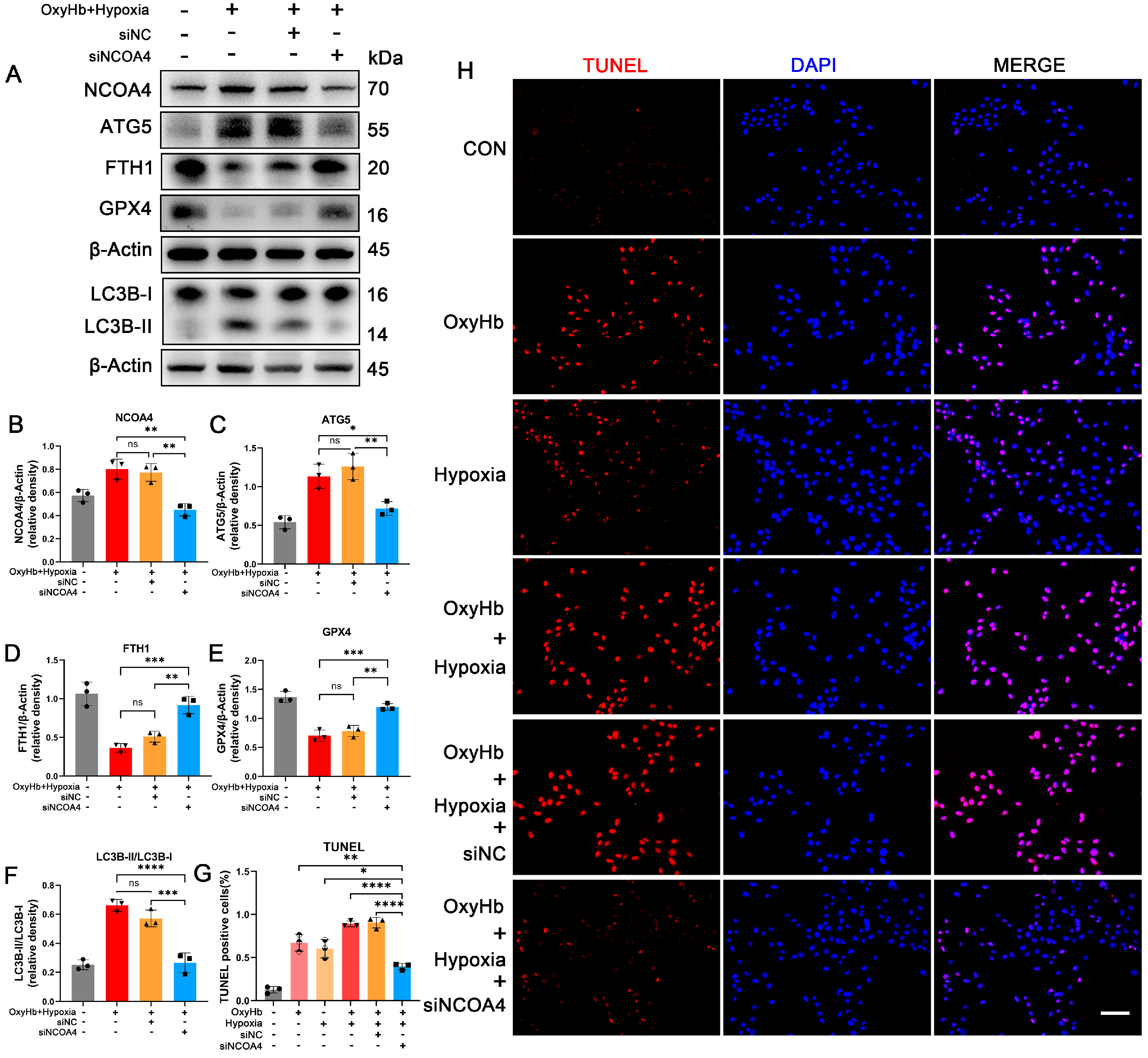
3.6. Knockdown of NCOA4 Effectively Inhibited Ferritinophagy during EBI In Vitro
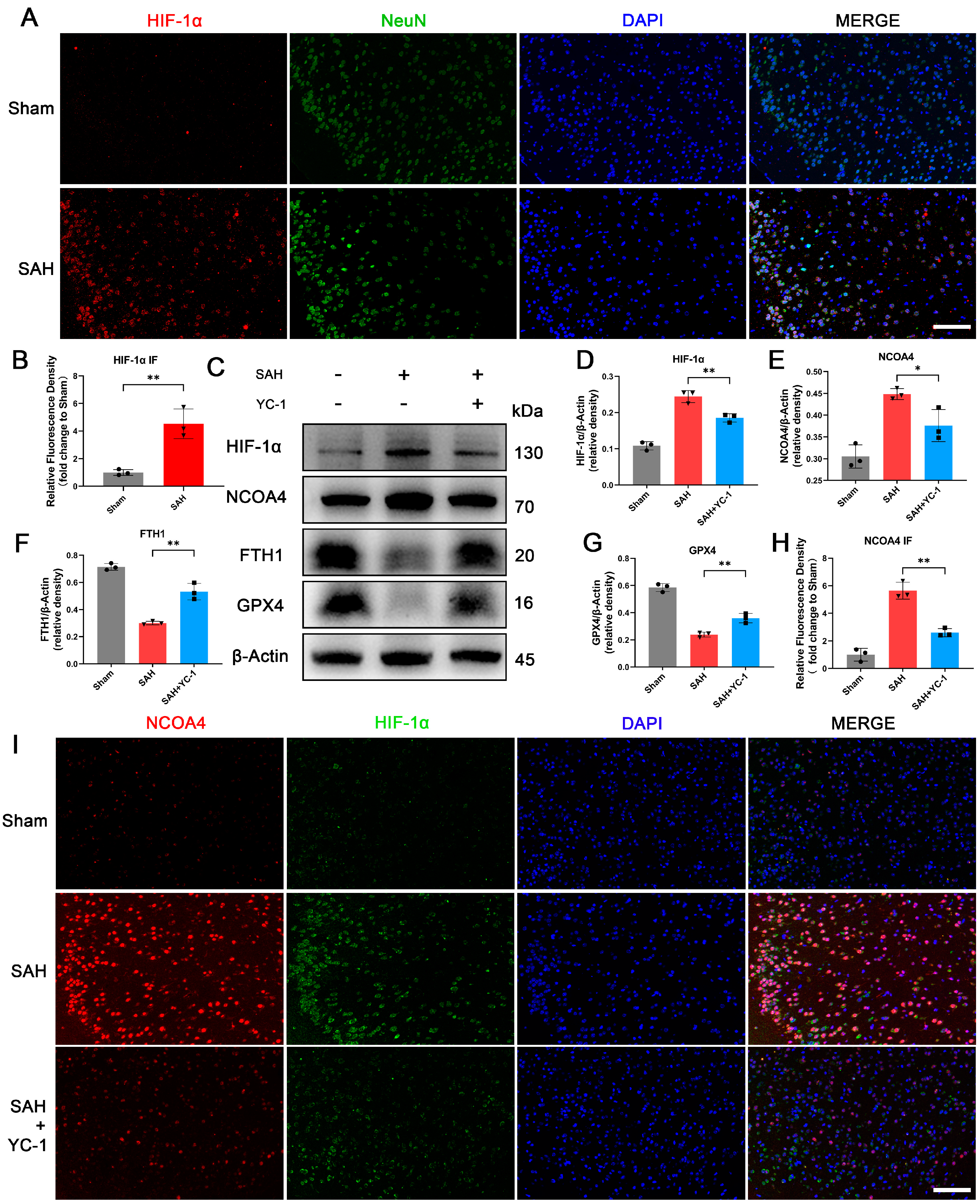
3.7. Administration of YC-1 for Hypoxia Pathway Inhibition Significantly Suppressed Ferritinophagy during EBI In Vivo
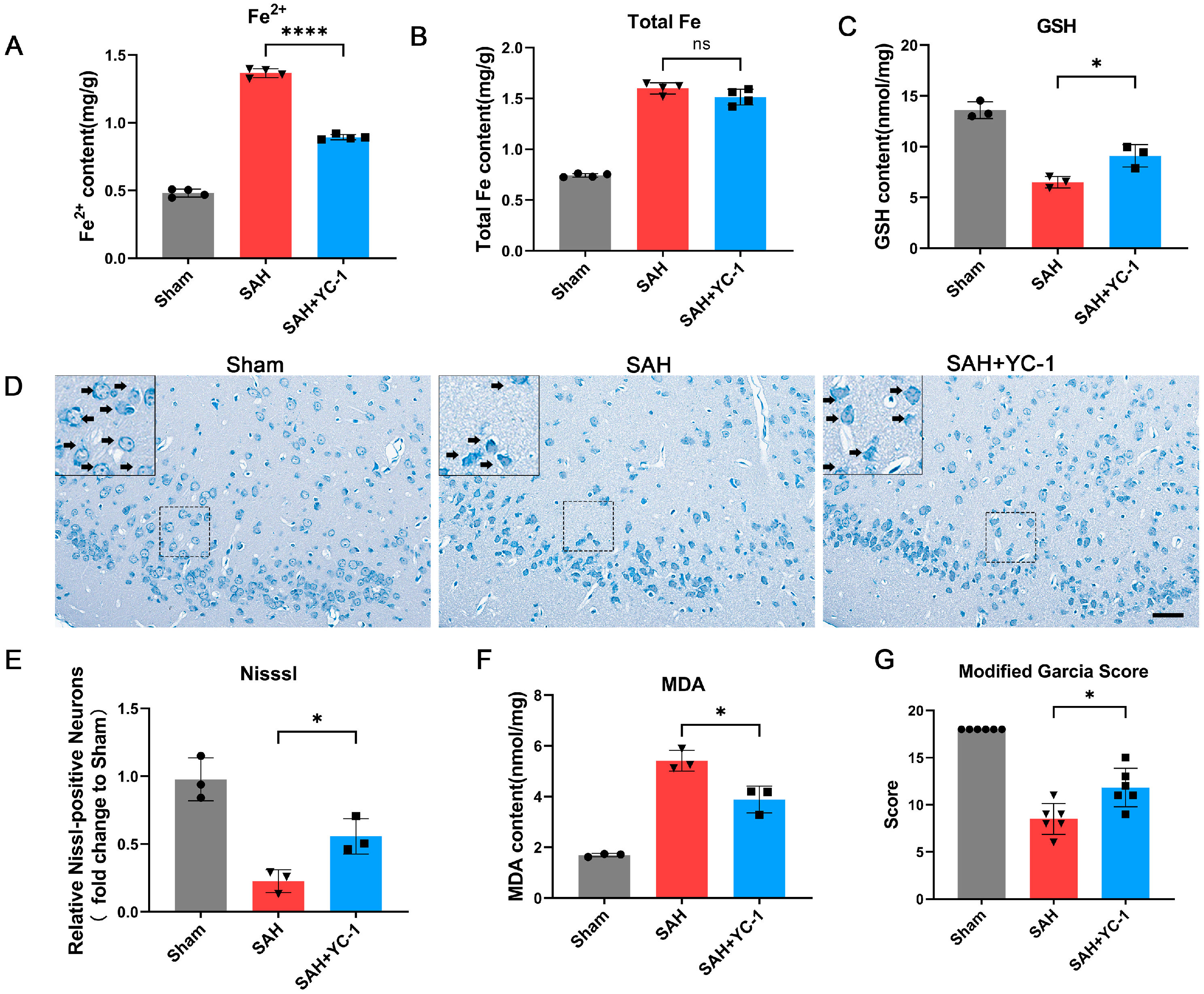
3.8. Pharmacological Inhibition of the Hypoxia Pathway Using YC-1 Significantly Enhances the Prognosis of SAH In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Claassen, J.; Park, S. Spontaneous Subarachnoid Haemorrhage. Lancet 2022, 400, 846–862. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, D.C.; Jayaraman, K.; Yuan, J.Y.; Diwan, D.; Vellimana, A.K.; Osbun, J.W.; Chatterjee, A.R.; Athiraman, U.; Dhar, R.; Zipfel, G.J. Early Brain Injury After Subarachnoid Hemorrhage: Incidence and Mechanisms. Stroke 2023, 54, 1426–1440. [Google Scholar] [CrossRef]
- Chen, Y.; Galea, I.; Macdonald, R.L.; Wong, G.K.C.; Zhang, J.H. Rethinking the Initial Changes in Subarachnoid Haemorrhage: Focusing on Real-Time Metabolism during Early Brain Injury. EBioMedicine 2022, 83, 104223. [Google Scholar] [CrossRef]
- Osgood, M.L. Aneurysmal Subarachnoid Hemorrhage: Review of the Pathophysiology and Management Strategies. Curr. Neurol. Neurosci. Rep. 2021, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N. Superficial Siderosis: A Clinical Review. Ann. Neurol. 2021, 89, 1068–1079. [Google Scholar] [CrossRef]
- Plesnila, N. Are We Looking Into an Iron Age for Subarachnoid Hemorrhage? Stroke 2022, 53, 1643–1644. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Cao, Y.; Xiao, W.; Liu, S.; Zeng, Y. Ferroptosis: Underlying Mechanism and the Crosstalk with Other Modes of Neuronal Death after Intracerebral Hemorrhage. Front. Cell. Neurosci. 2023, 17, 1080344. [Google Scholar] [CrossRef]
- Tao, Q.; Qiu, X.; Li, C.; Zhou, J.; Gu, L.; Zhang, L.; Pang, J.; Zhang, L.; Yin, S.; Jiang, Y.; et al. S100A8 Regulates Autophagy-Dependent Ferroptosis in Microglia after Experimental Subarachnoid Hemorrhage. Exp. Neurol. 2022, 357, 114171. [Google Scholar] [CrossRef]
- Ren, S.; Chen, Y.; Wang, L.; Wu, G. Neuronal Ferroptosis after Intracerebral Hemorrhage. Front. Mol. Biosci. 2022, 9, 966478. [Google Scholar] [CrossRef]
- Lee, S.; Hwang, N.; Seok, B.G.; Lee, S.; Lee, S.-J.; Chung, S.W. Autophagy Mediates an Amplification Loop during Ferroptosis. Cell Death Dis. 2023, 14, 464. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Zheng, B.; Zhou, X.; Pang, L.; Che, Y.; Qi, X. Baicalin Suppresses Autophagy-Dependent Ferroptosis in Early Brain Injury after Subarachnoid Hemorrhage. Bioengineered 2021, 12, 7794–7804. [Google Scholar] [CrossRef]
- McConnell, E.D.; Wei, H.S.; Reitz, K.M.; Kang, H.; Takano, T.; Vates, G.E.; Nedergaard, M. Cerebral Microcirculatory Failure after Subarachnoid Hemorrhage Is Reversed by Hyaluronidase. J. Cereb. Blood Flow Metab. 2016, 36, 1537–1552. [Google Scholar] [CrossRef]
- Hoffman, W.E.; Wheeler, P.; Edelman, G.; Charbel, F.T.; Torres, N.J.; Ausman, J.I. Hypoxic Brain Tissue Following Subarachnoid Hemorrhage. Anesthesiology 2000, 92, 442–446. [Google Scholar] [CrossRef]
- Milner, E.; Johnson, A.W.; Nelson, J.W.; Harries, M.D.; Gidday, J.M.; Han, B.H.; Zipfel, G.J. HIF-1α Mediates Isoflurane-Induced Vascular Protection in Subarachnoid Hemorrhage. Ann. Clin. Transl. Neurol. 2015, 2, 325–337. [Google Scholar] [CrossRef]
- Østergaard, L.; Aamand, R.; Karabegovic, S.; Tietze, A.; Blicher, J.U.; Mikkelsen, I.K.; Iversen, N.K.; Secher, N.; Engedal, T.S.; Anzabi, M.; et al. The Role of the Microcirculation in Delayed Cerebral Ischemia and Chronic Degenerative Changes after Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. 2013, 33, 1825–1837. [Google Scholar] [CrossRef]
- Diwan, D.; Vellimana, A.K.; Aum, D.J.; Clarke, J.; Nelson, J.W.; Lawrence, M.; Han, B.H.; Gidday, J.M.; Zipfel, G.J. Sirtuin 1 Mediates Protection Against Delayed Cerebral Ischemia in Subarachnoid Hemorrhage in Response to Hypoxic Postconditioning. J. Am. Heart Assoc. 2021, 10, e021113. [Google Scholar] [CrossRef] [PubMed]
- Vellimana, A.K.; Aum, D.J.; Diwan, D.; Clarke, J.V.; Nelson, J.W.; Lawrence, M.; Han, B.H.; Gidday, J.M.; Zipfel, G.J. SIRT1 Mediates Hypoxic Preconditioning Induced Attenuation of Neurovascular Dysfunction Following Subarachnoid Hemorrhage. Exp. Neurol. 2020, 334, 113484. [Google Scholar] [CrossRef] [PubMed]
- Helbok, R.; Schiefecker, A.J.; Beer, R.; Dietmann, A.; Antunes, A.P.; Sohm, F.; Fischer, M.; Hackl, W.O.; Rhomberg, P.; Lackner, P.; et al. Early Brain Injury after Aneurysmal Subarachnoid Hemorrhage: A Multimodal Neuromonitoring Study. Crit. Care 2015, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Schwarting, J.; Harapan, B.N.; Lin, X.; Plesnila, N.; Terpolilli, N.A. Nimodipine Reduces Microvasospasms After Experimental Subarachnoid Hemorrhage. Stroke 2023, 54, 2666–2670. [Google Scholar] [CrossRef]
- Daou, B.J.; Koduri, S.; Thompson, B.G.; Chaudhary, N.; Pandey, A.S. Clinical and Experimental Aspects of Aneurysmal Subarachnoid Hemorrhage. CNS Neurosci. Ther. 2019, 25, 1096–1112. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yan, J.; Ocak, U.; Lenahan, C.; Shao, A.; Tang, J.; Zhang, J.; Zhang, J.H. Melanocortin 1 Receptor Attenuates Early Brain Injury Following Subarachnoid Hemorrhage by Controlling Mitochondrial Metabolism via AMPK/SIRT1/PGC-1α Pathway in Rats. Theranostics 2021, 11, 522–539. [Google Scholar] [CrossRef] [PubMed]
- Broussy, S.; Laaroussi, H.; Vidal, M. Biochemical Mechanism and Biological Effects of the Inhibition of Silent Information Regulator 1 (SIRT1) by EX-527 (SEN0014196 or Selisistat). J. Enzym. Inhib. Med. Chem. 2020, 35, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, H.; Yue, L.; Zhang, J.; Li, X.; Wang, B.; Lin, Y.; Qu, Y. Melatonin Attenuates Early Brain Injury via the Melatonin Receptor/Sirt1/NF-κB Signaling Pathway Following Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2017, 54, 1612–1621. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Q.; Lu, Y.; Wan, J.; Dai, H.; Zhou, X.; Lv, S.; Chen, X.; Zhang, X.; Hang, C.; et al. Cerebroprotection by Salvianolic Acid B after Experimental Subarachnoid Hemorrhage Occurs via Nrf2- and SIRT1-Dependent Pathways. Free. Radic. Biol. Med. 2018, 124, 504–516. [Google Scholar] [CrossRef]
- Gao, S.-Q.; Liu, J.-Q.; Han, Y.-L.; Deji, Q.-Z.; Zhaba, W.-D.; Deng, H.-J.; Liu, X.-L.; Zhou, M.-L. Neuroprotective Role of Glutathione Peroxidase 4 in Experimental Subarachnoid Hemorrhage Models. Life Sci. 2020, 257, 118050. [Google Scholar] [CrossRef]
- Li, W.; Li, W.; Wang, Y.; Leng, Y.; Xia, Z. Inhibition of DNMT-1 Alleviates Ferroptosis through NCOA4 Mediated Ferritinophagy during Diabetes Myocardial Ischemia/Reperfusion Injury. Cell Death Discov. 2021, 7, 267. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Zhou, X.-M.; You, Z.-Q.; Xu, W.-D.; Fan, J.-M.; Chen, S.-J.; Han, Y.-L.; Wu, Q.; Zhang, X. Inhibition of AIM2 Inflammasome Activation Alleviates GSDMD-Induced Pyroptosis in Early Brain Injury after Subarachnoid Haemorrhage. Cell Death Dis. 2020, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Hao, G.; Eser, P.; Mo, J. Oxidative Stress and Intracranial Hypertension after Aneurysmal Subarachnoid Hemorrhage. Antioxidants 2022, 11, 2423. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, H.; Hu, Y.; Zhou, C.; Wu, J.; Wu, Y.; Wang, H.; Lenahan, C.; Huang, L.; Nie, S.; et al. Puerarin Attenuates Oxidative Stress and Ferroptosis via AMPK/PGC1α/Nrf2 Pathway after Subarachnoid Hemorrhage in Rats. Antioxidants 2022, 11, 1259. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Duan, J.; Du, L.; Xing, W.; Peng, X.; Zhao, Q. Inflammation and Immune Cell Abnormalities in Intracranial Aneurysm Subarachnoid Hemorrhage (SAH): Relevant Signaling Pathways and Therapeutic Strategies. Front. Immunol. 2022, 13, 1027756. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, R.L. Age and Outcome after Aneurysmal Subarachnoid Haemorrhage. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1143. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Song, C.; Zhang, J.; Zhao, X. ROS and Iron Homeostasis Dependent Ferroptosis Play a Vital Role in 5-Fluorouracil Induced Cardiotoxicity in Vitro and in Vivo. Toxicology 2022, 468, 153113. [Google Scholar] [CrossRef]
- Tian, Q.; Liu, S.; Han, S.-M.; Zhang, W.; Qin, X.-Y.; Chen, J.-H.; Liu, C.-L.; Guo, Y.-J.; Li, M.-C. The Mechanism and Relevant Mediators Associated with Neuronal Apoptosis and Potential Therapeutic Targets in Subarachnoid Hemorrhage. Neural Regen. Res. 2023, 18, 244–252. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Chung, C.-L.; Tsai, H.-P.; Tzou, R.-D.; Wu, S.-C.; Chai, C.-Y.; Lee, T.-C.; Kwan, A.-L. Impact of Hyperglycemia on Neuronal Apoptosis after Subarachnoid Hemorrhage in Rodent Brain: An Experimental Research. Int. J. Surg. 2020, 83, 246–252. [Google Scholar] [CrossRef]
- Chen, J.; Li, M.; Liu, Z.; Wang, Y.; Xiong, K. Molecular Mechanisms of Neuronal Death in Brain Injury after Subarachnoid Hemorrhage. Front. Cell. Neurosci. 2022, 16, 1025708. [Google Scholar] [CrossRef]
- Li, X.; Lozovatsky, L.; Sukumaran, A.; Gonzalez, L.; Jain, A.; Liu, D.; Ayala-Lopez, N.; Finberg, K.E. NCOA4 Is Regulated by HIF and Mediates Mobilization of Murine Hepatic Iron Stores after Blood Loss. Blood 2020, 136, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, G.; Chen, B.; Xu, L.; Ye, Y.; He, J.; Bao, Z.; Zhao, P.; Miao, Z.; Zhao, L.; et al. Nuclear Receptor Coactivator 4-Mediated Ferritinophagy Contributes to Cerebral Ischemia-Induced Ferroptosis in Ischemic Stroke. Pharmacol. Res. 2021, 174, 105933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 Induces NCOA4 Mediated Autophagy and Ferroptosis in Glioblastoma Cell Lines. Oncogene 2021, 40, 1425–1439. [Google Scholar] [CrossRef]
- Xiu, Z.; Zhu, Y.; Han, J.; Li, Y.; Yang, X.; Yang, G.; Song, G.; Li, S.; Li, Y.; Cheng, C.; et al. Caryophyllene Oxide Induces Ferritinophagy by Regulating the NCOA4/FTH1/LC3 Pathway in Hepatocellular Carcinoma. Front. Pharmacol. 2022, 13, 930958. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, B.; Xu, A.; Shen, J.; Li, K.; Hao, K.; Hao, R.; Yang, W.; Jiang, W.; Zheng, Y.; et al. TRIM7 Modulates NCOA4-Mediated Ferritinophagy and Ferroptosis in Glioblastoma Cells. Redox Biol. 2022, 56, 102451. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Deng, Y.; Zhao, J.; Liu, L.; Wang, J.; Chen, P.; Zhang, Q.; Sun, C.; Wang, Y.; Xiang, Y. Ferritinophagy Is Involved in Experimental Subarachnoid Hemorrhage-Induced Neuronal Ferroptosis. Neurochem. Res. 2022, 47, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Song, A.; Ma, M.; Wang, P.; Zhang, X.; Lu, C.; Zhang, J.; Zheng, S.; Jin, H. Curcumol Inhibits Ferritinophagy to Restrain Hepatocyte Senescence through YAP/NCOA4 in Non-Alcoholic Fatty Liver Disease. Cell Prolif. 2021, 54, e13107. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, S.; Tu, B.; Jiang, X.; Cheng, S.; Tang, Q.; Zhang, J.; Qin, X.; Wang, B.; Zou, Z.; et al. Arsenite Induces Ferroptosis in the Neuronal Cells via Activation of Ferritinophagy. Food Chem. Toxicol. 2021, 151, 112114. [Google Scholar] [CrossRef]
- Wu, H.; Liu, Q.; Shan, X.; Gao, W.; Chen, Q. ATM Orchestrates Ferritinophagy and Ferroptosis by Phosphorylating NCOA4. Autophagy 2023, 19, 2062–2077. [Google Scholar] [CrossRef]
- Deji, Q.-Z.; Wang, X.; Zhaba, W.-D.; Deng, H.-J.; Han, Y.-L.; Gao, S.-Q.; Liu, X.-L.; Zhou, M.-L. Activation of HIF-1α/VEGF-A Pathway by Deferoxamine Ameliorates Retinal Hypoxia in a Rat Subarachnoid Hemorrhage Model. Neuroreport 2022, 33, 690–696. [Google Scholar] [CrossRef]
- Sun, X.-G.; Chu, X.-H.; Godje Godje, I.S.; Liu, S.-Y.; Hu, H.-Y.; Zhang, Y.-B.; Zhu, L.-J.; Wang, H.; Sui, C.; Huang, J.; et al. Aerobic Glycolysis Induced by mTOR/HIF-1α Promotes Early Brain Injury After Subarachnoid Hemorrhage via Activating M1 Microglia. Transl. Stroke Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Das, N.K.; Jain, C.; Sankar, A.; Schwartz, A.J.; Santana-Codina, N.; Solanki, S.; Zhang, Z.; Ma, X.; Parimi, S.; Rui, L.; et al. Modulation of the HIF2α-NCOA4 Axis in Enterocytes Attenuates Iron Loading in a Mouse Model of Hemochromatosis. Blood 2022, 139, 2547–2552. [Google Scholar] [CrossRef] [PubMed]
- Weiland, J.; Beez, A.; Westermaier, T.; Kunze, E.; Sirén, A.-L.; Lilla, N. Neuroprotective Strategies in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2021, 22, 5442. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Q.; Xu, C.-Y.; Mao, S.; Jin, J.-J.; Gu, W.; Shi, Y.; Zou, C.-F.; Ye, L. Obstructive Sleep Apnea Aggravates Neuroinflammation and Pyroptosis in Early Brain Injury Following Subarachnoid Hemorrhage via ASC/HIF-1α Pathway. Neural Regen. Res. 2022, 17, 2537–2543. [Google Scholar] [CrossRef]
- Zhang, F.; Lin, B.; Huang, S.; Wu, P.; Zhou, M.; Zhao, J.; Hei, X.; Ke, Y.; Zhang, Y.; Huang, D. Melatonin Alleviates Retinal Ischemia–Reperfusion Injury by Inhibiting P53–Mediated Ferroptosis. Antioxidants 2023, 12, 1173. [Google Scholar] [CrossRef]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-Induced Damage in Cardiomyopathy: Oxidative-Dependent and Independent Mechanisms. Oxidative Med. Cell. Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef]
- He, J.; Li, Z.; Xia, P.; Shi, A.; FuChen, X.; Zhang, J.; Yu, P. Ferroptosis and Ferritinophagy in Diabetes Complications. Mol. Metab. 2022, 60, 101470. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Zhou, X.; Zou, Y.; Zhang, B.; Jian, Y.; Wu, Q.; Chen, S.; Zhang, X. Hypoxia Aggravates Neuron Ferroptosis in Early Brain Injury Following Subarachnoid Hemorrhage via NCOA4-Meditated Ferritinophagy. Antioxidants 2023, 12, 2097. https://doi.org/10.3390/antiox12122097
Yuan Z, Zhou X, Zou Y, Zhang B, Jian Y, Wu Q, Chen S, Zhang X. Hypoxia Aggravates Neuron Ferroptosis in Early Brain Injury Following Subarachnoid Hemorrhage via NCOA4-Meditated Ferritinophagy. Antioxidants. 2023; 12(12):2097. https://doi.org/10.3390/antiox12122097
Chicago/Turabian StyleYuan, Zixuan, Xiaoming Zhou, Yan Zou, Bingtao Zhang, Yao Jian, Qi Wu, Shujuan Chen, and Xin Zhang. 2023. "Hypoxia Aggravates Neuron Ferroptosis in Early Brain Injury Following Subarachnoid Hemorrhage via NCOA4-Meditated Ferritinophagy" Antioxidants 12, no. 12: 2097. https://doi.org/10.3390/antiox12122097