
Selenium Deficiency-Induced Oxidative Stress Causes Myocardial Injury in Calves by Activating Inflammation, Apoptosis, and Necroptosis


Abstract
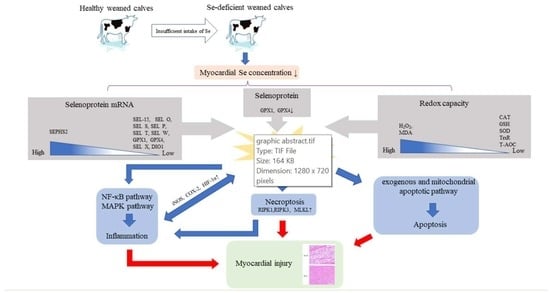
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Analysis of Se Concentrations in Tissues
2.3. TUNEL Staining and Histopathological Examination of the Myocardium
2.4. Redox Parameters and Inflammatory Cytokine Analysis
2.5. Real-Time PCR and Western Blot Analysis
2.6. Statistical Analysis
3. Results
3.1. Effect of Se Deficiency on Myocardial Se Concentration and Selenoprotein Expression in Weaned Calves
3.2. Histopathological Changes in the Myocardium of Weaned Calves under Se-Deficient Conditions
3.3. Effect of Se Deficiency on Myocardial Oxidative Stress Levels in Weaned Calves
3.4. The Apoptotic Pathways Were Activated in the Myocardium of Se-Deficient Calves
3.5. The Activated NF-κB and MAPK Pathways Induced Inflammation in the Myocardium of Se-Deficient Calves
3.6. Necroptosis Was Activated in the Myocardium of Se-Deficient Calves
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michael, R.; Bösl, K.T.; Oshima, M.; Nishimura, S.; Makoto, M. Early Embryonic Lethality Caused by Targeted Disruption of the Mouse Selenocysteine tRNA Gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [Green Version]
- Susan, J.; Tait, F.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in Human Health and Disease. Antioxid. Redox Signal 2011, 14, 1337–1383. [Google Scholar] [CrossRef]
- Yang, C.; Yao, H.; Wu, Y.; Sun, G.; Yang, W.; Li, Z.; Shang, L. Status and risks of selenium deficiency in a traditional selenium-deficient area in Northeast China. Sci. Total Environ. 2021, 762, 144103. [Google Scholar] [CrossRef]
- Mehdi, Y.; Dufrasne, I. Selenium in Cattle: A Review. Molecules 2016, 21, 545. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.J.; Myburgh, J.G. Investigation of stillbirths, perinatal mortality and weakness in beef calves with low-selenium whole blood concentrations. J. S. Afr. Vet. Assoc. 2016, 87, a1336. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Droz, B.; Greve, P.; Gottschalk, P.; Poffet, D.; McGrath, S.P.; Seneviratne, S.I.; Smith, P.; Winkel, L.H. Selenium deficiency risk predicted to increase under future climate change. Proc. Natl. Acad. Sci. USA 2017, 114, 2848–2853. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G.; Yang, K.-S.; Kang, S.W.; Woo, H.A.; Chang, T.-S. Controlled Elimination of Intracellular H2O2: Regulation of Peroxiredoxin, Catalase, and Glutathione Peroxidase via Post-translational Modification. Antioxid. Redox Signal. 2005, 7, 619–626. [Google Scholar] [CrossRef]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, A2, 811–820. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, L.; Yin, J.; Luo, Y.; Huang, S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol. 2009, 41, 1284–1295. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Pagliari, L.J.; Pinkoski, M.J.; Green, D.R. Apostosis Signaling: A Means to an End. Handb. Cell Signal. 2003, 3, 2535–2543. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Hulpiau, P.; Saeys, Y.; Bertrand, M.J.M.; Vandenabeele, P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016, 26, 721–732. [Google Scholar] [CrossRef]
- Agueda-Pinto, A.; Alves, L.Q.; Neves, F.; McFadden, G.; Jacobs, B.L.; Castro, L.F.C.; Rahman, M.M.; Esteves, P.J. Convergent Loss of the Necroptosis Pathway in Disparate Mammalian Lineages Shapes Viruses Countermeasures. Front. Immunol. 2021, 12, 747737. [Google Scholar] [CrossRef]
- Zarczynska, K.; Baumgartner, W.; Sobiech, P. Coagulology, biochemical profile and muscle pathology in calves diagnosed with nutritional muscular dystrophy. Pol. J. Vet. Sci. 2017, 20, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Fordyce, F.M. Selenium Deficiency and Toxicity in the Environment. In Essentials of Medical Geology; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Fu, Y.; Cheng, W.-H.; Porres, J.M.; Ross, D.A.; Lei, X.G. Knockout of cellular glutathione peroxidase gene renders mice susceptible to diquat-induced oxidative stress. Free. Radic. Biol. Med. 1999, 27, 605–611. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-A.; Kirnarsky, L.; Sherman, S.; Gladyshev, V.N. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. USA 2001, 98, 3673–3678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromer, S.; Johansson, L.; Bauer, H.; Arscott, L.D.; Rauch, S.; Ballou, D.P.; Arner, E.S.J. Active sites of thioredoxin reductases: Why selenoproteins? Proc. Natl. Acad. Sci. USA 2003, 100, 12618–12623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-M.; Turanov, A.A.; Carlson, B.A.; Yoo, M.-H.; Everley, R.A.; Nandakumar, R.; Sorokina, I.; Gygi, S.P.; Gladyshev vgladyshev, V.N.; Hatfield, D.L. Targeted insertion of cysteine by decoding UGA codons with mammalian selenocysteine machinery. Proc. Natl. Acad. Sci. USA 2015, 107, 21430–21434. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, P.R.; Hoge, S.C.; Li, P.A.; Hoffmann, F.W.; Hashimoto, A.C.; Berry, M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucleic Acids Res. 2007, 35, 3963–3973. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Sun, W.; Zhang, K.; Zhu, J.; Jia, X.; Guo, X.; Zhao, Q.; Tang, C.; Yin, J.; Zhang, J. Selenium deficiency induces spleen pathological changes in pigs by decreasing selenoprotein expression, evoking oxidative stress, and activating inflammation and apoptosis. J. Anim. Sci. Biotechnol. 2021, 12, 65. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, P.; Teng, T.; Shi, B.; Shan, A.; Lei, X.G. Effects of Dietary Selenium Deficiency or Excess on Selenoprotein Gene Expression in the Spleen Tissue of Pigs. Animals 2019, 9, 1122. [Google Scholar] [CrossRef] [Green Version]
- Hilal, T.; Killam, B.Y.; Grozdanović, M.; Dobosz-Bartoszek, M.; Loerke, J.; Bürger, J.; Mielke, T.; Copeland, P.R.; Simonović, M.; Spahn, C.M.T. Structure of the mammalian ribosome as it decodes the selenocysteine UGA codon. Science 2022, 376, 1338–1343. [Google Scholar] [CrossRef]
- Kristina, E.; Hill, P.R.L.; Raymond, F.B. Differential regulation of rat liver selenoprotein mRNAs in selenium deficiency. Biochem. Biophys. Res. Commun. 1992, 182, 260–263. [Google Scholar] [CrossRef]
- Wingler, K.; Böcher, M.; Flohé, L.; Kollmus, H.; Flohé, B. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 1999, 259, 149–157. [Google Scholar] [CrossRef]
- Müller, C.; Wingler, K.; Flohé, B. 3′UTRs of Glutathione Peroxidases Differentially Affect Selenium-Dependent mRNA Stability and Selenocysteine Incorporation Efficiency. Biol. Chem. 2003, 384, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Stoytchev, I.; Forry, E.P.; Berry, M.J. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol. Cell Biol. 2007, 27, 7848–7855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkov, K.V.; Kumaraswamy, E.; Zhou, Y.; Hatfield, D.L.; Gladyshev, V.N. Association between the 15-kDa selenoprotein and UDP-glucose: Glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 2001, 276, 15330–15336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Q.; Zhang, Q.; Lu, Y.; Zhang, Y.; Xu, S.; Li, S. Roles of selenoprotein S in reactive oxygen species-dependent neutrophil extracellular trap formation induced by selenium-deficient arteritis. Redox Biol. 2021, 44, 102003. [Google Scholar] [CrossRef]
- Burk, H. Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Austin, L.M.; Norsworthy, B.K. Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole-body selenium content. Biochim. Biophys. Acta 2006, 1760, 1789–1793. [Google Scholar] [CrossRef] [Green Version]
- Dikiy, A.; Novoselov, S.V.; Fomenko, D.E.; Sengupta, A.; Carlson, B.A.; Cerny, R.L.; Ginalski, K.; Grishin, N.V.; Hatfield, D.L.; Gladyshev, V.N. SelT, SelW, SelH, and Rdx12: Genomics and Molecular Insights into the Functions of Selenoproteins of a Novel Thioredoxin-like Family. Biochemistry 2007, 46, 6871–6882. [Google Scholar] [CrossRef]
- Lee, B.C.; Péterfi, Z.; Hoffmann, F.W.; Moore, R.E.; Kaya, A.; Avanesov, A.; Tarrago, L.; Zhou, Y.; Weerapana, E.; Fomenko, D.E.; et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell 2013, 51, 397–404. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Fei, Y.; Han, Y.; Lu, S. Selenoprotein O deficiencies suppress chondrogenic differentiation of ATDC5 cells. Cell Biol. Int. 2016, 40, 1033–1040. [Google Scholar] [CrossRef]
- Dentice, M.; Marsili, A.; Ambrosio, R.; Guardiola, O.; Sibilio, A.; Paik, J.H.; Minchiotti, G.; DePinho, R.A.; Fenzi, G.; Larsen, P.R.; et al. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J. Clin. Investig. 2010, 120, 4021–4030. [Google Scholar] [CrossRef]
- Xu, X.M.; Carlson, B.A.; Irons, R.; Mix, H.; Zhong, N.; Gladyshev, V.N.; Hatfield, D.L. Selenophosphate synthetase 2 is essential for selenoprotein biosynthesis. Biochem. J. 2007, 404, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhao, Q.; Zhang, K.; Sun, W.; Jia, X.; Yang, Y.; Yin, J.; Tang, C.; Zhang, J. Se deficiency induces renal pathological changes by regulating selenoprotein expression, disrupting redox balance, and activating inflammation. Metallomics 2020, 12, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free. Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Bonfoco, E.; Krainc, D.; Ankarcrona, M.; Nicotera, P.; Lipton, S.A. Apoptosis and necrosis: Two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.J. The involvement of l -γ-glutamyl- l -cysteinyl-glycine (glutathione/GSH) in the mechanism of redox signaling mediating MAPK p38 -dependent regulation of pro-inflammatory cytokine production. Biochem. Pharmacol. 2002, 63, 305–320. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Packer, M. Mutual Antagonism of Hypoxia-Inducible Factor Isoforms in Cardiac, Vascular, and Renal Disorders. JACC Basic Transl. Sci. 2020, 5, 961–968. [Google Scholar] [CrossRef]
- Luo, L.; Luo, G.; Fang, Q.; Sun, Z. Stable expression of hypoxia-inducible factor-1alpha in human renal proximal tubular epithelial cells promotes epithelial to mesenchymal transition. Transpl. Proc. 2014, 46, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, B.N.; Coleman, A.E.; Schmiedt, C.W.; Brown, C.A.; Rissi, D.R.; Stanton, J.B.; Giguère, S.; Berghaus, R.D.; Brown, S.A.; Tarigo, J.L. Profibrotic gene transcription in renal tissues from cats with ischemia-induced chronic kidney disease. Am. J. Vet. Res. 2020, 81, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.Z.; Li, C.; Chen, Q.; Jing, Y.; Carpenter, R.; Jiang, Y.; Kung, H.F.; Lai, L.; Jiang, B.H. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PLoS ONE 2011, 6, e19139. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Brenhouse, H.C.; Andersen, S.L. Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol. Psychiatry 2011, 70, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cok, S.J.; Morrison, A.R. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J. Biol. Chem. 2001, 276, 23179–23185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, Y.; Chen, B.; Zhao, B.; Gao, X.J. Selenium Deficiency Promotes Oxidative Stress-Induced Mastitis via Activating the NF-κB and MAPK Pathways in Dairy Cow. Biol. Trace Elem. Res. 2022, 200, 2716–2726. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Lei, L.; Wang, D.; Liu, Y. Selenium Deficiency Induces Apoptosis, Mitochondrial Dynamic Imbalance, and Inflammatory Responses in Calf Liver. Biol. Trace Elem. Res. 2022, 200, 4678–4689. [Google Scholar] [CrossRef]
- Jian, Z.; Liang, B.; Pan, X.; Xu, G.; Guo, S.S.; Li, T.; Zhou, T.; Xiao, Y.B.; Li, A.L. CUEDC2 modulates cardiomyocyte oxidative capacity by regulating GPX1 stability. EMBO Mol. Med. 2016, 8, 813–829. [Google Scholar] [CrossRef]
- Marchi, S.; Giorgi, C.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Missiroli, S.; Patergnani, S.; Poletti, F.; et al. Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 2012, 329635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tummers, B.; Green, D.R. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol. Rev. 2022, 102, 411–454. [Google Scholar] [CrossRef] [PubMed]
- Shubina, M.; Tummers, B.; Boyd, D.F.; Zhang, T.; Yin, C.; Gautam, A.; Guo, X.J.; Rodriguez, D.A.; Kaiser, W.J.; Vogel, P.; et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020, 217, e20191259. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.L.; Hu, H.; Dong, X.Q.; Zhang, J.; Wang, J.; Schwieters, C.D.; Liu, J.; Wu, G.X.; Li, B.; Lin, J.Y.; et al. The amyloid structure of mouse RIPK3 (receptor interacting protein kinase 3) in cell necroptosis. Nat. Commun. 2021, 12, 1627. [Google Scholar] [CrossRef]
- Weinlich, R.; Green, D.R. The two faces of receptor interacting protein kinase-1. Mol. Cell 2014, 56, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Serial Number | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|---|
| DIO1 | NM_001122593.2 | 85F: CTCCTGACGCTGTTTCCCAG | 149R: GAAGTGTGGGTTCCTGGTCA |
| DIO2 | NM_001010992.7 | 558F: GAACCAGGAAGACCGATGCG | 670R: ACACGTTCAAAGGCTACCCC |
| DIO3 | NM_001010993.3 | 107F: AGACTTCCTGTGCATCCGTA | 363R: AGTCGAGGATGTGCTGGTTC |
| TXNRD1 | NM_174625.5 | 20F: TCCCGGAGCCCTATGACTAC | 219R: GGGCTTGACCTAACAACGCT |
| TXNRD2 | NM_174626.3 | 357F: TCAGAACCACGTGAAGTCCC | 560R: GATCCCATATTCCAAGGCAC |
| TXNRD3 | NM_001192109.2 | 1289F: AAGGGACAGAGACAATCGAG | 1377R: CACTGACGTTCACACCAATC |
| GPX1 | NM_174076.3 | 89F: AGCCCTTCAACCTGTCCTCC | 302R: CGTACTTCAGGCAATTCAGG |
| GPX2 | NM_001163139.2 | 137F: ACTTCACCCAACTCAACGAG | 340R: TTCAGGTAGGCAAAGACAGG |
| GPX3 | BC149266.1 | 252F: ACTGCAGGAAGAGCTTGAAC | 460R: GAGGTAGGAGGACAGGAGTT |
| GPX4 | NM_174770.4 | 13F: CGTCTGTACCGCCTGCTCAA | 212R: GTCAGTCTTGCCTCATTGCG |
| GPX6 | NM_001163142.2 | 246F: GAATGCACTACAGGAGGAGC | 496R: TCCCAGAAGAGCTGACTTGA |
| SEL H | NM_001164092.1 | 192F: CGAGCTGACGGTGAAGGTGA | 344R: GTACTTCTTCAGCTCCTCCA |
| SEL I | NM_001075257.2 | 408F: TGTTGTCACCGTGTATTCCA | 619R: AGGAAAGGTTCATACCAGGC |
| SEL K | NM_001037489.3 | 81F: CTGGGGAATAGCTGAGTTTG | 251R: GGCCATTGGAGGAGGATTAG |
| SEL M | NM_001163171.3 | 175F: CAGGACATCCCACTCTACCA | 255R: ATTCGCTCCAGTTCCTCAAAG |
| SEL N | NM_001114976.3 | 251F: ACATGTACATCAGCCCCGAG | 451R: CAGTTGCGGAGGCCAGACAG |
| SEL O | NM_001163193.3 | 762F: CACGTTCCTCAGGTTTGGAT | 862R: CTGATGACGTAGTCGAGCAT |
| SEL P | NM_174459.3 | 65F: AGGGCCAAAGCTCCTATTGT | 271R: CGAGAAGAGATTCCTTGATG |
| SEL S | NM_001046114.3 | 145F: GACAGAAGATCGAAATGTGG | 307R: TCCACCACCTTCACCAGACA |
| SEL T | NM_001103103.2 | 423F: CCAGTGTATGTCAACAGGTG | 538R: TGCACGTTGAGCTTCATTTC |
| SEL V | NM_001163244.2 | 842F: TAGCTTCCATCAGTGGGAAC | 1071R: GTAGCTTGACCTCATCCACA |
| SEL W | NM_001163225.1 | 22F: GTTTATTGTGGCGCTTGAGG | 216R: CGGCCACCAGCTTCAGAAAC |
| SEL X | BC105188.1 | 7F: TTCTGCAGCTTCTTCGGAGG | 168R: GATTGTGCTCTGGCCGCTTG |
| SEL 15 | BT021503.1 | 271F: TGTGGATGAAAATTGGGGAG | 394R: GCAATGTTCCCACTGTCGTC |
| SEPHS2 | NM_001114732.3 | 965F: GCTTTGGCATTCTGGGTCAC | 1203R: CGACGATCCAGGCTTGATGA |
| Bcl-2 | NM_001166486.1 | 408F: CATCGTGGCCTTCTTTGAGT | 592R: CAGGAGAAATCAAACAGGGG |
| Caspase 3 | NM_001077840.1 | 357F: AAGCCATGGTGAAGAAGGAA | 529R: GCCATGTCATCCTCAGCACC |
| Caspase 7 | XM_002698509.5 | 611F: ACACTGATGCTAATCCCCGT | 748R: AGGCTCTTCCCATGCTCATT |
| Caspase 8 | NM_001045970.2 | 1283F: TCCAGTCACTTTGCCAGAAT | 1411R: CGCAGTGTGAAAGTAGGTTG |
| Caspase 9 | NM_001205504.2 | 267F: GAAGACCAGCAGACAAGCAG | 418R: TCAGTGAATCCTCCAGAACC |
| Bak | NM_001077918.1 | 389F: CTCTGCTGGGCTTTGGCTAC | 592R: ACAAACTGGCCCAACAAAAC |
| Bax | NM_173894.1 | 72F: CCTTTTGCTTCAGGGTTTCAT | 265R: ACTCGGAAAAAGACCTCTCG |
| IL-1β | NM_174093.1 | 546F: GTCTTGTGTGAAAAAAGGTG | 715R: ACGGGCCTTTCTTCGATTTG |
| IL-6 | NM_173923.2 | 381F: CTACCTCCAGAACGAGTATG | 481R: GTGGCTGGAGTGGTTATTAG |
| IL-8 | BC103310.1 | 114F: AACACATTCCACACCTTTCC | 261R: TCACAAATACCTGCACAACC |
| IL-10 | NM_174088.1 | 251F: CGGAAATGATCCAGTTTTACC | 457R: CTCATGGCTTTGTAGACACC |
| IL-12 | NM_174356.1 | 702F: CAAACCAGACCCACCCAAGA | 866R: GGCTGAGGTTTGGTCCATGA |
| TNF-α | NM_173966.3 | 75F: GGGCTCCAGAAGTTGCTTGT | 175R: TGGGGACTGCTCTTCCCTCTG |
| TNFR1 | NM_174674.2 | 266F: ACACTGCCTTGGAGAACCAT | 402R: AGCCAGTTTCACCCCAGTAT |
| TGF β1 | NM_001166068.1 | 327F: CCGCGTGCTAATGGTGGAAT | 444R: CGTCTGCCCGAGAGAGCAAC |
| P50 | NM_001076409.1 | 720F: TGACAGCAAAGCCCCCAATG | 985R: CTCCGAAGCTGGACGAACAC |
| P65 | NM_001080242.2 | 402F: CCAGACCAACAACAACCCCT | 452R: GACGGCATTCAGGTCGTAGT |
| iNOS | DQ676956.1 | 117F: GACACAGGATGACCCCAAAC | 316R: GACTTGCAAGAGAGATCCCC |
| HIF-1α | NM_174339.3 | 17F: GCGCGAACGACAAGAAAAAG | 214R: AAATCACCAGCATCCAGAAG |
| COX-2 | AF004944.1 | 96F: AGCTCTTCCTCCTGTGCCTG | 283R: GCTGGTCCTCGTTCAAAATC |
| ERK2 | NM_175793.2 | 182F: ACCAGACGTACTGCCAGAGA | 440R: GGAAGGTTTGAGGTCACGGT |
| JNK | NM_001192974.2 | 8F: GAAGCAAGCGTGACAGCAAT | 96R: TTCCTTGGGCTCCTGAACCT |
| P38 | NM_001102174.1 | 505F: TTCGGACTGGCTCGACATAC | 721R: TAATGAGATAAGCAGGGGGAGT |
| cFLIP | NM_001012281.1 | 220F: ATGGATACCCCGACAGTGGA | 360R: TGGCTATCTTGCTTCGACCC |
| RIPK1 | NM_001035012.1 | 1359F: CCAACCCCAGTCACCATACT | 1595R: ACTGTCGCCAATCTGAATGC |
| RIPK3 | NM_001101884.2 | 594F: CTCCAGGGCCAGTGATGTCTA | 690R: AATGATGTCTGGGGCACTGAGG |
| MLKL | XM_024978879.1 | 488F: TGGAGGAAACCATCGAAGCC | 762R: GCAGGATGTTGGGGGAATCA |
| GAPDH | NM_001034034.2 | 8F: AGGTCGGAGTGAACGGATT | 194R: GGCCTTTCCATTGATGACGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, L.; Mu, J.; Zheng, Y.; Liu, Y. Selenium Deficiency-Induced Oxidative Stress Causes Myocardial Injury in Calves by Activating Inflammation, Apoptosis, and Necroptosis. Antioxidants 2023, 12, 229. https://doi.org/10.3390/antiox12020229
Lei L, Mu J, Zheng Y, Liu Y. Selenium Deficiency-Induced Oxidative Stress Causes Myocardial Injury in Calves by Activating Inflammation, Apoptosis, and Necroptosis. Antioxidants. 2023; 12(2):229. https://doi.org/10.3390/antiox12020229
Chicago/Turabian StyleLei, Lei, Jing Mu, Yingce Zheng, and Yun Liu. 2023. "Selenium Deficiency-Induced Oxidative Stress Causes Myocardial Injury in Calves by Activating Inflammation, Apoptosis, and Necroptosis" Antioxidants 12, no. 2: 229. https://doi.org/10.3390/antiox12020229