
Protection against Doxorubicin-Induced Cardiotoxicity by Ergothioneine

 , ,
, , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
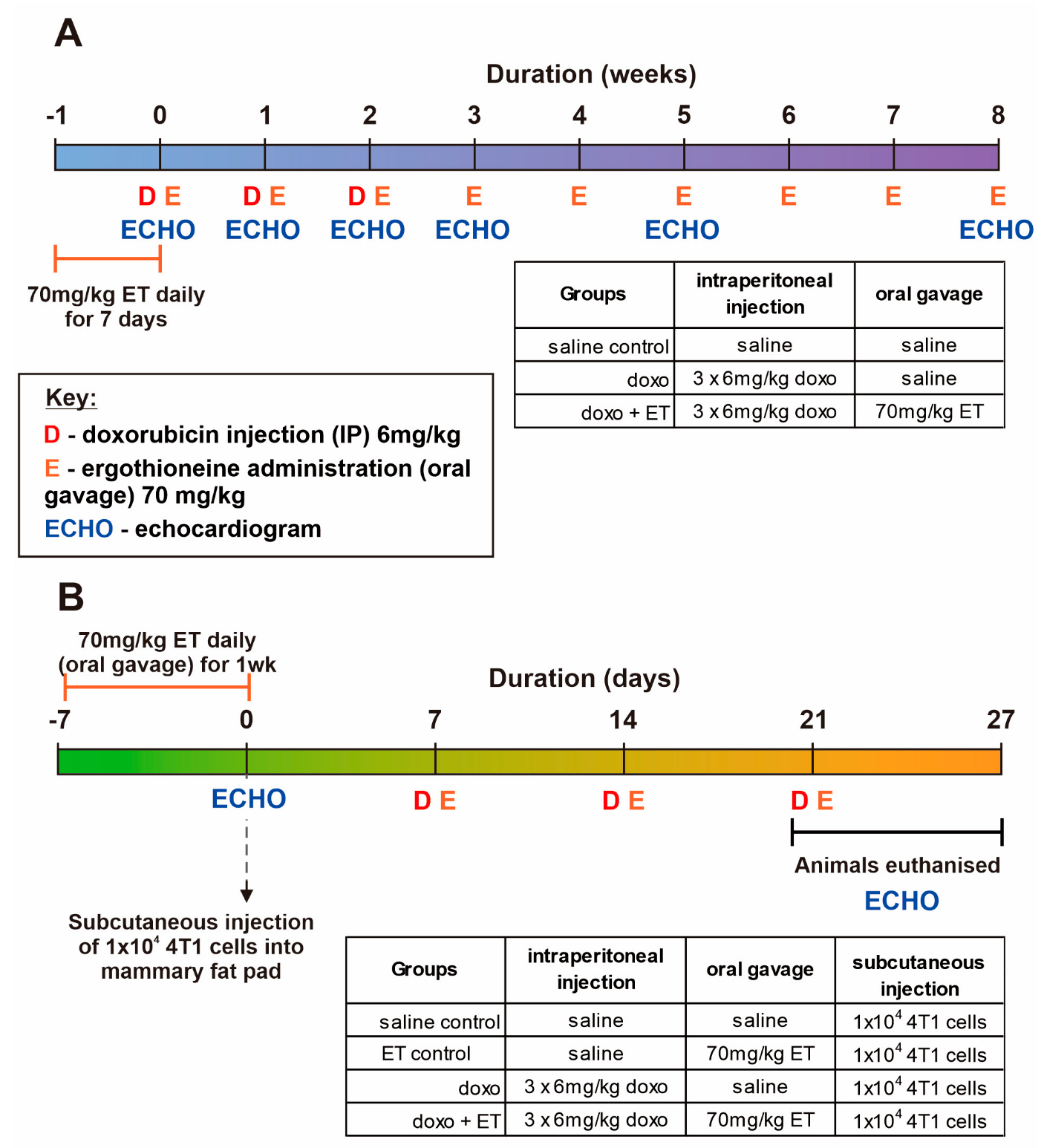
2.2. Animal Studies
2.3. Doxorubicin Cardiotoxicity Model
2.4. Mouse Breast Cancer Model
2.5. Echo Measurement and Assessment
2.6. Quantitative PCR of Mouse Heart RNA
2.7. Extraction of Blood and Tissues ET and its Metabolites
2.8. Quantifying Biomarkers of Oxidative Damage to RNA and DNA
2.9. Protein Carbonylation ELISA
2.10. Liquid Chromatography–Mass Spectrometry Analysis
2.11. Statistical Analysis
3. Results
3.1. Animal Weights in Doxorubicin Treated Mice
3.2. Echocardiographic Assessment of Cardiac Function in Doxorubicin-Treated Mice
3.3. Expression of ET Transporter mRNA in Heart
3.4. Levels of Ergothioneine and Hercynine in Heart Tissues
3.5. Levels of Ergothioneine and Hercynine in Plasma
3.6. Oxidative Damage Biomarkers
3.7. Breast Cancer Model
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer 5’→3’ | Reverse Primer 5’→3’ |
|---|---|---|
| OCTN1 | AAGTCCTCTTTGCAACCATGGC | CTGTGTTGTTTGCCATTGTGGG |
| β-ACTIN | TGACAGGATGCAGAAGGAGA | CGCTCAGGAGGAGCAATG |
| Targets | MRM Transition | Fragmentor Voltage (V) | Collision Energy (eV) |
|---|---|---|---|
| ET | 230 → 186 | 103 | 9 |
| ET-d9 | 239 → 195 | 98 | 9 |
| Hercynine | 198 → 95 | 94 | 21 |
| Hercynine-d9 | 207 → 95 | 103 | 9 |
| 8OHdG | 284 → 168 | 80 | 10 |
| 8OHdG-13C2,15N1 | 287 → 171 | 80 | 10 |
| 8OHG | 300 → 168 | 85 | 10 |
| 8OHG-13C1,15N2 | 303 → 171 | 85 | 10 |
| dG | 268 → 152 | 70 | 6 |
| dG-13C1,15N2 | 271 → 155 | 65 | 5 |
| Guanosine | 284 → 152 | 70 | 10 |
| Guanosine-15N5 | 289 → 157 | 85 | 10 |


References
- Raj, S.; Franco, V.I.; Lipshultz, S.E. Anthracycline-induced cardiotoxicity: A review of pathophysiology, diagnosis, and treatment. Curr. Treat Options Cardiovasc. Med. 2014, 16, 315. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, I.; Aimo, A.; Grigoratos, C.; Castiglione, V.; Gentile, F.; Saccaro, L.; Arzilli, C.; Cardinale, D.; Passino, C.; Emdin, M. Oxidative stress and inflammation: Determinants of anthracycline cardiotoxicity and possible therapeutic targets. Heart Fail Rev. 2021, 26, 881–890. [Google Scholar] [CrossRef]
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity from Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Muller, I.; Jenner, A.; Bruchelt, G.; Niethammer, D.; Halliwell, B. Effect of concentration on the cytotoxic mechanism of doxorubicin--apoptosis and oxidative DNA damage. Biochem. Biophys. Res. Commun. 1997, 230, 254–257. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a specific complex between adriamycin and negatively-charged phospholipids. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1980, 597, 1–14. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Prasad, S.V.N.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dalan, R.; Hu, Z.; Wang, J.; Chew, N.; Poh, K.; Tan, R.; Soong, T.; Dai, Y.; Ye, L.; et al. Reactive Oxygen Species Scavenging Nanomedicine for the Treatment of Ischemic Heart Disease. Adv. Mater. 2022, 34, e2202169. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M. Free Radicals in Biology and Medicine, 5th ed.; Clarendon Press: Oxford, UK, 2015. [Google Scholar]
- Akolkar, G.; da Silva Dias, D.; Ayyappan, P.; Bagchi, A.K.; Jassal, D.S.; Salemi, V.M.C.; Irigoyen, M.C.; De Angelis, K.; Singal, P. Vitamin C mitigates oxidative/nitrosative stress and inflammation in doxorubicin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H795–H809. [Google Scholar] [CrossRef] [PubMed]
- Bjelogrlic, S.K.; Radic, J.; Jovic, V.; Radulovic, S. Activity of d,l-alpha-tocopherol (vitamin E) against cardiotoxicity induced by doxorubicin and doxorubicin with cyclophosphamide in mice. Basic Clin. Pharmacol. Toxicol. 2005, 97, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Arica, V.; Demir, I.H.; Tutanc, M.; Basarslan, F.; Arica, S.; Karcoiglu, M.; Ozturk, H.; Nacar, A. N-acetylcysteine prevents doxorubucin-induced cardiotoxicity in rats. Hum. Exp. Toxicol. 2013, 32, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalcanti, I.D.L.; Soares, J.C.S.; Medeiros, S.M.d.F.R.d.S.; Cavalcanti, I.; Lira Nogueira, M.C.d.B. Can antioxidant vitamins avoid the cardiotoxicity of doxorubicin in treating breast cancer? PharmaNutrition 2021, 16, 100259. [Google Scholar] [CrossRef]
- Ambrosone, C.B.; Zirpoli, G.R.; Hutson, A.D.; McCann, W.E.; McCann, S.E.; Barlow, W.E.; Kelly, K.M.; Cannioto, R.; Sucheston-Campbell, L.; Hershman, D.; et al. Dietary Supplement Use During Chemotherapy and Survival Outcomes of Patients with Breast Cancer Enrolled in a Cooperative Group Clinical Trial (SWOG S0221). J. Clin. Oncol. 2020, 38, 804–814. [Google Scholar] [CrossRef]
- Lawenda, B.D.; Kelly, K.M.; Ladas, E.J.; Sagar, S.M.; Vickers, A.; Blumberg, J.B. Should Supplemental Antioxidant Administration Be Avoided During Chemotherapy and Radiation Therapy? JNCI J. Natl. Cancer Inst. 2008, 100, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.V.; Le Gal, K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.; Bergh, P.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4, e127647. [Google Scholar] [CrossRef]
- Buss, J.L.; Hasinoff, B. The one-ring open hydrolysis product intermediates of the cardioprotective agent ICRF-187 (dexrazoxane) displace iron from iron-anthracycline complexes. Agents Actions 1993, 40, 86–95. [Google Scholar] [CrossRef]
- Shaikh, F.; Dupuis, L.; Alexander, S.; Gupta, A.; Mertens, L.; Nathan, P. Cardioprotection and Second Malignant Neoplasms Associated with Dexrazoxane in Children Receiving Anthracycline Chemotherapy: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2016, 108, djv357. [Google Scholar] [CrossRef] [Green Version]
- Cheah, I.K.; Halliwell, B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta 2012, 1822, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Cheah, I.K.; Halliwell, B. Ergothioneine, recent developments. Redox Biol. 2021, 42, 101868. [Google Scholar] [CrossRef]
- Melville, D.B. Ergothioneine. Vitam. Horm. 1959, 17, 155–204. [Google Scholar]
- Halliwell, B.; Cheah, I.; Tang, R.M.Y. Ergothioneine—A diet-derived antioxidant with therapeutic potential. FEBS Lett. 2018, 592, 3357–3366. [Google Scholar] [CrossRef] [Green Version]
- Tang, R.M.Y.; Cheah, I.; Yew, T.; Halliwell, B. Distribution and accumulation of dietary ergothioneine and its metabolites in mouse tissues. Sci. Rep. 2018, 8, 1601. [Google Scholar] [CrossRef] [Green Version]
- Tucker, R.A.J.; Cheah, I.; Halliwell, B. Specificity of the ergothioneine transporter natively expressed in HeLa cells. Biochem. Biophys. Res. Commun. 2019, 513, 22–27. [Google Scholar] [CrossRef]
- Grundemann, D.; Hartmann, L.; Flogel, S. The ergothioneine transporter (ETT): Substrates and locations, an inventory. FEBS Lett. 2022, 596, 1252–1269. [Google Scholar] [CrossRef]
- Akanmu, D.; Cecchini, R.; Aruoma, O.I.; Halliwell, B. The antioxidant action of ergothioneine. Arch. Biochem. Biophys. 1991, 288, 10–16. [Google Scholar] [CrossRef]
- Koh, S.S.; Ooi, S.; Lui, N.; Qiong, C.; Ho, L.; Cheah, I.; Halliwell, B.; Herr, D.; Ong, W. Effect of Ergothioneine on 7-Ketocholesterol-Induced Endothelial Injury. Neuromolecular Med. 2021, 23, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Motohash, N.; Mori, I.; Sugiura, Y.; Tanaka, H. Metal-complexes of ergothioneine. Chem. Pharm. Bull. 1974, 22, 654–657. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.Z.; Mao, L.; Fan, R.; Zhu, J.; Zhang, Y.; Wang, J.; Kalyanaraman, B.; Frei, B. Ergothioneine prevents copper-induced oxidative damage to DNA and protein by forming a redox-inactive ergothioneine-copper complex. Chem. Res. Toxicol. 2011, 24, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S. The unusual amino acid L-ergothioneine is a physiologic cytoprotectant. Cell Death Differ. 2010, 17, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.D.; McCarthy, F.; Manna, S.; Groarke, E.; Kell, D.; Kenny, L.; McCarthy, C. L-(+)-Ergothioneine Significantly Improves the Clinical Characteristics of Preeclampsia in the Reduced Uterine Perfusion Pressure Rat Model. Hypertension 2020, 75, 561–568. [Google Scholar] [CrossRef]
- Smith, E.; Ottosson, F.; Hellstrand, S.; Ericson, U.; Orho-Melander, M.; Fernandez, C.; Melander, O. Ergothioneine is associated with reduced mortality and decreased risk of cardiovascular disease. Heart 2020, 106, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Dare, A.; Elrashedy, A.; Channa, M.; Nadar, A. Cardioprotective Effects and in-silico Antioxidant Mechanism of L-Ergothioneine in Experimental Type-2 Diabetic Rats. Cardiovasc. Hematol. Agents Med. Chem. 2022, 20, 133–147. [Google Scholar] [CrossRef]
- Gökçe, G.; Arun, M.; Ertuna, E. Ergothioneine prevents endothelial dysfunction induced by mercury chloride. Exp. Ther. Med. 2018, 15, 4697–4702. [Google Scholar] [CrossRef] [Green Version]
- Li, R.W.; Yang, C.; Sit, A.; Kwan, Y.; Lee, S.; Hoi, M.; Chan, S.; Hausman, M.; Vanhoutte, P.; Leung, G. Uptake and protective effects of ergothioneine in human endothelial cells. J. Pharmacol. Exp. Ther. 2014, 350, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Sakrak, Ö.; Kerem, M.; Bedirli, A.; Pasaoğlu, H.; Alper, M.; Ofluoğlu, E.; Yılmaz, T. Ergothioneine prevents acute lung injury in mesenteric ischemia and reperfusion injury in rats. J. Crit. Care 2008, 23, 268–269. [Google Scholar] [CrossRef]
- Bedirli, A.; Sakrak, O.; Muhtaroglu, S.; Soyuer, I.; Guler, I.; Erdogan, A.R.; Sozuer, E. Ergothioneine pretreatment protects the liver from ischemia-reperfusion injury caused by increasing hepatic heat shock protein 70. J. Surg Res. 2004, 122, 96–102. [Google Scholar] [CrossRef]
- Sakrak, O.; Kerem, M.; Bedirli, A.; Pasaoglu, H.; Akyurek, N.; Ofluoglu, E.; Gultekin, F. Ergothioneine modulates proinflammatory cytokines and heat shock protein 70 in mesenteric ischemia and reperfusion injury. J. Surg Res. 2008, 144, 36–42. [Google Scholar] [CrossRef]
- Martin, K.R. The bioactive agent ergothioneine, a key component of dietary mushrooms, inhibits monocyte binding to endothelial cells characteristic of early cardiovascular disease. J. Med. Food 2010, 13, 1340–1346. [Google Scholar] [CrossRef]
- Arduini, A.; Eddy, L.; Hochstein, P. The reduction of ferryl myoglobin by ergothioneine: A novel function for ergothioneine. Arch. Biochem. Biophys. 1990, 281, 41–43. [Google Scholar] [CrossRef]
- Sansbury, B.E.; DeMartino, A.; Xie, Z.; Brooks, A.; Brainard, R.; Watson, L.; DeFilippis, A.; Cummins, T.; Harbeson, M.; Brittian, K.; et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ. Heart Fail. 2014, 7, 634–642. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Cheah, I.; Drum, C. Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem. Biophys. Res. Commun. 2016, 470, 245–250. [Google Scholar] [CrossRef]
- Cheah, I.K.; Tang, R.; Ye, P.; Yew, T.; Lim, K.; Halliwell, B. Liver ergothioneine accumulation in a guinea pig model of non-alcoholic fatty liver disease. A possible mechanism of defence? Free Radic. Res. 2016, 50, 14–25. [Google Scholar] [CrossRef]
- Wang, J.W.; Fontes, M.; Wang, X.; Chong, S.; Kessler, E.; Zhang, Y.; de Haan, J.; Arslan, F.; de Jager, S.; Timmers, L.; et al. Leukocytic Toll-Like Receptor 2 Deficiency Preserves Cardiac Function and Reduces Fibrosis in Sustained Pressure Overload. Sci. Rep. 2017, 7, 9193. [Google Scholar] [CrossRef]
- de Kleijn, D.P.V.; Chong, S.; Wang, X.; Yatim, S.; Fairhurst, A.; Vernooij, F.; Zharkova, O.; Chan, M.; Foo, R.; Timmers, L.; et al. Toll-like receptor 7 deficiency promotes survival and reduces adverse left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2019, 115, 1791–1803. [Google Scholar] [CrossRef]
- Tei, C. New non-invasive index for combined systolic and diastolic ventricular function. J. Cardiol. 1995, 26, 135–136. [Google Scholar]
- Salemi, V.M.; Pires, M.; Cestari, I.; Cestari, I.; Picard, M.; Leirner, A.; Mady, C. Echocardiographic assessment of global ventricular function using the myocardial performance index in rats with hypertrophy. Artif. Organs 2004, 28, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.; Davies, K.; Davies, M.; Dick, T.; Finkel, T.; Forman, H.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Cheah, I.K.; Tang, R.; Yew, T.; Lim, K.; Halliwell, B. Administration of pure ergothioneine to healthy human subjects; Uptake, metabolism and effects on biomarkers of oxidative damage and inflammation. Antioxid. Redox Signal. 2017, 26, 193–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaorsky, N.G.; Churilla, T.; Egleston, B.; Fisher, S.; Ridge, J.; Horwitz, E.; Meyer, J. Causes of death among cancer patients. Ann. Oncol. 2017, 28, 400–407. [Google Scholar] [CrossRef]
- Schairer, C.; Mink, P.; Carroll, L.; Devesa, S. Probabilities of death from breast cancer and other causes among female breast cancer patients. J. Natl. Cancer Inst. 2004, 96, 1311–1321. [Google Scholar] [CrossRef] [Green Version]
- Florescu, M.; Cinteza, M.; Vinereanu, D. Chemotherapy-induced Cardiotoxicity. Maedica (Bucur) 2013, 8, 59–67. [Google Scholar]
- Grenier, M.A.; Lipshultz, S. Epidemiology of anthracycline cardiotoxicity in children and adults. Semin. Oncol. 1998, 25 (Suppl. 10), 72–85. [Google Scholar]
- Gianni, L.; Salvatorelli, E.; Minotti, G. Anthracycline cardiotoxicity in breast cancer patients: Synergism with trastuzumab and taxanes. Cardiovasc. Toxicol. 2007, 7, 67–71. [Google Scholar] [CrossRef]
- Halliwell, B.; Tang, R.; Cheah, I. Diet-Derived Antioxidants: The Special Case of Ergothioneine. Annu. Rev. Food Sci. Technol. 2023, 14. [Google Scholar] [CrossRef]
- Kato, Y.; Kubo, Y.; Iwata, D.; Kato, S.; Sudo, T.; Sugiura, T.; Kagaya, T.; Wakayama, T.; Hirayama, A.; Sugimoto, M.; et al. Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm. Res. 2010, 27, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, C.; Bach, M.; Bauer, T.; da Ponte, J.C.; Schomig, E.; Grundemann, D. Knockout of the ergothioneine transporter ETT in zebrafish results in increased 8-oxoguanine levels. Free Radic. Biol. Med. 2015, 83, 178–185. [Google Scholar] [CrossRef]
- Cheah, I.K.; Ong, R.; Gruber, J.; Yew, T.; Ng, L.; Chen, C.; Halliwell, B. Knockout of a putative ergothioneine transporter in Caenorhabditis elegans decreases lifespan and increases susceptibility to oxidative damage. Free Radic. Res. 2013, 47, 1036–1045. [Google Scholar] [CrossRef]
- Tei, C.; Ling, L.; Hodge, D.; Bailey, K.; Oh, J.; Rodeheffer, R.; Tajik, A.; Seward, J. New index of combined systolic and diastolic myocardial performance: A simple and reproducible measure of cardiac function—A study in normals and dilated cardiomyopathy. J. Cardiol. 1995, 26, 357–366. [Google Scholar]
- Sofia, R.; Melita, V.; De Vita, A.; Ruggiero, A.; Romano, A.; Attina, G.; Birritella, L.; Lamendola, P.; Lombardo, A.; Lanza, G.; et al. Cardiac Surveillance for Early Detection of Late Subclinical Cardiac Dysfunction in Childhood Cancer Survivors after Anthracycline Therapy. Front. Oncol. 2021, 11, 624057. [Google Scholar] [CrossRef]
- Dawson, D.; Lygate, C.; Saunders, J.; Schneider, J.; Ye, X.; Hulbert, K.; Noble, J.; Neubauer, S. Quantitative 3-dimensional echocardiography for accurate and rapid cardiac phenotype characterization in mice. Circulation 2004, 110, 1632–1637. [Google Scholar] [CrossRef]
- Chong, S.Y.; Zharkova, O.; Yatim, S.; Wang, X.; Lim, X.; Huang, C.; Tan, C.; Jiang, J.; Ye, L.; Tan, M.; et al. Tissue factor cytoplasmic domain exacerbates post-infarct left ventricular remodeling via orchestrating cardiac inflammation and angiogenesis. Theranostics 2021, 11, 9243–9261. [Google Scholar] [CrossRef]
- Obayashi, K.; Kurihara, K.; Okano, Y.; Masaki, H.; Yarosh, D. L-Ergothioneine scavenges superoxide and singlet oxygen and suppresses TNF-alpha and MMP-1 expression in UV-irradiated human dermal fibroblasts. J. Cosmet. Sci. 2005, 56, 17–27. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Whiteman, M.; England, T.; Halliwell, B. Antioxidant action of ergothioneine: Assessment of its ability to scavenge peroxynitrite. Biochem. Biophys. Res. Commun. 1997, 231, 389–391. [Google Scholar] [CrossRef]
- Chaudiere, J.; Ferrari-Iliou, R. Intracellular antioxidants: From chemical to biochemical mechanisms. Food Chem. Toxicol. 1999, 37, 949–962. [Google Scholar] [CrossRef]
- Colognato, R.; Laurenza, I.; Fontana, I.; Coppede, F.; Siciliano, G.; Coecke, S.; Aruoma, O.; Benzi, L.; Migliore, L. Modulation of hydrogen peroxide-induced DNA damage, MAPKs activation and cell death in PC12 by ergothioneine. Clin. Nutr. 2006, 25, 135–145. [Google Scholar] [CrossRef]
- Aruoma, O.I.; Spencer, J.; Mahmood, N. Protection against oxidative damage and cell death by the natural antioxidant ergothioneine. Food Chem. Toxicol. 1999, 37, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Gokce, G.; Arun, M. Ergothioneine produces relaxation in isolated rat aorta by inactivating superoxide anion. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3339–3345. [Google Scholar] [PubMed]
- Servillo, L.; D’Onofrio, N.; Casale, R.; Cautela, D.; Giovane, A.; Castaldo, D.; Balestrieri, M. Ergothioneine products derived by superoxide oxidation in endothelial cells exposed to high-glucose. Free Radic. Biol. Med. 2017, 108, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Servillo, L.; Castaldo, D.; Casale, R.; D’Onofrio, N.; Giovane, A.; Cautela, D.; Balestrieri, M. An uncommon redox behavior sheds light on the cellular antioxidant properties of ergothioneine. Free Radic. Biol. Med. 2015, 79, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Ando, C.; Morimitsu, Y. A proposed antioxidation mechanism of ergothioneine based on the chemically derived oxidation product hercynine and further decomposition products. Biosci. Biotechnol. Biochem. 2021, 85, 1175–1182. [Google Scholar] [CrossRef]
- Myers, C.E.; Gianni, L.; Simone, C.; Klecker, R.; Greene, R. Oxidative destruction of erythrocyte ghost membranes catalyzed by the doxorubicin-iron complex. Biochemistry 1982, 21, 1707–1712. [Google Scholar] [CrossRef]
- Hershko, C.; Link, G.; Tzahor, M.; Kaltwasser, J.; Athias, P.; Grynberg, A.; Pinson, A. Anthracycline toxicity is potentiated by iron and inhibited by deferoxamine: Studies in rat heart cells in culture. J. Lab. Clin. Med. 1993, 122, 245–251. [Google Scholar]
- Panjrath, G.S.; Patel, V.; Valdiviezo, C.; Narula, N.; Narula, J.; Jain, D. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll Cardiol 2007, 49, 2457–2464. [Google Scholar] [CrossRef] [Green Version]
- Arkadopoulos, N.; Nastos, C.; Kalimeris, K.; Economou, E.; Theodoraki, K.; Kouskouni, E.; Pafiti, A.; Kostopanagiotou, G.; Smyrniotis, V. Iron chelation for amelioration of liver ischemia-reperfusion injury. Hemoglobin 2010, 34, 265–277. [Google Scholar] [CrossRef]
- Koyama, T.; Tsubota, A.; Sawano, T.; Tawa, M.; Watanabe, B.; Hiratake, J.; Nakagawa, K.; Matsumura, Y.; Ohkita, M. Involvement of gamma-Glutamyl Transpeptidase in Ischemia/Reperfusion-Induced Cardiac Dysfunction in Isolated Rat Hearts. Biol. Pharm. Bull. 2019, 42, 1947–1952. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Evans, J.; Robins, S.; Wilson, P.; Albano, I.; Fox, C.; Wang, T.; Benjamin, E.; D’Agostino, R.B.; Vasan, R. Gamma glutamyl transferase and metabolic syndrome, cardiovascular disease, and mortality risk: The Framingham Heart Study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Ndrepepa, G.; Colleran, R.; Kastrati, A. Gamma-glutamyl transferase and the risk of atherosclerosis and coronary heart disease. Clin. Chim. Acta 2018, 476, 130–138. [Google Scholar] [CrossRef]
- Salama, S.A.; Abd-Allah, G.; Mohamadin, A.; Elshafey, M.; Gad, H. Ergothioneine mitigates cisplatin-evoked nephrotoxicity via targeting Nrf2, NF-kappaB, and apoptotic signaling and inhibiting gamma-glutamyl transpeptidase. Life Sci. 2021, 278, 119572. [Google Scholar] [CrossRef]
- Brancaccio, M.; Russo, M.; Masullo, M.; Palumbo, A.; Russo, G.; Castellano, I. Sulfur-containing histidine compounds inhibit gamma-glutamyl transpeptidase activity in human cancer cells. J. Biol. Chem. 2019, 294, 14603–14614. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Lo, H.; Korivi, M.; Tsai, Y.; Tang, M.; Yang, H. Dermato-protective properties of ergothioneine through induction of Nrf2/ARE-mediated antioxidant genes in UVA-irradiated Human keratinocytes. Free Radic. Biol. Med. 2015, 86, 102–117. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Martino, E.; Balestrieri, A.; Mele, L.; Cautela, D.; Castaldo, D.; Balestrieri, M. Diet-derived ergothioneine induces necroptosis in colorectal cancer cells by activating the SIRT3/MLKL pathway. FEBS Lett. 2022, 596, 1313–1329. [Google Scholar] [CrossRef]
- Turck, D.; Bresson, J.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.; Mangelsdorf, I.; McArdle, H.; Naska, A.; et al. Safety of synthetic l-ergothioneine (Ergoneine®) as a novel food pursuant to Regulation (EC) No 258/97. EFSA J. 2016, 14, 4629. [Google Scholar] [CrossRef] [Green Version]
- Turck, D.; Bresson, J.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.; Mangelsdorf, I.; McArdle, H.; Naska, A.; et al. Statement on the safety of synthetic l-ergothioneine as a novel food—Supplementary dietary exposure and safety assessment for infants and young children, pregnant and breastfeeding women. EFSA J. 2017, 15, 5060. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheah, I.K.; Tang, R.M.Y.; Wang, X.; Sachaphibulkij, K.; Chong, S.Y.; Lim, L.H.K.; Wang, J.-W.; Halliwell, B. Protection against Doxorubicin-Induced Cardiotoxicity by Ergothioneine. Antioxidants 2023, 12, 320. https://doi.org/10.3390/antiox12020320
Cheah IK, Tang RMY, Wang X, Sachaphibulkij K, Chong SY, Lim LHK, Wang J-W, Halliwell B. Protection against Doxorubicin-Induced Cardiotoxicity by Ergothioneine. Antioxidants. 2023; 12(2):320. https://doi.org/10.3390/antiox12020320
Chicago/Turabian StyleCheah, Irwin K., Richard M. Y. Tang, Xiaoyuan Wang, Karishma Sachaphibulkij, Suet Yen Chong, Lina H. K. Lim, Jiong-Wei Wang, and Barry Halliwell. 2023. "Protection against Doxorubicin-Induced Cardiotoxicity by Ergothioneine" Antioxidants 12, no. 2: 320. https://doi.org/10.3390/antiox12020320