
Mild Traumatic Brain Injury Induces Mitochondrial Calcium Overload and Triggers the Upregulation of NCLX in the Hippocampus




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Antibodies and Reagents
2.3. Mild Traumatic Brain Injury Induction
2.4. Mitochondrial Isolation
2.5. Immunoblot
2.6. Indirect Mitochondrial Membrane Potential Measurement
2.7. Intramitochondrial Calcium Levels
2.8. Calcium Uptake Assays
2.9. Swelling Assay
2.10. Electron Microscopy
2.11. Statistical Analysis
3. Results
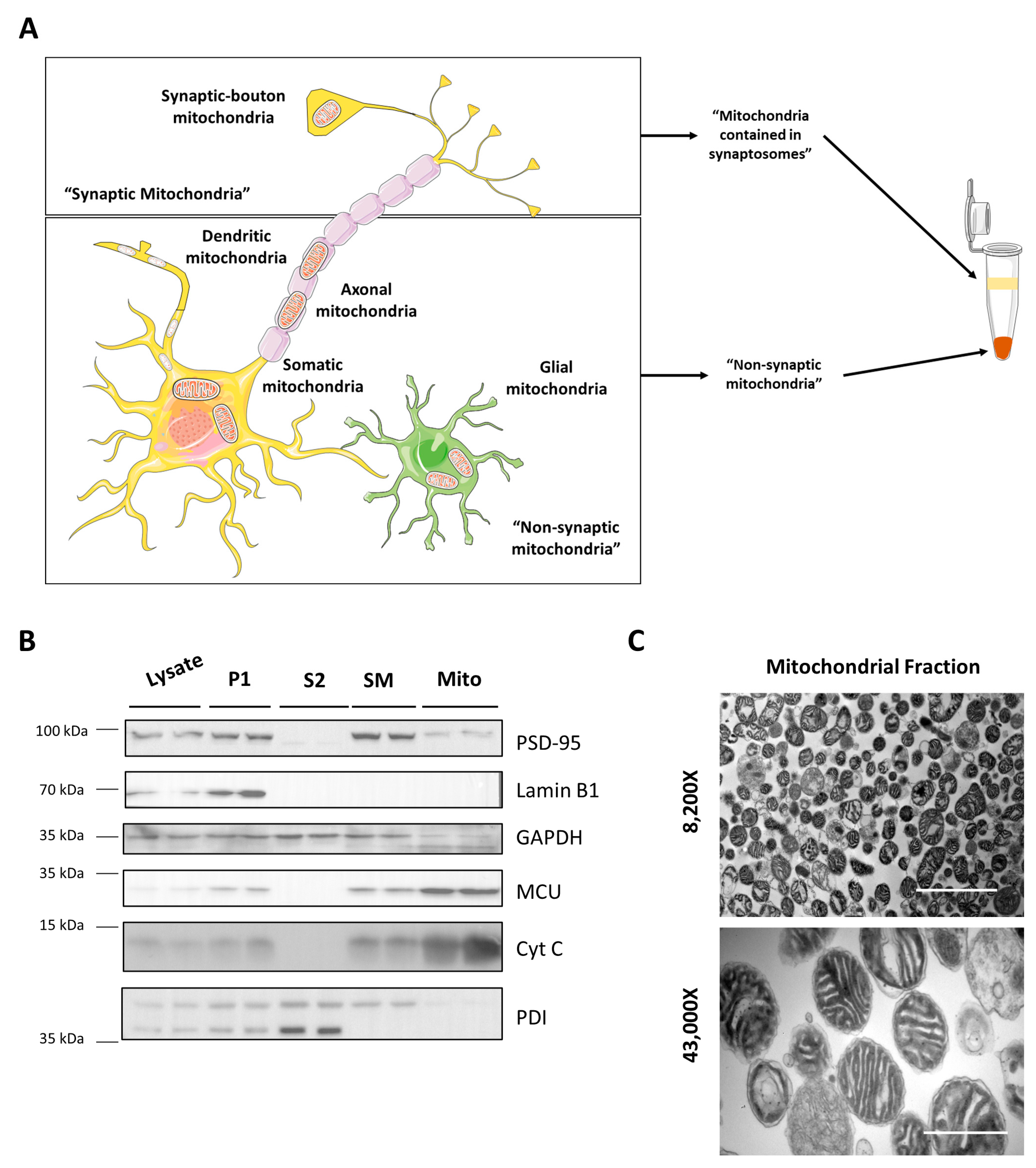
3.1. Mitochondrial Isolation
3.2. Mitochondrial Membrane Potential Decreases Soon after mTBI
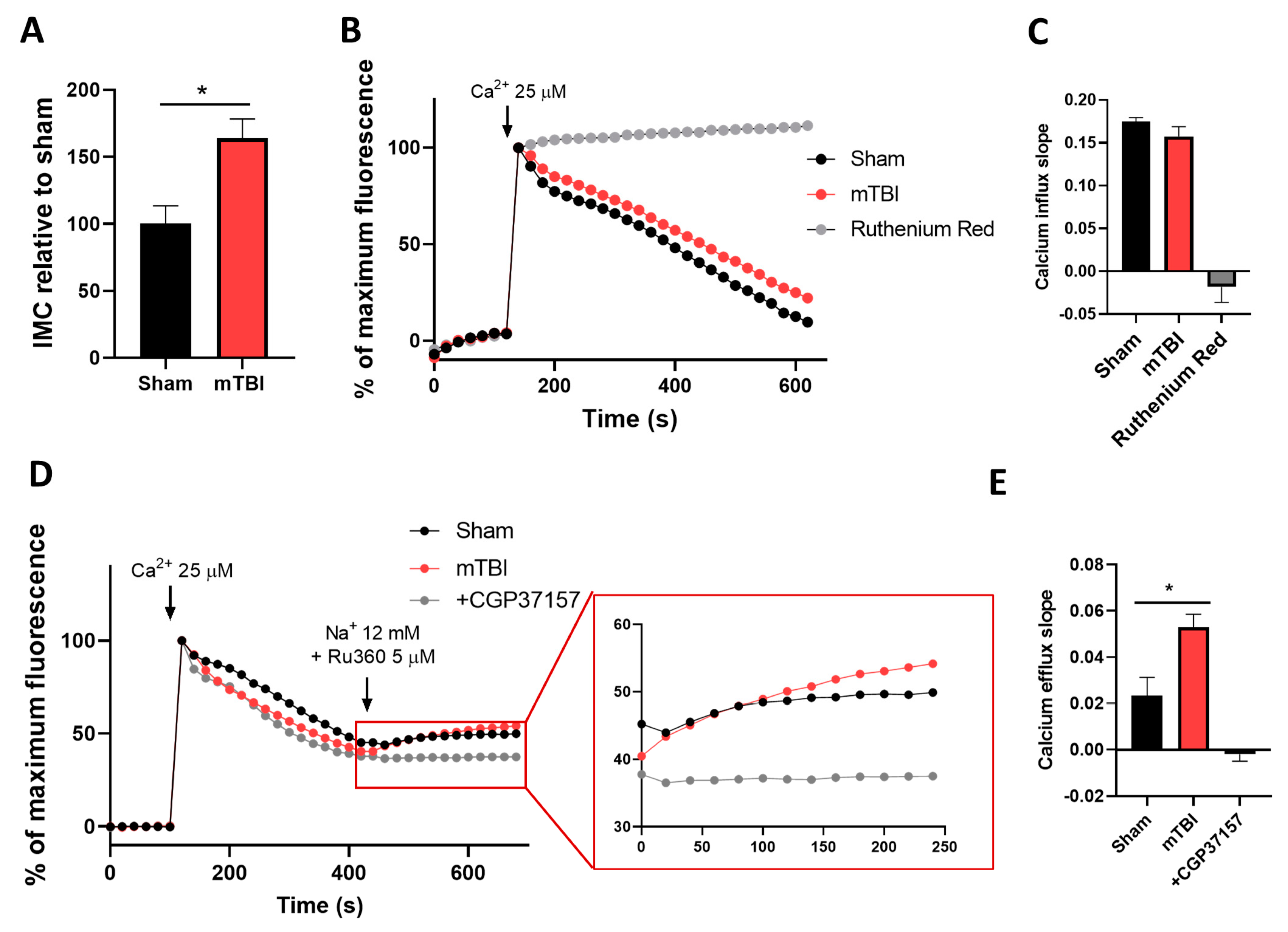
3.3. mTBI Increases Intramitochondrial Calcium Content and Calcium Efflux in Hippocampal Mitochondria
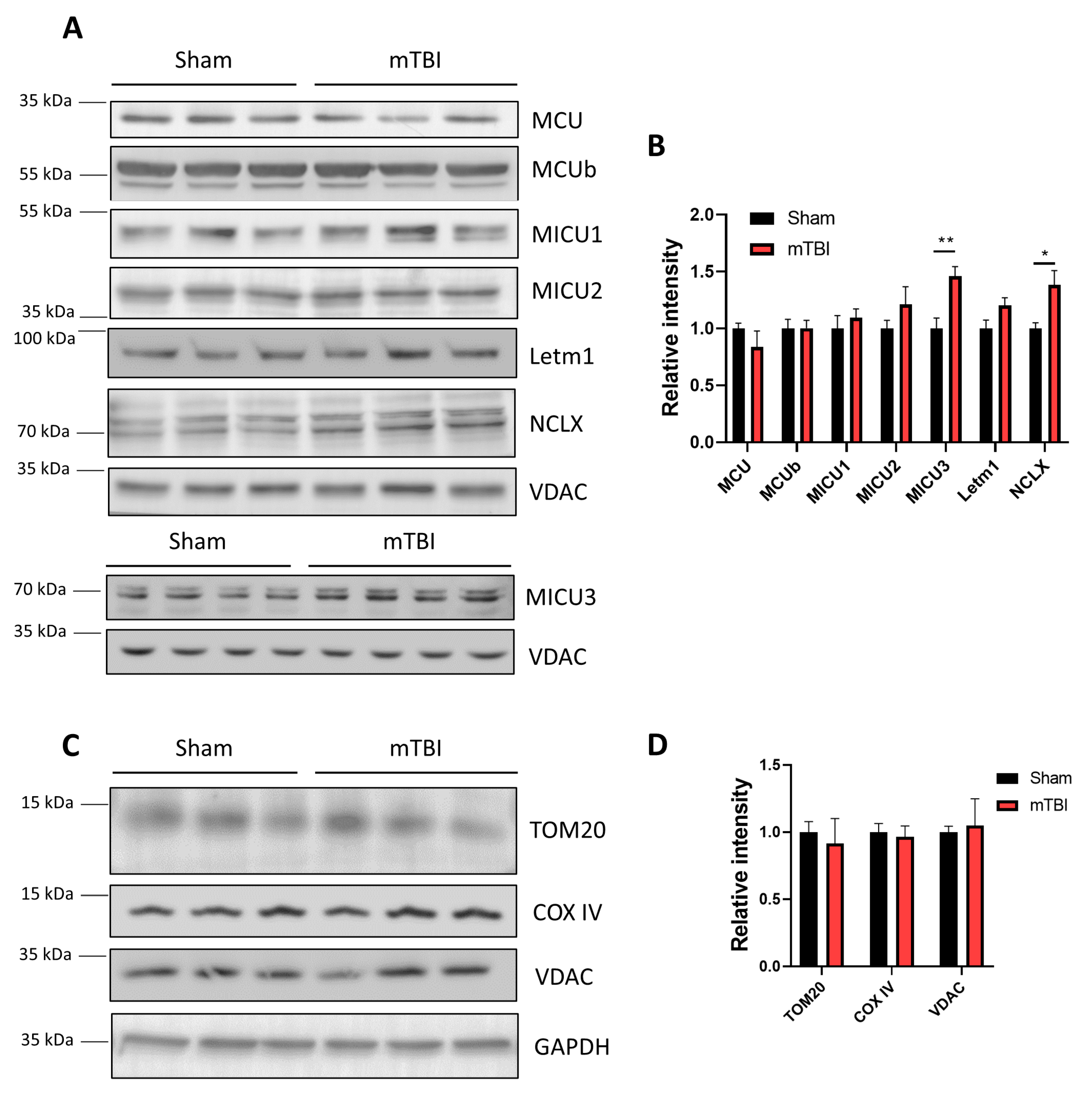
3.4. mTBI Increases the Protein Levels of the Mitochondrial Sodium/Calcium Exchanger NCLX in Hippocampal Mitochondria
3.5. mTBI Increases the Calcium Retention Capacity in Hippocampal Mitochondria
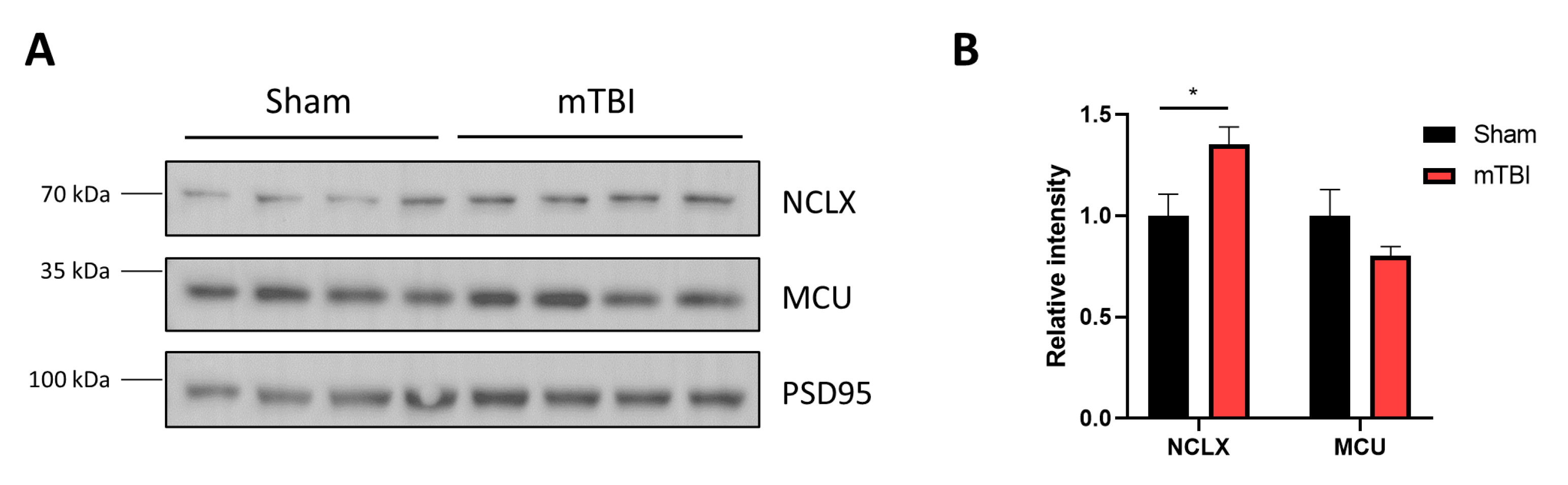
3.6. mTBI Increases NCLX Protein Levels in Mitochondria Contained in Synaptosomes
4. Discussion
5. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blennow, K.; Brody, D.L.; Kochanek, P.M.; Levin, H.; Mckee, A.; Ribbers, G.M.; Yaffe, K.; Zetterberg, H. Traumatic brain injuries. Nat. Rev. Dis. Primers 2016, 2, 16084. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Mollayeva, T.; Mollayeva, S.; Colantonio, A. Traumatic brain injury: Sex, gender and intersecting vulnerabilities. Nat. Rev. Neurol. 2018, 14, 711–722. [Google Scholar] [CrossRef]
- Mccrory, P.; Meeuwisse, W.; Aubry, M.; Cantu, B.; Dvorak, J.; Echemendia, R.; Engebretsen, L.; Johnston, K.; Kutcher, J.; Raftery, M.; et al. Consensus statement on Concus-sion in—The 4th International Conference on Concussion in Sport held in Zurich, November 2012. Phys. Ther. Sport 2013, 14, e1–e13. [Google Scholar] [CrossRef]
- Silverberg, N.D.; Duhaime, A.C.; Iaccarino, M.A. Mild Traumatic Brain Injury in 2019–2020. JAMA 2020, 323, 177–178. [Google Scholar] [CrossRef]
- Gupta, R.; Sen, N. Traumatic brain injury: A risk factor for neurodegenerative diseases. Rev. Neurosci. 2016, 27, 93–100. [Google Scholar] [CrossRef]
- Rule, G.T.; Bocchieri, R.T.; Burns, J.M.; Young, L.A. Biophysical Mechanisms of Traumatic Brain Injuries. Semin. Neurol. 2015, 35, 005–011. [Google Scholar] [CrossRef]
- Sharp, D.J.; Scott, G.; Leech, R. Network dysfunction after traumatic brain injury. Nat. Rev. Neurol. 2014, 10, 156–166. [Google Scholar] [CrossRef]
- Cominski, T.P.; Jiao, X.; Catuzzi, J.E.; Stewart, A.L.; Pang, K.C.H. The Role of the Hippocampus in Avoidance Learning and Anxiety Vulnerability. Front. Behav. Neurosci. 2014, 8, 273. [Google Scholar] [CrossRef]
- Carlesimo, G.A. The temporal lobes and memory. Handb. Clin. Neurol. 2022, 187, 319–337. [Google Scholar]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol. Life Sci. 2004, 61, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Acosta, S.A.; Tajiri, N.; Shinozuka, K.; Ishikawa, H.; Grimmig, B.; Diamond, D.M.; Sanberg, P.R.; Bickford, P.C.; Kaneko, Y.; Borlongan, C.V. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 2013, 8, e53376. [Google Scholar] [CrossRef]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Ohta, M.; Higashi, Y.; Yawata, T.; Kitahara, M.; Nobumoto, A.; Ishida, E.; Tsuda, M.; Fujimoto, Y.; Shimizu, K. Attenuation of axonal injury and oxidative stress by edaravone protects against cognitive impairments after traumatic brain injury. Brain Res. 2013, 1490, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Li, L.; Liu, H.; Lu, W.; Ling, X.; Gong, M. Rutaecarpine Attenuates Oxidative Stress-Induced Traumatic Brain Injury and Reduces Secondary Injury via the PGK1/KEAP1/NRF2 Signaling Pathway. Front. Pharmacol. 2022, 13, 807125. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Joseph, B.; Spry, M.; Vekaria, H.J.; Saatman, K.E.; Sullivan, P.G. Acute Mito-chondrial Impairment Underlies Prolonged Cellular Dysfunction after Repeated Mild Traumatic Brain Injuries. J. Neurotrauma 2019, 36, 1252–1263. [Google Scholar]
- Gilmer, L.K.; Roberts, K.N.; Joy, K.; Sullivan, P.G.; Scheff, S.W. Early mitochondrial dysfunction after cortical contusion injury. J. Neurotrauma 2009, 26, 1271–1280. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury pos-sibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar]
- Kulbe, J.R.; Hill, R.L.; Singh, I.N.; Wang, J.A.; Hall, E.D. Synaptic Mitochondria Sustain More Damage than Non-Synaptic Mitochondria after Traumatic Brain Injury and Are Protected by Cyclosporine A. J. Neurotrauma 2017, 34, 1291–1301. [Google Scholar] [CrossRef]
- Readnower, R.D.; Pandya, J.D.; McEwen, M.L.; Pauly, J.R.; Springer, J.E.; Sullivan, P.G. Post-Injury Administration of the Mitochondrial Permeability Transition Pore Inhibitor, NIM811, Is Neuroprotective and Improves Cognition after Traumatic Brain Injury in Rats. J. Neurotrauma 2011, 28, 1845–1853. [Google Scholar] [CrossRef]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dys-function in Chronic and Acute Diseases. Cell Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [PubMed]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential com-ponent of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar]
- Nita, L.I.; Hershfinkel, M.; Sekler, I. Life after the birth of the mitochondrial Na+/Ca2+ exchanger, NCLX. Sci. China Life Sci. 2015, 58, 59–65. [Google Scholar]
- Yalamanchili, K.; Afzal, N.; Boyman, L.; Mannella, C.A.; Lederer, W.J.; Jafri, M.S. Under-standing the Dynamics of the Transient and Permanent Opening Events of the Mitochondrial Permeability Transition Pore with a Novel Stochastic Model. Membranes 2022, 12, 494. [Google Scholar]
- Kilbourne, M.; Kuehn, R.; Tosun, C.; Caridi, J.; Keledjian, K.; Bochicchio, G.; Scalea, T.; Gerzanich, V.; Simard, J.M. Novel Model of Frontal Impact Closed Head Injury in the Rat. J. Neurotrauma 2009, 26, 2233–2243. [Google Scholar] [CrossRef]
- Mira, R.G.; Lira, M.; Quintanilla, R.A.; Cerpa, W. Alcohol consumption during adolescence alters the hippocampal response to traumatic brain injury. Biochem. Biophys. Res. Commun. 2020, 528, 514–519. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Cerpa, W. Regulation of Phosphorylated State of NMDA Receptor by STEP61 Phosphatase after Mild-Traumatic Brain Injury: Role of Oxidative Stress. Antioxidants 2021, 10, 1575. [Google Scholar] [CrossRef]
- Lira, M.; Zamorano, P.; Cerpa, W. Exo70 intracellular redistribution after repeated mild traumatic brain injury. Biol. Res. 2021, 54, 5. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Zhang, S.F.; Thomas, B.; Ten, V.; Starkov, A.A. Isolation and Functional Assessment of Mitochondria from Small Amounts of Mouse Brain Tissue. Neurodegener. Methods Protoc. 2011, 793, 311–324. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Lampl, T.; Crum, J.A.; Davis, T.A.; Milligan, C.; Del Gaizo Moore, V. Isolation and functional analysis of mitochondria from cultured cells and mouse tissue. J. Vis. Exp. 2015, 23, e52076. [Google Scholar]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mito-chondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.-X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef]
- Lai, J.C.K.; Walsh, J.M.; Dennis, S.C.; Clark, J.B. Synaptic and non-synaptic mitochondria from rat brain: Isolation and characterization. J. Neurochem. 1977, 28, 625–631. [Google Scholar] [CrossRef]
- Rudolf, R.; Mongillo, M.; Magalhaes, P.J.; Pozzan, T. In vivo monitoring of Ca(2+) uptake into mi-tochondria of mouse skeletal muscle during contraction. J. Cell Biol. 2004, 166, 527–536. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpen-Ter, A.C.; Kolmetzky, D.; Gao, E.; Van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Dröse, S.; Brandt, U.; Schulz, K.; Jendrach, M.; Leuner, K.; Eckert, A.; Müller, W. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Wu, T.; Shao, Y.; Luo, L.; Zhou, L.; Ou, S.; Tang, H.; Huang, W.; Guo, K.; et al. Studies on APP metabolism related to age-associated mitochondrial dysfunction in APP/PS1 transgenic mice. Aging 2019, 11, 10242–10251. [Google Scholar] [CrossRef]
- Stavsky, A.; Stoler, O.; Kostic, M.; Katoshevsky, T.; Assali, E.A.; Savic, I.; Amitai, Y.; Prokisch, H.; Leiz, S.; Daumer-HAAS, C.; et al. Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 2021, 4, 666. [Google Scholar] [CrossRef]
- Di Pietro, V.; Lazzarino, G.; Amorini, A.M.; Signoretti, S.; Hill, L.J.; Porto, E.; Tavazzi, B.; Lazzarino, G.; Belli, A. Fusion or Fission: The Destiny of Mitochondria in Traumatic Brain Injury of Different Severities. Sci. Rep. 2017, 7, 9189. [Google Scholar] [CrossRef]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Wojtyniak, P.; Boratynska-Jasinska, A.; Serwach, K.; Gruszczynska-Biegala, J.; Zablocka, B.; Jaworski, J.; Kawalec, M. Mitofusin 2 Integrates Mitochondrial Network Remodelling, Mitophagy and Re-newal of Respiratory Chain Proteins in Neurons after Oxygen and Glucose Deprivation. Mol. Neurobiol. 2022, 59, 6502–8618. [Google Scholar] [CrossRef]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacin, M.; Zorzano, A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mi-tochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Kostic, M.; Katoshevski, T.; Sekler, I. Allosteric Regulation of NCLX by Mitochondrial Membrane Potential Links the Metabolic State and Ca2+ Signaling in Mitochondria. Cell Rep. 2018, 25, 3465–3475.e4. [Google Scholar] [CrossRef]
- Kostic, M.; Ludtmann, M.H.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef]
- Atkins, C.M.; Oliva, A.A.; Alonso, O.F.; Pearse, D.D.; Bramlett, H.M.; Dietrich, W.D. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 2007, 208, 145–158. [Google Scholar] [CrossRef]
- Hagenston, A.M.; Yan, J.; Bas-Orth, C.; Tan, Y.; Sekler, I.; Bading, H. Disrupted expression of mi-tochondrial NCLX sensitizes neuroglial networks to excitotoxic stimuli and renders synaptic activity toxic. J. Biol. Chem. 2022, 298, 101508. [Google Scholar] [CrossRef]
- Parnis, J.; Montana, V.; Delgado-Martinez, I.; Matyash, V.; Parpura, V.; Kettenmann, H.; Sekler, I.; Nolte, C. Mitochondrial Exchanger NCLX Plays a Major Role in the Intracellular Ca2+ Signaling, Gliotransmission, and Proliferation of Astrocytes. J. Neurosci. 2013, 33, 7206–7219. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2018, 26, 179–195. [Google Scholar] [CrossRef]
- Sun, J.; Jacobs, K.M. Knockout of Cyclophilin-D Provides Partial Amelioration of Intrinsic and Synaptic Prop-erties Altered by Mild Traumatic Brain Injury. Front. Syst. Neurosci. 2016, 10, 63. [Google Scholar] [CrossRef]
- Duan, C.; Kuang, L.; Hong, C.; Xiang, X.; Liu, J.; Li, Q.; Peng, X.; Zhou, Y.; Wang, H.; Liu, L.; et al. Mitochondrial Drp1 recognizes and induces excessive mPTP opening after hypoxia through BAX-PiC and LRRK2-HK2. Cell Death Dis. 2021, 12, 1050. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mira, R.G.; Quintanilla, R.A.; Cerpa, W. Mild Traumatic Brain Injury Induces Mitochondrial Calcium Overload and Triggers the Upregulation of NCLX in the Hippocampus. Antioxidants 2023, 12, 403. https://doi.org/10.3390/antiox12020403
Mira RG, Quintanilla RA, Cerpa W. Mild Traumatic Brain Injury Induces Mitochondrial Calcium Overload and Triggers the Upregulation of NCLX in the Hippocampus. Antioxidants. 2023; 12(2):403. https://doi.org/10.3390/antiox12020403
Chicago/Turabian StyleMira, Rodrigo G., Rodrigo A. Quintanilla, and Waldo Cerpa. 2023. "Mild Traumatic Brain Injury Induces Mitochondrial Calcium Overload and Triggers the Upregulation of NCLX in the Hippocampus" Antioxidants 12, no. 2: 403. https://doi.org/10.3390/antiox12020403
APA StyleMira, R. G., Quintanilla, R. A., & Cerpa, W. (2023). Mild Traumatic Brain Injury Induces Mitochondrial Calcium Overload and Triggers the Upregulation of NCLX in the Hippocampus. Antioxidants, 12(2), 403. https://doi.org/10.3390/antiox12020403