
Di-(2-ethylhexyl) Phthalate Limits the Lipid-Lowering Effects of Simvastatin by Promoting Protein Degradation of Low-Density Lipoprotein Receptor: Role of PPARγ-PCSK9 and LXRα-IDOL Signaling Pathways


 , , and
, , and 
Abstract
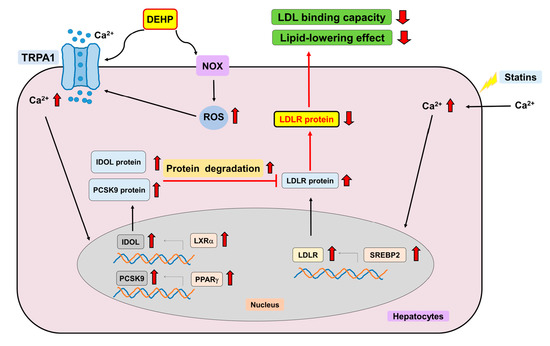
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. LDL Binding Assay
2.4. Cholesterol Measurement
2.5. Detection of Ca2+ Influx
2.6. Western Blot Analysis
2.7. Transfection of Small Interfering RNAs (siRNAs)
2.8. Measurement of Intracellular ROS Levels
2.9. Determination of NADP+/NADPH Ratio
2.10. Determination of PPARγ and LXRα Activity
2.11. Statistical Analysis
3. Results
3.1. DEHP and Its Metabolites Limit the Lipid-Lowering Effect of Simvastatin in Huh7 Cells
3.2. PPARγ-PCSK9 and LXRα-IDOL Cascades Are Essential for DEHP Inhibiting the Simvastatin-Conferred Lipid-Lowering Effects in Huh7 Cells
3.3. DEHP Inhibits the Simvastatin-Elicited Lipid-Lowering Effect by Activating the NOX-ROS Signaling Pathway
3.4. DEHP Induces the Activation of the NOX-ROS-TRPA1-Ca2+ Signaling Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tickner, J.A.; Schettler, T.; Guidotti, T.; McCally, M.; Rossi, M. Health risks posed by use of Di-2-ethylhexyl phthalate (DEHP) in PVC medical devices: A critical review. Am. J. Ind. Med. 2001, 39, 100–111. [Google Scholar] [CrossRef]
- Van Vliet, E.D.; Reitano, E.M.; Chhabra, J.S.; Bergen, G.P.; Whyatt, R.M. A review of alternatives to di (2-ethylhexyl) phthalate-containing medical devices in the neonatal intensive care unit. J. Perinatol. 2011, 31, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Rowdhwal, S.S.S.; Chen, J. Toxic effects of di-2-ethylhexyl phthalate: An overview. Biomed. Res. Int. 2018, 2018, 1750368. [Google Scholar] [CrossRef]
- Hab, S.A.; Naz, F.; Baig, J.A.; Afridi, H.I.; Surhio, M.A.; Talpur, M.K. Leaching and exposure of phthalates from medical devices; health impacts and regulations. Environ. Contam. Rev. (ECR) 2018, 1, 13–21. [Google Scholar] [CrossRef]
- Posnack, N.G. The adverse cardiac effects of Di(2-ethylhexyl) phthalate and Bisphenol A. Cardiovasc. Toxicol. 2014, 14, 339–357. [Google Scholar] [CrossRef]
- Zhao, J.F.; Hsiao, S.H.; Hsu, M.H.; Pao, K.C.; Kou, Y.R.; Shyue, S.K.; Lee, T.S. Di-(2-ethylhexyl) phthalate accelerates atherosclerosis in apolipoprotein E-deficient mice. Arch. Toxicol. 2016, 90, 181–190. [Google Scholar] [CrossRef]
- Guo, B.C.; Kuo, K.L.; Chen, C.H.; Chen, S.L.; Tsou, T.C.; Lee, T.S. Di-(2-ethylhexyl) phthalate limits the pleiotropic effects of statins in chronic kidney disease patients undergoing dialysis and endothelial cells. Environ. Pollut. 2020, 267, 115548. [Google Scholar] [CrossRef]
- Mariana, M.; Cairrao, E. Phthalates implications in the cardiovascular system. J. Cardiovasc. Dev. Dis. 2020, 7, 26. [Google Scholar] [CrossRef]
- Nässberger, L.; Arbin, A.; Ostelius, J. Exposure of patients to phthalates from polyvinyl chloride tubes and bags during dialysis. Nephron. 1987, 45, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Mettang, T.; Thomas, S.; Kiefer, T.; Fischer, F.P.; Kuhlmann, U.; Wodarz, R.; Rettenmeier, A.W. Uraemic pruritus and exposure to di(2-ethylhexyl) phthalate (DEHP) in haemodialysis patients. Nephrol. Dial. Transplant. 1996, 11, 2439–2443. [Google Scholar] [CrossRef]
- Faouzi, M.A.; Dine, T.; Gressier, B.; Kambia, K.; Luyckx, M.; Pagniez, D.; Brunet, C.; Cazin, M.; Belabed, A.; Cazin, J.C. Exposure of hemodialysis patients to di-2-ethylhexyl phthalate. Int. J. Pharm. 1999, 180, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E.S.; Deisenhofer, J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science. 2001, 292, 1160–1164. [Google Scholar] [CrossRef]
- Yokoyama, M.; Seo, T.; Park, T.; Yokoyama, M.; Seo, T.; Park, T.; Yagyu, H.; Hu, Y.; Son, N.H.; Augustus, A.S.; et al. Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J. Lipid. Res. 2007, 48, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Furuya, Y.; Sekine, Y.; Kato, H.; Miyazawa, Y.; Koike, H.; Suzuki, K. Low-density lipoprotein receptors play an important role in the inhibition of prostate cancer cell proliferation by statins. Prostate Int. 2016, 4, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Chen, Y.; Xu, C.B. Statins and New-onset diabetes mellitus: LDL receptor may provide a key link. Front. Pharmacol. 2017, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Statins and cardiovascular diseases: From cholesterol lowering to pleiotropy. Curr. Pharm. Des. 2009, 15, 467–478. [Google Scholar] [CrossRef]
- Nelson, R.H. Hyperlipidemia as a risk factor for cardiovascular disease. Prim. Care 2013, 40, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Lefer, D.J. Statins as potent antiinflammatory drugs. Circulation 2002, 106, 2041–2042. [Google Scholar] [CrossRef]
- Davignon, J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 2004, 109, III39–III43. [Google Scholar] [CrossRef]
- Jain, M.K.; Ridker, P.M. Anti-inflammatory effects of statins: Clinical evidence and basic mechanisms. Nat. Rev. Drug Discov. 2005, 4, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic effects of statins on the cardiovascular system. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory action of statins in cardiovascular disease: The role of inflammasome and toll-like receptor pathways. Clin. Rev. Allergy Immunol. 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed]
- Drüeke, T.B.; Massy, Z.A. Atherosclerosis in CKD: Differences from the general population. Nat. Rev. Nephrol. 2010, 6, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in chronic kidney disease: More, less, or just different? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef] [PubMed]
- Deedwania, P.C. Statins in chronic kidney disease: Cardiovascular risk and kidney function. Postgrad. Med. 2014, 126, 29–36. [Google Scholar] [CrossRef]
- Obialo, C.I.; Ofili, E.O.; Norris, K.C. Statins and cardiovascular disease outcomes in chronic kidney disease: Reaffirmation vs. repudiation. Int. J. Environ. Res. Public. Health 2018, 15, 2733. [Google Scholar] [CrossRef]
- Hwang, S.D.; Kim, K.; Kim, Y.J.; Lee, S.W.; Lee, J.H.; Song, J.H. Effect of statins on cardiovascular complications in chronic kidney disease patients: A network meta-analysis. Medicine 2020, 99, e20061. [Google Scholar] [CrossRef]
- Fellström, B.C.; Jardine, A.G.; Schmieder, R.E.; Holdaas, H.; Bannister, K.; Beutler, J.; Chae, D.W.; Chevaile, A.; Cobbe, S.M.; Grönhagen-Riska, C.; et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N. Engl. J. Med. 2009, 360, 1395–1407. [Google Scholar] [CrossRef]
- Constantine, E.K.; Eddy, D.; Evdokia, S.; Rosmery, M.; Peter, D.M.; Eliscer, G. Effects of statins on cardiovascular outcomes in patients with chronic kidney disease. Clin. Med. Insights Ther. 2017, 9, 1–5. [Google Scholar]
- Meents, J.E.; Ciotu, C.I.; Fischer, M.J.M. TRPA1: A molecular view. J. Neurophysiol. 2019, 121, 427–443. [Google Scholar] [CrossRef]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian transient receptor potential TRPA1 channels: From structure to disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Mori, Y.; Takahashi, N.; Polat, O.K.; Kurokawa, T.; Takeda, N.; Inoue, M. Redox-sensitive transient receptor potential channels in oxygen sensing and adaptation. Pflugers Arch. 2016, 468, 85–97. [Google Scholar] [CrossRef]
- Arenas, O.M.; Zaharieva, E.E.; Para, A.; Vásquez-Doorman, C.; Petersen, C.P.; Gallio, M. Activation of planarian TRPA1 by reactive oxygen species reveals a conserved mechanism for animal nociception. Nat. Neurosci. 2017, 20, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, S.R.; Chen, H.; Pan, H.L. Endogenous transient receptor potential ankyrin 1 and vanilloid 1 activity potentiates glutamatergic input to spinal lamina I neurons in inflammatory pain. J. Neurochem. 2019, 149, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Souza Monteiro de Araujo, D.; Nassini, R.; Geppetti, P.; De Logu, F. TRPA1 as a therapeutic target for nociceptive pain. Expert. Opin. Ther. Targets. 2020, 24, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.F.; Shyue, S.K.; Kou, Y.R.; Lu, T.M.; Lee, T.S. Transient receptor potential Ankyrin 1 channel involved in atherosclerosis and macrophage-foam cell formation. Int. J. Biol. Sci. 2016, 12, 812–823. [Google Scholar] [CrossRef]
- Tian, C.; Han, X.; He, L.; Tang, F.; Huang, R.; Lin, Z.; Li, S.; Deng, S.; Xu, J.; Huang, H.; et al. Transient receptor potential ankyrin 1 contributes to the ATP-elicited oxidative stress and inflammation in THP-1-derived macrophage. Mol. Cell Biochem. 2020, 473, 179–192. [Google Scholar] [CrossRef]
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am. J. Kidney Dis. 1998, 32, S112–S119. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Del Vecchio, L.; Manzoni, C. Morbidity and mortality on maintenance haemodialysis. Contrib. Nephrol. 1998, 124, 166–201. [Google Scholar]
- Parfrey, P.S.; Foley, R.N. The clinical epidemiology of cardiac disease in chronic renal failure. J. Am. Soc. Nephrol. 1999, 10, 1606–1615. [Google Scholar] [CrossRef]
- Merrill, J.P. Dialysis versus transplantation in the treatment of end-stage renal disease. Annu. Rev. Med. 1978, 29, 343–358. [Google Scholar] [CrossRef]
- Jha, V. Current status of end-stage renal disease care in India and Pakistan. Kidney Int. 2013, 3, 157–160. [Google Scholar] [CrossRef]
- Zazzeroni, L.; Pasquinelli, G.; Nanni, E.; Cremonini, V.; Rubbi, I. Comparison of quality of life in patients undergoing hemodialysis and peritoneal dialysis: A systematic review and meta-analysis. Kidney Blood Press Res. 2017, 42, 717–727. [Google Scholar] [CrossRef]
- Mettang, T.; Thomas, S.; Kiefer, T.; Fischer, F.P.; Kuhlmann, U.; Wodarz, R.; Rettenmeier, A.W. The fate of leached di(2-ethylhexyl) phthalate in patients undergoing CAPD treatment. Perit. Dial. Int. 1996, 16, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.J.; Luo, B.B.; Feng, Q.W.; Wang, Q.N. Effects of developmental exposure to DEHP on learning and memory of mice. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 2017, 35, 241–245. [Google Scholar] [PubMed]
- Wen, Z.J.; Wang, Z.Y.; Zhang, Y.F. Adverse cardiovascular effects and potential molecular mechanisms of DEHP and its metabolites-a review. Sci. Total Environ. 2022, 847, 157443. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Almeida, S.O.; Budoff. M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc. Med. 2019, 29, 451–455. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Pansini, F.; Perkovic, V.; Manno, C.; Pellegrini, F.; Johnson, D.W.; Craig, J.C.; Strippoli, G.F. HMG CoA reductase inhibitors (statins) for people with chronic kidney disease not requiring dialysis. Cochrane Database Syst. Rev. 2009, 2, CD007784. [Google Scholar]
- Koren, M.J.; Davidson, M.H.; Wilson, D.J.; Fayyad, R.S.; Zuckerman, A.; Reed, D.P.; ALLIANCE Investigators. Focused atorvastatin therapy in managed-care patients with coronary heart disease and CKD. Am. J. Kidney Dis. 2009, 53, 741–750. [Google Scholar] [CrossRef]
- Shurraw, S.; Tonelli, M. Statins for treatment of dyslipidemia in chronic kidney disease. Perit. Dial. Int. 2006, 26, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Krane, V.; März, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E.; German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Craig, J.C.; Navaneethan, S.D.; Tonelli, M.; Pellegrini, F.; Strippoli, G.F. Benefits and harms of statin therapy for persons with chronic kidney disease: A systematic review and meta-analysis. Ann. Intern. Med. 2012, 157, 263–275. [Google Scholar] [CrossRef]
- Sorrentino, V.; Zelcer, N. Post-transcriptional regulation of lipoprotein receptors by the E3-ubiquitin ligase inducible degrader of the low-density lipoprotein receptor. Curr. Opin. Lipidol. 2012, 23, 213–219. [Google Scholar] [CrossRef] [PubMed]
- van Loon, N.M.; van Wouw, S.A.E.; Ottenhoff, R.; Nelson, J.K.; Kingma, J.; Scheij, S.; Moeton, M.; Zelcer, N. Regulation of intestinal LDLR by the LXR-IDOL axis. Atherosclerosis 2020, 315, 1–9. [Google Scholar] [CrossRef]
- Zelcer, N.; Hong, C.; Boyadjian, R.; Tontonoz, P. LXR regulates cholesterol uptake through idol-dependent ubiquitination of the LDL receptor. Science 2009, 325, 100–104. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, Y.; Hu, W.; Li, X.; Yang, X.; Zhou, X.; Yin, Z.; Kong, D.; Yao, Z.; Hajjar, D.P.; et al. Peroxisome proliferator-activated receptor γ activation by ligands and dephosphorylation induces proprotein convertase subtilisin kexin type 9 and low-density lipoprotein receptor expression. J. Biol. Chem. 2012, 287, 23667–23677. [Google Scholar] [CrossRef]
- Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, reactive oxygen species and regulation of vascular cell fate. Antioxidants 2017, 6, 90. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, C.; Ye, Y.; Zeng, J.; Zhu, J.; Li, Y.; Wang, W.; Zhang, W.; Chen, Y.; Xie, H.; et al. The increase of ROS caused by the interference of DEHP with JNK/p38/p53 pathway as the reason for hepatotoxicity. Int. J. Environ. Res. Public Health 2019, 16, 356. [Google Scholar] [CrossRef]
- Tripathi, A.; Pandey, V.; Sahu, A.N.; Singh, A.; Dubey, P.K. Di-(2-ethylhexyl) phthalate (DEHP) inhibits steroidogenesis and induces mitochondria-ROS mediated apoptosis in rat ovarian granulosa cells. Toxicol. Res. 2019, 8, 81–394. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, L.; Sun, X.; Li, J.; Wang, N.; Liu, X.; Yao, X.; Zhang, C.; Deng, H.; Wang, S.; et al. DEHP induces ferroptosis in testes via p38α-lipid ROS circulation and destroys the BTB integrity. Food Chem. Toxicol. 2022, 164, 113046. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Kurokawa, T.; Mori, Y. Sensing of redox status by TRP channels. Cell Calcium. 2016, 60, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Takahashi, N.; Kurokawa, T.; Kiyonaka, S. TRP channels in oxygen physiology: Distinctive functional properties and roles of TRPA1 in O2 sensing. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 464–482. [Google Scholar] [CrossRef]
- Kuczera, P.; Więcek, A.; Adamczak, M. Impaired fertility in women and men with chronic kidney disease. Adv. Clin. Exp. Med. 2022, 31, 187–195. [Google Scholar] [CrossRef]
- Edey, M.M. Male Sexual dysfunction and chronic kidney disease. Front. Med. 2017, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- Romejko, K.; Rymarz, A.; Sadownik, H.; Niemczyk, S. Testosterone deficiency as one of the major endocrine disorders in chronic kidney disease. Nutrients 2022, 14, 3438. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, P.; Schmidt, R.J. Sexual function in chronic kidney disease. Adv. Chronic. Kidney Dis. 2007, 14, 119–125. [Google Scholar] [CrossRef]
- Beverly, B.E.; Lambright, C.S.; Furr, J.R.; Sampson, H.; Wilson, V.S.; McIntyre, B.S.; Foster, P.M.; Travlos, G.; Gray, L.E., Jr. Simvastatin and dipentyl phthalate lower ex vivo testicular testosterone production and exhibit additive effects on testicular testosterone and gene expression via distinct mechanistic pathways in the fetal rat. Toxicol. Sci. 2014, 141, 524–537. [Google Scholar] [CrossRef]
- Sundararaj, S.C.; Thomas, M.V.; Peyyala, R.; Dziubla, T.D.; Puleo, D.A. Design of a multiple drug delivery system directed at periodontitis. Biomaterials 2013, 34, 8835–8842. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, B.-C.; Kuo, K.-L.; Huang, J.-W.; Chen, C.-H.; Tarng, D.-C.; Lee, T.-S. Di-(2-ethylhexyl) Phthalate Limits the Lipid-Lowering Effects of Simvastatin by Promoting Protein Degradation of Low-Density Lipoprotein Receptor: Role of PPARγ-PCSK9 and LXRα-IDOL Signaling Pathways. Antioxidants 2023, 12, 477. https://doi.org/10.3390/antiox12020477
Guo B-C, Kuo K-L, Huang J-W, Chen C-H, Tarng D-C, Lee T-S. Di-(2-ethylhexyl) Phthalate Limits the Lipid-Lowering Effects of Simvastatin by Promoting Protein Degradation of Low-Density Lipoprotein Receptor: Role of PPARγ-PCSK9 and LXRα-IDOL Signaling Pathways. Antioxidants. 2023; 12(2):477. https://doi.org/10.3390/antiox12020477
Chicago/Turabian StyleGuo, Bei-Chia, Ko-Lin Kuo, Jenq-Wen Huang, Chia-Hui Chen, Der-Cherng Tarng, and Tzong-Shyuan Lee. 2023. "Di-(2-ethylhexyl) Phthalate Limits the Lipid-Lowering Effects of Simvastatin by Promoting Protein Degradation of Low-Density Lipoprotein Receptor: Role of PPARγ-PCSK9 and LXRα-IDOL Signaling Pathways" Antioxidants 12, no. 2: 477. https://doi.org/10.3390/antiox12020477
APA StyleGuo, B.-C., Kuo, K.-L., Huang, J.-W., Chen, C.-H., Tarng, D.-C., & Lee, T.-S. (2023). Di-(2-ethylhexyl) Phthalate Limits the Lipid-Lowering Effects of Simvastatin by Promoting Protein Degradation of Low-Density Lipoprotein Receptor: Role of PPARγ-PCSK9 and LXRα-IDOL Signaling Pathways. Antioxidants, 12(2), 477. https://doi.org/10.3390/antiox12020477