
Mitochondrial Open Reading Frame of the 12S rRNA Type-c: Potential Therapeutic Candidate in Retinal Diseases
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
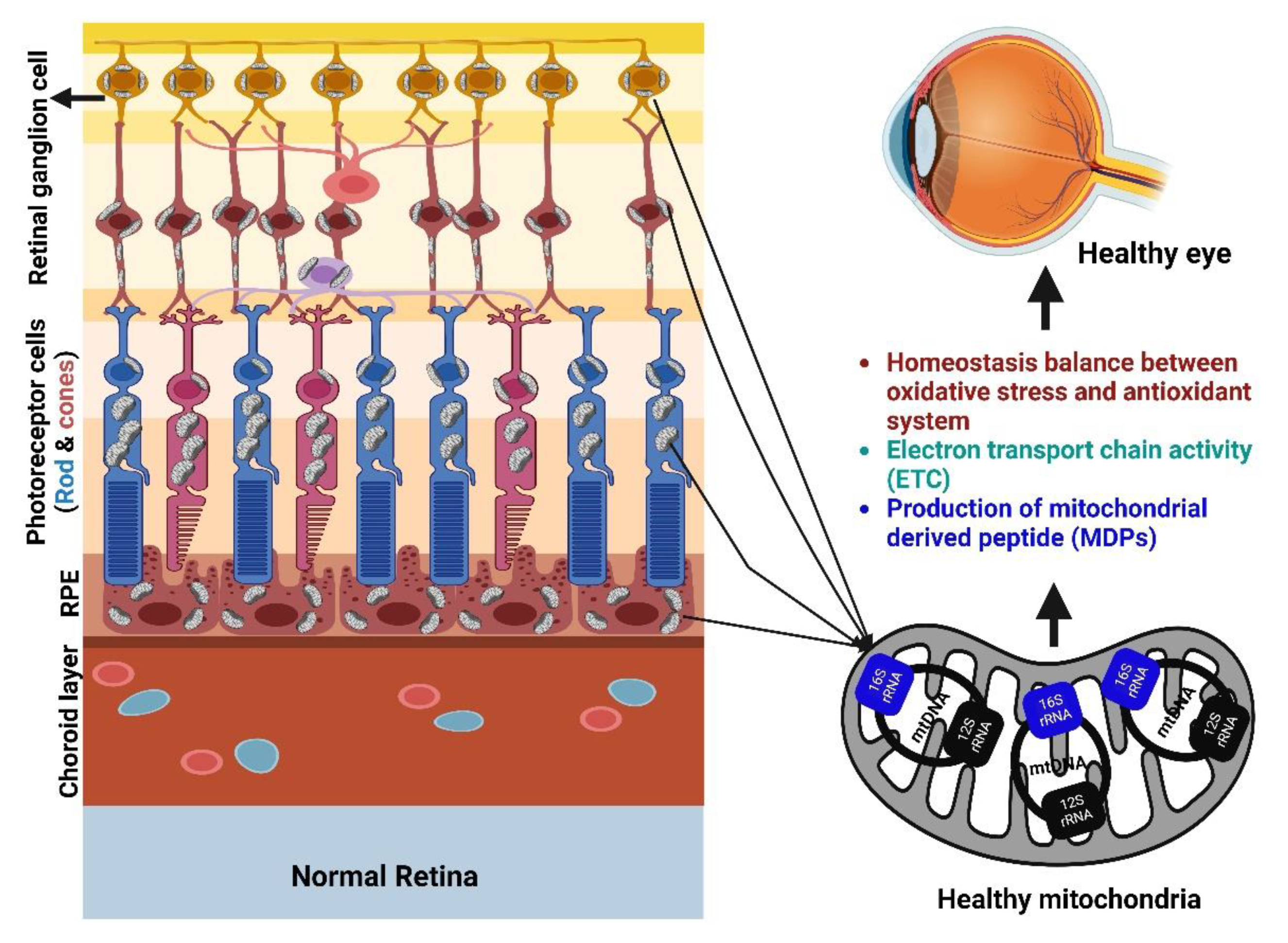
2. Role of Mitochondria in the Retina
3. MOTS-c and Its Role in Mitochondrial Biogenesis and Mitophagy
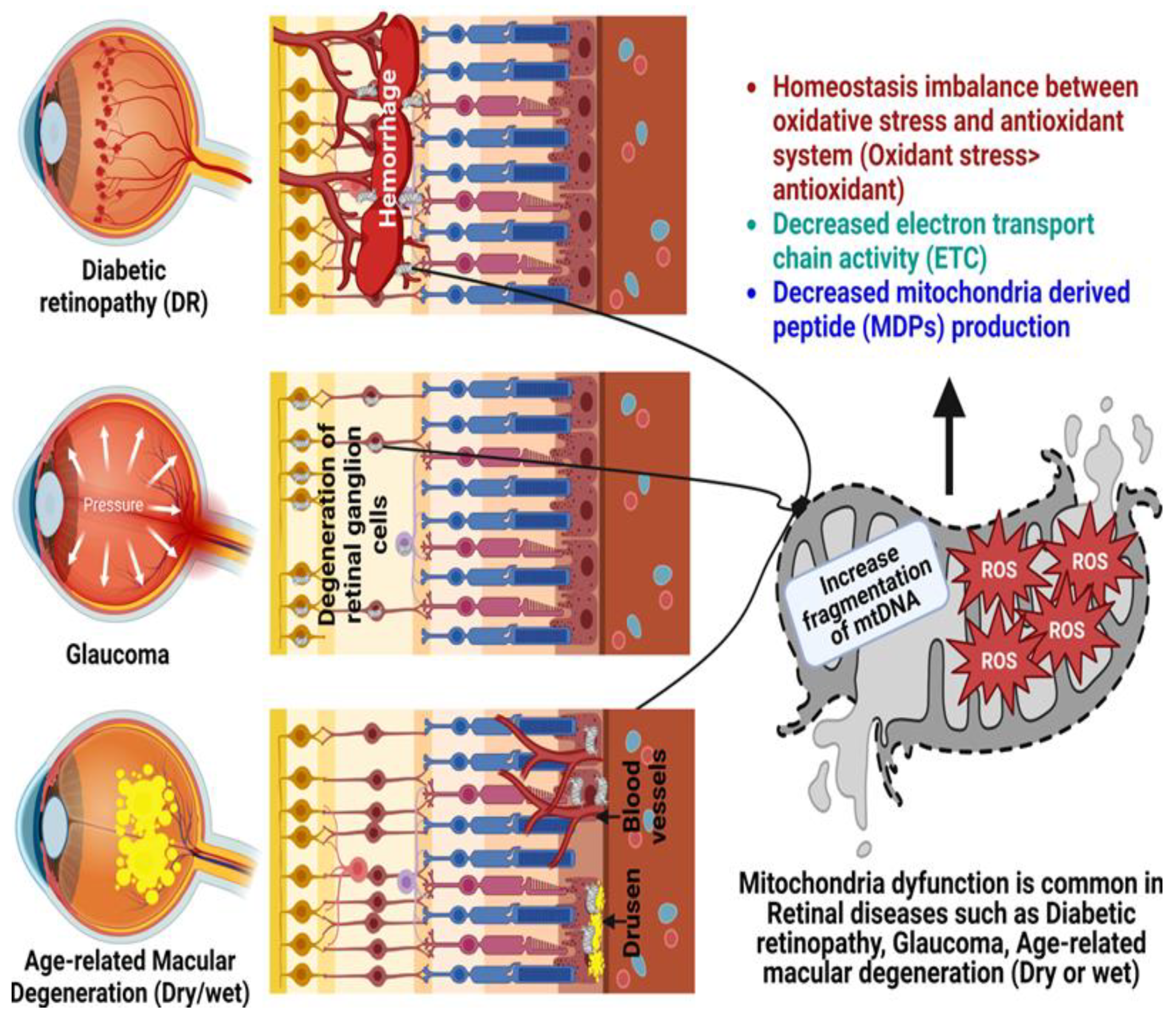
4. Age-Related Retinal Diseases
4.1. Glaucoma
4.2. Diabetic Retinopathy
4.3. Age-Related Macular Degeneration
5. Future Directions
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Miller, B.; Kumagai, H.; Silverstein, A.R.; Flores, M.; Yen, K. Mitochondrial-derived peptides in aging and age-related diseases. Geroscience 2021, 43, 1113–1121. [Google Scholar] [CrossRef]
- Mohtashami, Z.; Singh, M.K.; Salimiaghdam, N.; Ozgul, M.; Kenney, M.C. MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 1991. [Google Scholar] [CrossRef]
- Sreekumar, P.G.; Kannan, R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox Biol. 2020, 37, 101663. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, L.; Zhuang, Z.; Hu, X.; Dong, D. Mitochondrial-Derived Peptides in Diabetes and Its Complications. Front. Endocrinol. 2021, 12, 808120. [Google Scholar] [CrossRef]
- Nashine, S.; Kenney, M.C. Effects of Mitochondrial-Derived Peptides (MDPs) on Mitochondrial and Cellular Health in AMD. Cells 2020, 9, 1102. [Google Scholar] [CrossRef]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Carrella, S.; Massa, F.; Indrieri, A. The Role of MicroRNAs in Mitochondria-Mediated Eye Diseases. Front. Cell Dev. Biol. 2021, 9, 653522. [Google Scholar] [CrossRef]
- Schrier, S.A.; Falk, M.J. Mitochondrial disorders and the eye. Curr. Opin. Ophthalmol. 2011, 22, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Bourne, R.; Steinmetz, J.D.; Flaxman, S.; Briant, P.S.; Taylor, H.R.; Resnikoff, S.; Tareque, M.I. Trends in prevalence of blindness and distance and near vision impairment over 30 years: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e130–e143. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef] [Green Version]
- Ames, A., 3rd; Li, Y.Y.; Heher, E.C.; Kimble, C.R. Energy metabolism of rabbit retina as related to function: High cost of Na+ transport. J. Neurosci. 1992, 12, 840–853. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Medrano, C.J.; Fox, D.A. Oxygen consumption in the rat outer and inner retina: Light- and pharmacologically-induced inhibition. Exp. Eye Res. 1995, 61, 273–284. [Google Scholar] [CrossRef]
- Sung, C.H.; Chuang, J.Z. The cell biology of vision. J. Cell Biol. 2010, 190, 953–963. [Google Scholar] [CrossRef] [Green Version]
- Eells, J.T. Mitochondrial Dysfunction in the Aging Retina. Biology 2019, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [Green Version]
- Kooragayala, K.; Gotoh, N.; Cogliati, T.; Nellissery, J.; Kaden, T.R.; French, S.; Balaban, R.; Li, W.; Covian, R.; Swaroop, A. Quantification of Oxygen Consumption in Retina Ex Vivo Demonstrates Limited Reserve Capacity of Photoreceptor Mitochondria. Investig. Ophthalmol. Vis. Sci. 2015, 56, 8428–8436. [Google Scholar] [CrossRef] [Green Version]
- Al-Enezi, M.; Al-Saleh, H.; Nasser, M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 2008, 15, 81–86. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Geng, Z.; Khattak, S.; Ji, X.; Wu, D.; Dang, Y. Role of Oxidative Stress in Retinal Disease and the Early Intervention Strategies: A Review. Oxid. Med. Cell. Longev. 2022, 2022, 7836828. [Google Scholar] [CrossRef]
- Prithivirajsingh, S.; Story, M.D.; Bergh, S.A.; Geara, F.B.; Ang, K.K.; Ismail, S.M.; Stevens, C.W.; Buchholz, T.A.; Brock, W.A. Accumulation of the common mitochondrial DNA deletion induced by ionizing radiation. FEBS Lett. 2004, 571, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longev. 2017, 2017, 9208489. [Google Scholar] [CrossRef] [Green Version]
- Monsalve, M.; Borniquel, S.; Valle, I.; Lamas, S. Mitochondrial dysfunction in human pathologies. Front. Biosci. 2007, 12, 1131–1153. [Google Scholar] [CrossRef] [Green Version]
- Barak, A.; Morse, L.S.; Goldkorn, T. Ceramide: A potential mediator of apoptosis in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 247–254. [Google Scholar]
- Merry, T.L.; Chan, A.; Woodhead, J.S.T.; Reynolds, J.C.; Kumagai, H.; Kim, S.J.; Lee, C. Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 319, e659–e666. [Google Scholar] [CrossRef]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell. Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell. Metab. 2018, 28, 516–524.e7. [Google Scholar] [CrossRef] [Green Version]
- Lesnik, C.; Cohen, Y.; Atir-Lande, A.; Schuldiner, M.; Arava, Y. OM14 is a mitochondrial receptor for cytosolic ribosomes that supports co-translational import into mitochondria. Nat. Commun. 2014, 5, 5711. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef] [Green Version]
- Ibsen, K.H. The Crabtree effect: A review. Cancer Res. 1961, 21, 829–841. [Google Scholar]
- Lee, C. Nuclear transcriptional regulation by mitochondrial-encoded MOTS-c. Mol. Cell. Oncol. 2019, 6, 1549464. [Google Scholar] [CrossRef] [Green Version]
- Zarse, K.; Ristow, M. A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell. Metab. 2015, 21, 355–356. [Google Scholar] [CrossRef] [Green Version]
- Fuku, N.; Pareja-Galeano, H.; Zempo, H.; Alis, R.; Arai, Y.; Lucia, A.; Hirose, N. The mitochondrial-derived peptide MOTS-c: A player in exceptional longevity? Aging Cell 2015, 14, 921–923. [Google Scholar] [CrossRef]
- Sun, H.; Guo, X.; Wang, Z.; Wang, P.; Zhang, Z.; Dong, J.; Zhuang, R.; Zhou, Y.; Ma, G.; Cai, W. Alphalipoic Acid Prevents Oxidative Stress and Peripheral Neuropathy in Nab-Paclitaxel-Treated Rats through the Nrf2 Signalling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 3142732. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, A.R.; Larrick, J.W. Mitochondrial-Derived Peptides Exacerbate Senescence. Rejuvenation Res. 2018, 21, 369–373. [Google Scholar] [CrossRef]
- Zhang, H.; He, S.; Spee, C.; Ishikawa, K.; Hinton, D.R. SIRT1 mediated inhibition of VEGF/VEGFR2 signaling by Resveratrol and its relevance to choroidal neovascularization. Cytokine 2015, 76, 549–552. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Fulco, M.; Sartorelli, V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle 2008, 7, 3669–3679. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Res 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T. mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology 2018, 64, 127–134. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [Green Version]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Bao, X.L.; Cong, Y.Y.; Fan, B.; Li, G.Y. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Yang, B.; Yu, Q.; Chang, B.; Guo, Q.; Xu, S.; Yi, X.; Cao, S. MOTS-c interacts synergistically with exercise intervention to regulate PGC-1α expression, attenuate insulin resistance and enhance glucose metabolism in mice via AMPK signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166126. [Google Scholar] [CrossRef]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell. Biol. 2002, 158, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Luo, J.; Zhang, H. Role of Sirtuin 1 in the pathogenesis of ocular disease (Review). Int. J. Mol. Med. 2018, 42, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Xinqiang, Y.; Quan, C.; Yuanyuan, J.; Hanmei, X. Protective effect of MOTS-c on acute lung injury induced by lipopolysaccharide in mice. Int. Immunopharmacol. 2020, 80, 106174. [Google Scholar] [CrossRef]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef]
- Priest, C.; Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 2019, 1, 1177–1188. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Lai, R.W.; Woodhead, J.S.T.; Joly, J.H.; Mitchell, C.J.; Cameron-Smith, D.; Lu, R.; Cohen, P.; Graham, N.A.; Benayoun, B.A.; et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 2021, 12, 470. [Google Scholar] [CrossRef]
- Li, H.; Ren, K.; Jiang, T.; Zhao, G.J. MOTS-c attenuates endothelial dysfunction via suppressing the MAPK/NF-κB pathway. Int. J. Cardiol. 2018, 268, 40. [Google Scholar] [CrossRef]
- Slee, A.D. Exploring metabolic dysfunction in chronic kidney disease. Nutr. Metab. 2012, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Cavallotti, C.; Artico, M.; Pescosolido, N.; Leali, F.M.; Feher, J. Age-related changes in the human retina. Can. J. Ophthalmol. 2004, 39, 61–68. [Google Scholar] [CrossRef]
- Samuel, M.A.; Zhang, Y.; Meister, M.; Sanes, J.R. Age-related alterations in neurons of the mouse retina. J. Neurosci. 2011, 31, 16033–16044. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Sobczuk, P.; Pawlowska, E.; Kaarniranta, K. Interplay between aging and other factors of the pathogenesis of age-related macular degeneration. Ageing Res. Rev. 2022, 81, 101735. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Lewin, A.S.; Boulton, M.E. The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res. 2010, 44, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Yücel, Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007, 18, 110–114. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, A.C.; Liu, J. Neurodegeneration and Neuroprotection in Glaucoma. Yale J. Biol. Med. 2016, 89, 73–79. [Google Scholar]
- Agarwal, R.; Gupta, S.K.; Agarwal, P.; Saxena, R.; Agrawal, S.S. Current concepts in the pathophysiology of glaucoma. Indian J. Ophthalmol. 2009, 57, 257–266. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A. Oxidative stress and glaucoma: Injury in the anterior segment of the eye. Prog. Brain Res. 2008, 173, 385–407. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef] [Green Version]
- Lo Faro, V.; Nolte, I.M.; Ten Brink, J.B.; Snieder, H.; Jansonius, N.M.; Bergen, A.A. Mitochondrial Genome Study Identifies Association Between Primary Open-Angle Glaucoma and Variants in MT-CYB, MT-ND4 Genes and Haplogroups. Front. Genet. 2021, 12, 781189. [Google Scholar] [CrossRef]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Aslan, M.; Dogan, S.; Kucuksayan, E. Oxidative stress and potential applications of free radical scavengers in glaucoma. Redox Rep. 2013, 18, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Adornetto, A.; Rombolà, L.; Morrone, L.A.; Nucci, C.; Corasaniti, M.T.; Bagetta, G.; Russo, R. Natural Products: Evidence for Neuroprotection to Be Exploited in Glaucoma. Nutrients 2020, 12, 3158. [Google Scholar] [CrossRef]
- Oikawa, K.; Ver Hoeve, J.N.; Teixeira, L.B.C.; Snyder, K.C.; Kiland, J.A.; Ellinwood, N.M.; McLellan, G.J. Sub-region-Specific Optic Nerve Head Glial Activation in Glaucoma. Mol. Neurobiol. 2020, 57, 2620–2638. [Google Scholar] [CrossRef]
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Dégardin, J.; Godefroy, D.; Rostène, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J. Neuroinflamm. 2016, 13, 44. [Google Scholar] [CrossRef] [Green Version]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration. Exp. Neurol. 2020, 328, 113247. [Google Scholar] [CrossRef]
- Guo, X.; Dason, E.S.; Zanon-Moreno, V.; Jiang, Q.; Nahirnyj, A.; Chan, D.; Flanagan, J.G.; Sivak, J.M. PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am. J. Pathol. 2014, 184, 1017–1029. [Google Scholar] [CrossRef]
- Zuo, L.; Khan, R.S.; Lee, V.; Dine, K.; Wu, W.; Shindler, K.S. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5097–5102. [Google Scholar] [CrossRef]
- Balaiya, S.; Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Sirtuins Expression and Their Role in Retinal Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 3187594. [Google Scholar] [CrossRef] [Green Version]
- Balaiya, S.; Khetpal, V.; Chalam, K.V. Hypoxia initiates sirtuin1-mediated vascular endothelial growth factor activation in choroidal endothelial cells through hypoxia inducible factor-2α. Mol. Vis. 2012, 18, 114–120. [Google Scholar]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Sykiotis, G.P. Keap1/Nrf2 Signaling Pathway. Antioxidants 2021, 10, 828. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, S.; Pan, Y.; Jin, M.; Li, J.; Luo, Y.; Sun, X.; Li, G. Diabetic retinopathy: Involved cells, biomarkers, and treatments. Front. Pharmacol. 2022, 13, 953691. [Google Scholar] [CrossRef]
- Nian, S.; Lo, A.C.Y.; Mi, Y.; Ren, K.; Yang, D. Neurovascular unit in diabetic retinopathy: Pathophysiological roles and potential therapeutical targets. Eye Vis. 2021, 8, 15. [Google Scholar] [CrossRef]
- Kenney, M.; Falatoonzadeh, P.; Atilano, S.; Chwa, M.; Caceres-del-Carpio, J.; Malik, D.; Kuppermann, B. African-origin mitochondrial DNA variants as a contributing factor to susceptibilities for diabetes and age-related diseases. Int. J. Diabetes Clin. Res. 2016, 3, 53. [Google Scholar]
- Dolinko, A.H.; Chwa, M.; Atilano, S.R.; Kenney, M.C. African and Asian Mitochondrial DNA Haplogroups Confer Resistance Against Diabetic Stresses on Retinal Pigment Epithelial Cybrid Cells In Vitro. Mol. Neurobiol. 2020, 57, 1636–1655. [Google Scholar] [CrossRef]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef] [Green Version]
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy—Ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489–499. [Google Scholar] [CrossRef]
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef]
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants 2020, 9, 905. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, K.; Molina, A.J.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Romeo, G.; Liu, W.H.; Asnaghi, V.; Kern, T.S.; Lorenzi, M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 2002, 51, 2241–2248. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol. 2005, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Kanwar, M.; Chan, P.S.; Kern, T.S.; Kowluru, R.A. Oxidative damage in the retinal mitochondria of diabetic mice: Possible protection by superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Miller, B.; Mehta, H.H.; Xiao, J.; Wan, J.; Arpawong, T.E.; Yen, K.; Cohen, P. The mitochondrial-derived peptide MOTS-c is a regulator of plasma metabolites and enhances insulin sensitivity. Physiol. Rep. 2019, 7, e14171. [Google Scholar] [CrossRef] [Green Version]
- Ramanjaneya, M.; Bettahi, I.; Jerobin, J.; Chandra, P.; Abi Khalil, C.; Skarulis, M.; Atkin, S.L.; Abou-Samra, A.B. Mitochondrial-Derived Peptides Are Down Regulated in Diabetes Subjects. Front. Endocrinol. 2019, 10, 331. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genomics Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31. [Google Scholar] [CrossRef]
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [Green Version]
- Bohr, V.A.; Stevnsner, T.; de Souza-Pinto, N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002, 286, 127–134. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease. Antioxidants 2021, 10, 653. [Google Scholar] [CrossRef]
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001. [Google Scholar] [CrossRef]
- Khan, A.H.; Pierce, C.O.; De Salvo, G.; Griffiths, H.; Nelson, M.; Cree, A.J.; Menon, G.; Lotery, A.J. The effect of systemic levels of TNF-alpha and complement pathway activity on outcomes of VEGF inhibition in neovascular AMD. Eye 2021, 36, 2192–2199. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [Green Version]
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. Endoplasmic reticulum-mitochondrial crosstalk: A novel role for the mitochondrial peptide humanin. Neural Regen. Res. 2017, 12, 35–38. [Google Scholar] [CrossRef]
- Nashine, S.; Cohen, P.; Wan, J.; Kenney, M.C. Effect of Humanin G (HNG) on inflammation in age-related macular degeneration (AMD). Aging 2022, 14, 4247–4269. [Google Scholar] [CrossRef]
- Chan, T.C.; Wilkinson Berka, J.L.; Deliyanti, D.; Hunter, D.; Fung, A.; Liew, G.; White, A. The role of reactive oxygen species in the pathogenesis and treatment of retinal diseases. Exp. Eye Res. 2020, 201, 108255. [Google Scholar] [CrossRef]
- Pardue, M.T.; Allen, R.S. Neuroprotective strategies for retinal disease. Prog. Retin. Eye Res. 2018, 65, 50–76. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohtashami, Z.; Singh, M.K.; Neto, F.T.; Salimiaghdam, N.; Hasanpour, H.; Kenney, M.C. Mitochondrial Open Reading Frame of the 12S rRNA Type-c: Potential Therapeutic Candidate in Retinal Diseases. Antioxidants 2023, 12, 518. https://doi.org/10.3390/antiox12020518
Mohtashami Z, Singh MK, Neto FT, Salimiaghdam N, Hasanpour H, Kenney MC. Mitochondrial Open Reading Frame of the 12S rRNA Type-c: Potential Therapeutic Candidate in Retinal Diseases. Antioxidants. 2023; 12(2):518. https://doi.org/10.3390/antiox12020518
Chicago/Turabian StyleMohtashami, Zahra, Mithalesh Kumar Singh, Farid Thomaz Neto, Nasim Salimiaghdam, Hossein Hasanpour, and M. Cristina Kenney. 2023. "Mitochondrial Open Reading Frame of the 12S rRNA Type-c: Potential Therapeutic Candidate in Retinal Diseases" Antioxidants 12, no. 2: 518. https://doi.org/10.3390/antiox12020518