
DJ-1 Deficiency Protects against Sepsis-Induced Myocardial Depression
 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bioinformatics Analysis
2.2. Human Samples
2.3. Animals
2.4. CLP Model of Sepsis
2.5. Echo and Hemodynamic Monitoring
2.6. Tissue Collection
2.7. Cell Culture
2.8. Real-Time Quantitative RT-PCR
2.9. Histopathology Assessment
2.10. Transmission Electron Microscopy (TEM)
2.11. TUNEL Staining
2.12. Western Blot
2.13. Serum Biochemistry Markers
2.14. Levels of Inflammatory Mediators
2.15. Tissue ROS Measurements
2.16. Statistical Analyses
3. Results
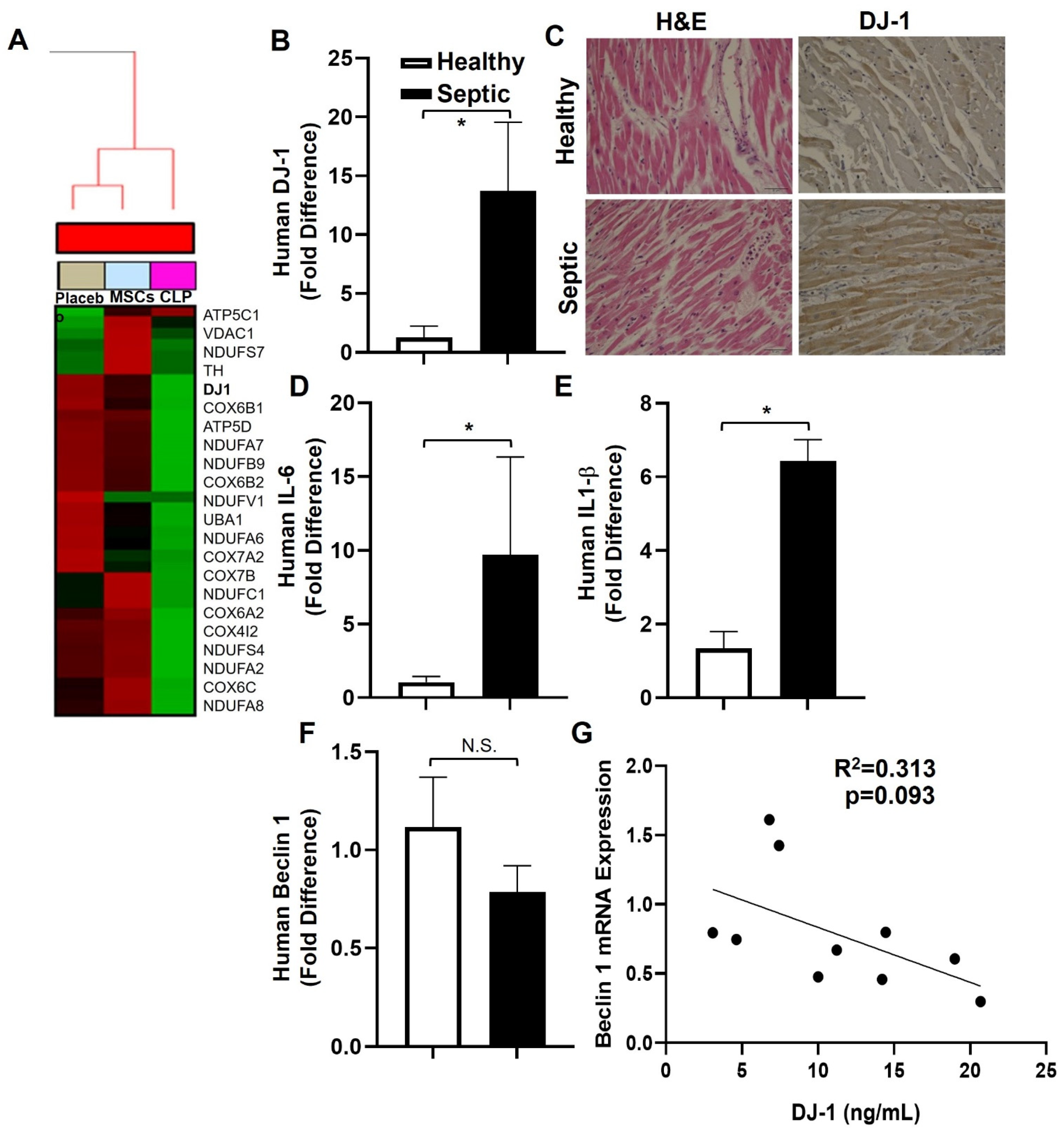
3.1. Bioinformatic Analysis
3.2. D-1-, IL-6, and IL-1β Are Increased in Human Septic Hearts
3.3. DJ-1 Deficiency Improves Survival Post-CLP
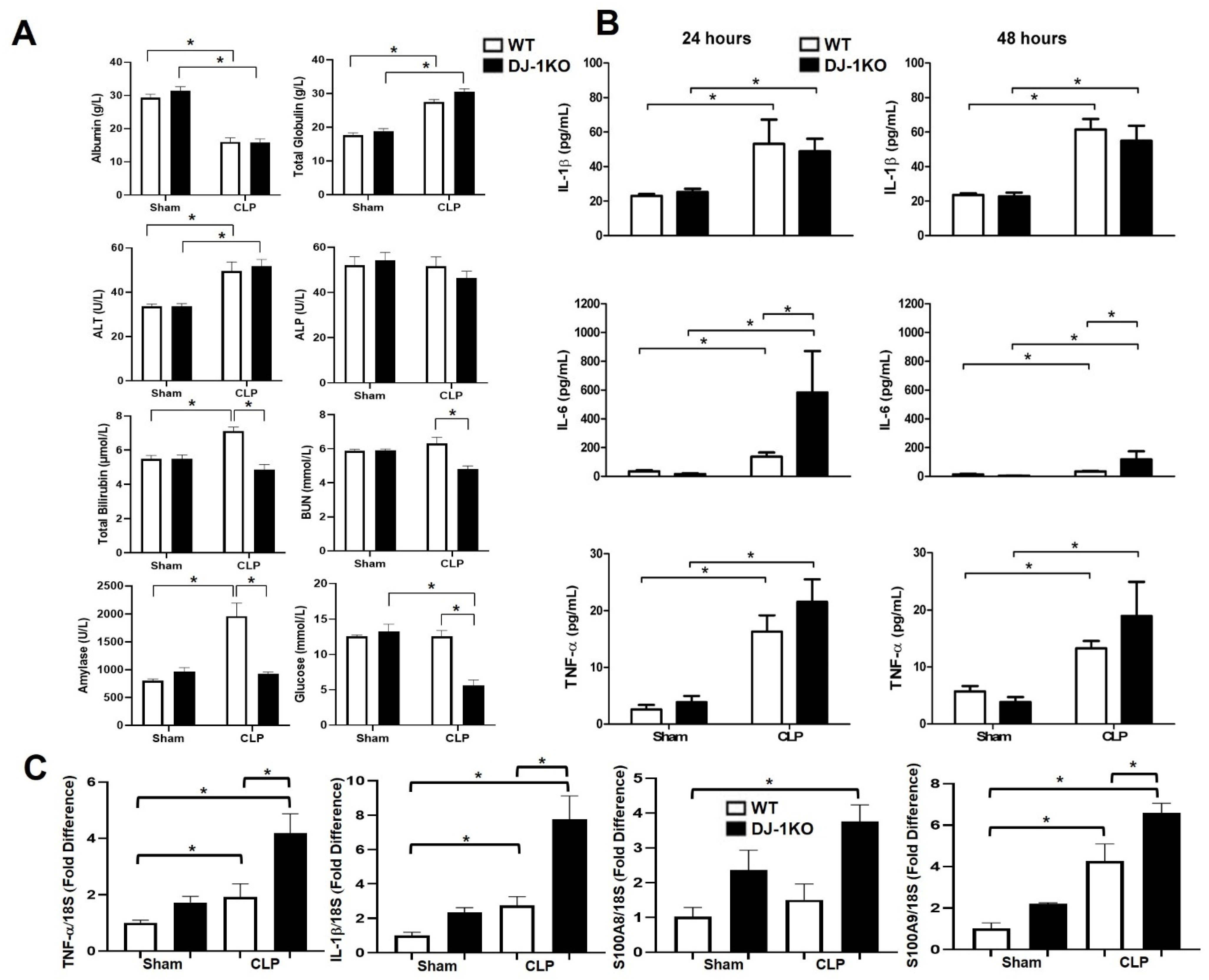
3.4. DJ-1 Deletion Exacerbates Inflammation Post-CLP
3.5. DJ-1 Deficiency Protects against CLP-Induced Myocardial Depression
3.6. DJ-1 Deficiency Suppresses Nitrative Stress Post-CLP
3.7. DJ-1 Deficiency Increases Antioxidant Pathways Post-CLP
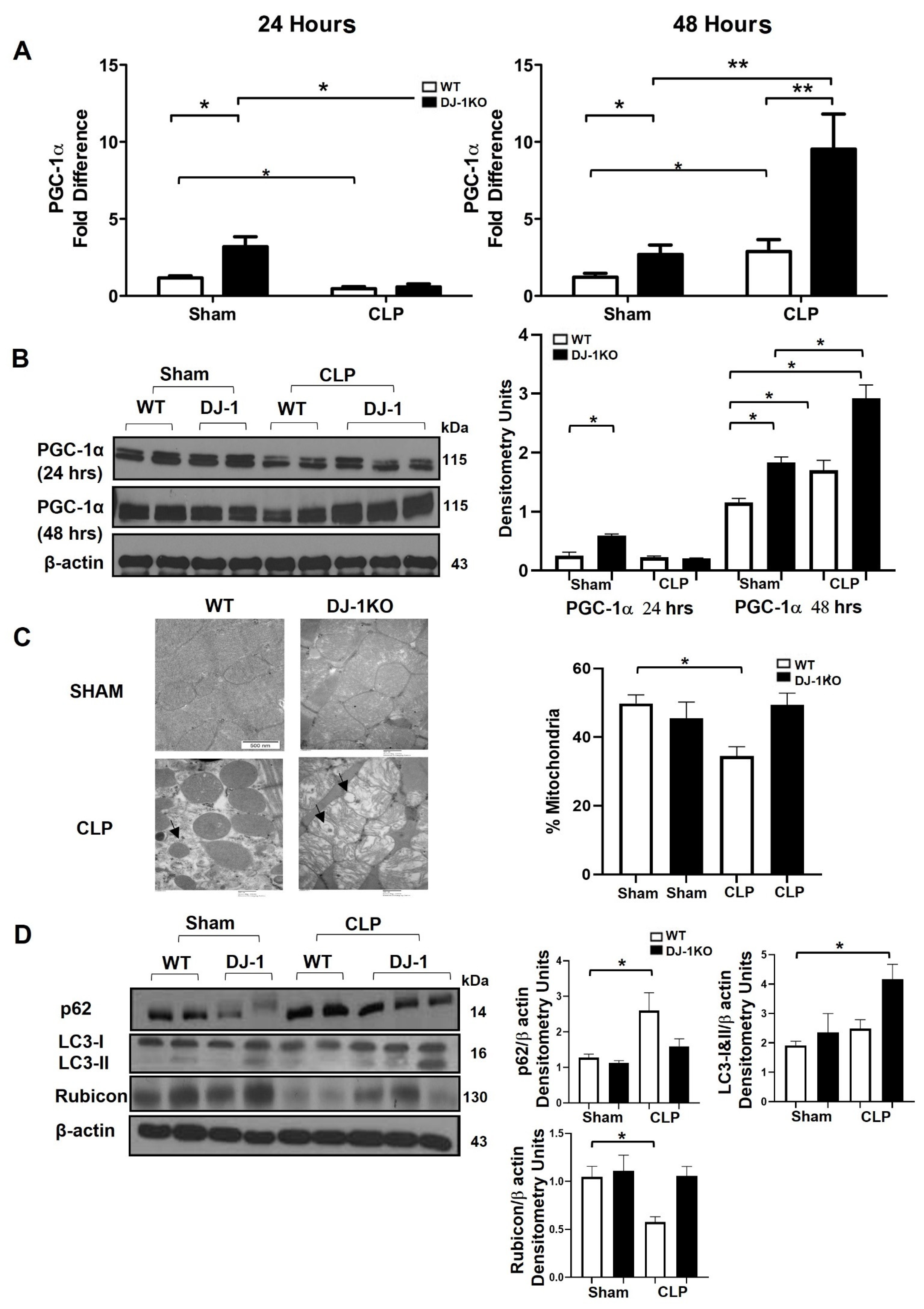
3.8. DJ-1 Deficiency Exacerbates Mitochondrial Biogenesis and Attenuates Apoptosis Post-CLP
3.9. DJ-1 Deficient Cardiomyocytes Have an Attenuated Hypertrophic Phenotype and Less Apoptosis despite a Marked Inflammation in Response to LPS
4. Discussions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Cinel, I.; Dellinger, R.P. Advances in pathogenesis and management of sepsis. Curr. Opin. Infect. Dis. 2007, 20, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Kennedy, J.N.; Wang, S.; Chang, C.-C.H.; Elliott, C.; Xu, Z.; Berry, S.; Clermont, G.; Cooper, G.; Gomez, H.; et al. Derivation, Validation, and Potential Treatment Implications of Novel Clinical Phenotypes for Sepsis. JAMA 2019, 321, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Seymour, C.W.; Gesten, F.; Prescott, H.C.; Friedrich, M.E.; Iwashyna, T.J.; Phillips, G.S.; Lemeshow, S.; Osborn, T.; Terry, K.M.; Levy, M.M. Time to Treatment and Mortality during Mandated Emergency Care for Sepsis. N. Engl. J. Med. 2017, 376, 2235–2244. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, Y.; Ito, T.; Nakahara, M.; Yamaguchi, K.; Yasuda, T. Sepsis-induced myocardial dysfunction: Pathophysiology and management. J. Intensiv. Care 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, E.; Fiaccadori, E.; Donadello, K.; Taccone, F.S.; Franchi, F.; Scolletta, S. Myocardial depression in sepsis: From pathogenesis to clinical manifestations and treatment. J. Crit. Care 2014, 29, 500–511. [Google Scholar] [CrossRef]
- Smeding, L.; Leong-Poi, H.; Hu, P.; Shan, Y.; Haitsma, J.J.; Horvath, E.; Furmli, S.; Masoom, H.; Kuiper, J.W.; Slutsky, A.S.; et al. Salutary effect of resveratrol on sepsis-induced myocardial depression. Crit. Care Med. 2012, 40, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, C.C.; Gattas, D.J.; Tsoporis, J.N.; Smeding, L.; Kabir, G.; Masoom, H.; Parker, T.G. Sepsis-induced myocardial depression is associated with transcriptional changes in energy metabolism and contractile related genes: A physiological and gene expression-based approach. Crit. Care Med. 2010, 38, 894–902. [Google Scholar] [CrossRef]
- Fernandes, C.J., Jr.; de Assuncao, M.S. Myocardial dysfunction in sepsis: A large, unsolved puzzle. Crit. Care Res. Pract. 2012, 2012, 896430. [Google Scholar] [CrossRef]
- Kumar, A.; Roberts, D.; Wood, K.E.; Light, B.; Parrillo, J.E.; Sharma, S.; Suppes, R.; Feinstein, D.; Zanotti, S.; Taiberg, L.; et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit. Care Med. 2006, 34, 1589–1596. [Google Scholar] [CrossRef]
- Lai, C.-C.; Lee, M.-T.G.; Lee, W.-C.; Chao, C.C.-T.; Hsu, T.-C.; Lee, S.-H.; Lee, C.-C. Susceptible period for cardiovascular complications in patients recovering from sepsis. Can. Med. Assoc. J. 2018, 190, E1062–E1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prescott, H.C.; Angus, D.C. Postsepsis Morbidity. JAMA 2018, 319, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nduka, O.O.; Parrillo, J.E. The Pathophysiology of Septic Shock. Crit. Care Clin. 2009, 25, 677–702. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, K.; Hammarqvist, F.; Strigård, K.; Hultenby, K.; Ljungqvist, O.; Wernerman, J.; Rooyackers, O. Derangements in mitochondrial metabolism in intercostal and leg muscle of critically ill patients with sepsis-induced multiple organ failure. Am. J. Physiol. Metab. 2006, 291, E1044–E1050. [Google Scholar] [CrossRef] [Green Version]
- Crouser, E.D.; Julian, M.W.; Blaho, D.V.; Pfeiffer, D.R. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit. Care Med. 2002, 30, 276–284. [Google Scholar] [CrossRef]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Schouten, M.; Wiersinga, W.J.; Levi, M.; van der Poll, T. Inflammation, endothelium, and coagulation in sepsis. J. Leukoc. Biol. 2008, 83, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.; Ariga, H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Hamakubo, T.; Yoshida, Y.; Ogawa, Y.; Hara, Y.; Fujimura, H.; Imai, Y.; Iwanari, H.; Mochizuki, Y.; Shichiri, M.; et al. Preparation and application of monoclonal antibodies against oxidized DJ-1. Significant elevation of oxidized DJ-1 in erythrocytes of early-stage Parkinson disease patients. Neurosci. Lett. 2009, 465, 1–5. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. DJ-1 Up-regulates Glutathione Synthesis during Oxidative Stress and Inhibits A53T α-Synuclein Toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxidative Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billia, F.; Hauck, L.; Grothe, D.; Konecny, F.; Rao, V.; Kim, R.H.; Mak, T.W. Parkinson-susceptibility gene DJ-1/PARK7 protects the murine heart from oxidative damage in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6085–6090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongworth, R.K.; Mukherjee, U.A.; Hall, A.R.; Astin, R.; Ong, S.-B.; Yao, Z.; Dyson, A.; Szabadkai, G.; Davidson, S.M.; Yellon, D.M.; et al. DJ-1 protects against cell death following acute cardiac ischemia–reperfusion injury. Cell Death Dis. 2014, 5, e1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Lambert, J.P.; Nicholson, C.K.; Kim, J.J.; Wolfson, D.W.; Cho, H.C.; Husain, A.; Naqvi, N.; Chin, L.-S.; Li, L.; et al. DJ-1 protects the heart against ischemia–reperfusion injury by regulating mitochondrial fission. J. Mol. Cell. Cardiol. 2016, 97, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Amatullah, H.; Shan, Y.; Beauchamp, B.L.; Gali, P.L.; Gupta, S.; Maron-Gutierrez, T.; Dos Santos, C.C. DJ-1/PARK7 Impairs Bacterial Clearance in Sepsis. Am. J. Respir. Crit. Care Med. 2017, 195, 889–905. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Gupta, S.; Amatullah, H.; Tsoporis, J.N.; Wei, K.; Monteiro, A.P.T.; Ektesabi, A.M.; Varkouhi, A.K.; Vaswani, C.M.; Formosa, A.; Fabro, A.T.; et al. DJ-1 binds to Rubicon to Impair LC-3 Associated Phagocytosis. Cell Death Differ. 2022, 29, 2024–2033. [Google Scholar] [CrossRef]
- dos Santos, C.C.; Murthy, S.; Hu, P.; Shan, Y.; Haitsma, J.J.; Mei, S.H.; Liles, W.C. Network analysis of transcriptional responses inducedby mesenchymal stem cell treatment of experimental sepsis. Am. J. Pathol. 2012, 18, 1681–1692. [Google Scholar] [CrossRef]
- Kim, R.H.; Smith, P.D.; Aleyasin, H.; Hayley, S.; Mount, M.P.; Pownall, S.; Wakeham, A.; You-Ten, A.J.; Kalia, S.K.; Horne, P.; et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 5215–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoporis, J.; Izhar, S.; Desjardins, J.-F.; Leong-Poi, H.; Parker, T. Conditional Cardiac Overexpression of S100A6 Attenuates Myocyte Hypertrophy and Apoptosis Following Myocardial Infarction. Curr. Pharm. Des. 2014, 20, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Tsoporis, J.N.; Izhar, S.; Leong-Poi, H.; Desjardins, J.F.; Huttunen, H.J.; Parker, T.G. S100B interaction with the receptor for advanced glycation end products (RAGE): A novel receptor-mediated mechanism for myocyte apoptosis postinfarction. Circ. Res. 2010, 106, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Most, P.; Seifert, H.; Gao, E.; Funakoshi, H.; Völkers, M.; Heierhorst, J.; Remppis, A.; Pleger, S.T.; DeGeorge, B.R.; Eckhart, A.D.; et al. Cardiac S100A1 Protein Levels Determine Contractile Performance and Propensity Toward Heart Failure After Myocardial Infarction. Circulation 2006, 114, 1258–1268. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. et Biophys. Acta (BBA)-Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320. [Google Scholar] [CrossRef] [Green Version]
- Mei, S.H.J.; Haitsma, J.J.; Dos Santos, C.C.; Deng, Y.; Lai, P.F.H.; Slutsky, A.S.; Liles, W.C.; Stewart, D.J. Mesenchymal Stem Cells Reduce Inflammation while Enhancing Bacterial Clearance and Improving Survival in Sepsis. Am. J. Respir. Crit. Care Med. 2010, 182, 1047–1057. [Google Scholar] [CrossRef]
- Rudiger, A.; Singer, M. Mechanisms of sepsis-induced cardiac dysfunction. Crit. Care Med. 2007, 35, 1599–1608. [Google Scholar] [CrossRef]
- Fernandes, C.J.; Akamine, N.; Knobel, E. Myocardial depression in sepsis. Shock 2008, 30 (Suppl. 1), 14–17. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, S.; Zhang, Y.; Yang, Y.; Escano, C.; Asico, L.; Jones, J.E.; Armando, I.; Jose, P.A. Role of Renal DJ-1 in the Pathogenesis of Hypertension Associated with Increased Reactive Oxygen Species Production. Hypertension 2012, 59, 446–452. [Google Scholar] [CrossRef]
- Aleyasin, H.; Rousseaux, M.W.C.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc. Natl. Acad. Sci. USA 2007, 104, 18748–18753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGarvey, N.C.; Suliman, H.B.; Bartz, R.R.; Fu, P.; Withers, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2012, 18, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme Oxygenase-1 Regulates Cardiac Mitochondrial Biogenesis via Nrf2-Mediated Transcriptional Control of Nuclear Respiratory Factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-S.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Pai, P.Y.; Hsueh, H.W.; Yuan, S.S.; Hsieh, Y.C. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg 2011, 253, 1190–1200. [Google Scholar] [CrossRef]
- Gunst, J.; Derese, I.; Aertgeerts, A.; Ververs, E.-J.; Wauters, A.; Berghe, G.V.D.; Vanhorebeek, I. Insufficient Autophagy Contributes to Mitochondrial Dysfunction, Organ Failure, and Adverse Outcome in an Animal Model of Critical Illness. Crit. Care Med. 2013, 41, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Carchman, E.H.; Rao, J.; Loughran, P.A.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-1–mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011, 53, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, W.; Watanabe, E.; Fujimura, L.; Watanabe-Takano, H.; Yoshidome, H.; Swanson, P.E.; Tokuhisa, T.; Oda, S.; Hatano, M. Kinetics and protective role of autophagy in a mouse cecal ligation and puncture-induced sepsis. Crit. Care 2013, 17, R160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, W.-S.; Chen, Y.-H.; Chiang, P.-C.; Hsiao, H.-W.; Chuang, S.-M.; Lue, S.-I.; Hsu, C. Suppression of Autophagy in Rat Liver at Late Stage of Polymicrobial Sepsis. Shock 2011, 35, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, Y.; Taneike, M.; Ueda, H.; Kitazume-Taneike, R.; Murakawa, T.; Sugihara, R.; Yorifuji, H.; Nishida, H.; Mine, K.; Hioki, A.; et al. Rubicon-regulated beta-1 adrenergic receptor recycling protects the heart from pressure overload. Sci. Rep. 2022, 12, 41. [Google Scholar] [CrossRef]
- Matsuda, J.; Takahashi, A.; Takabatake, Y.; Sakai, S.; Minami, S.; Yamamoto, T.; Fujimura, R.; Namba-Hamano, T.; Yonishi, H.; Nakamura, J.; et al. Metabolic effects of RUBCN/Rubicon deficiency in kidney proximal tubular epithelial cells. Autophagy 2020, 16, 1889–1904. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsoporis, J.N.; Amatullah, H.; Gupta, S.; Izhar, S.; Ektesabi, A.M.; Vaswani, C.M.; Desjardins, J.-F.; Kabir, G.; Teixera Monteiro, A.P.; Varkouhi, A.K.; et al. DJ-1 Deficiency Protects against Sepsis-Induced Myocardial Depression. Antioxidants 2023, 12, 561. https://doi.org/10.3390/antiox12030561
Tsoporis JN, Amatullah H, Gupta S, Izhar S, Ektesabi AM, Vaswani CM, Desjardins J-F, Kabir G, Teixera Monteiro AP, Varkouhi AK, et al. DJ-1 Deficiency Protects against Sepsis-Induced Myocardial Depression. Antioxidants. 2023; 12(3):561. https://doi.org/10.3390/antiox12030561
Chicago/Turabian StyleTsoporis, James N., Hajera Amatullah, Sahil Gupta, Shehla Izhar, Amin M. Ektesabi, Chirag M. Vaswani, Jean-Francois Desjardins, Golam Kabir, Ana Paula Teixera Monteiro, Amir K. Varkouhi, and et al. 2023. "DJ-1 Deficiency Protects against Sepsis-Induced Myocardial Depression" Antioxidants 12, no. 3: 561. https://doi.org/10.3390/antiox12030561