
24-Hydroxycholesterol Induces Tau Proteasome-Dependent Degradation via the SIRT1/PGC1α/Nrf2 Pathway: A Potential Mechanism to Counteract Alzheimer’s Disease

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Autopsy Specimens of AD Brains
2.2. Cell Culture and Treatments
2.3. RNA Extraction, cDNA Synthesis, and Real-Time RT-PCR
2.4. Protein Extraction and Western Blotting
2.5. Immunofluorescence Confocal Microscopy
2.6. Small Interfering RNA Transfection
2.7. Proteasome Activity Assay
2.8. Cell Viability Assay
2.9. Statistical Analysis
3. Results
3.1. Over-Expression and Deacetylation of PGC1α by 24-OHC
3.2. 24-OHC Regulates the Expression of Nrf2 and, through SIRT1, Its Deacetylation
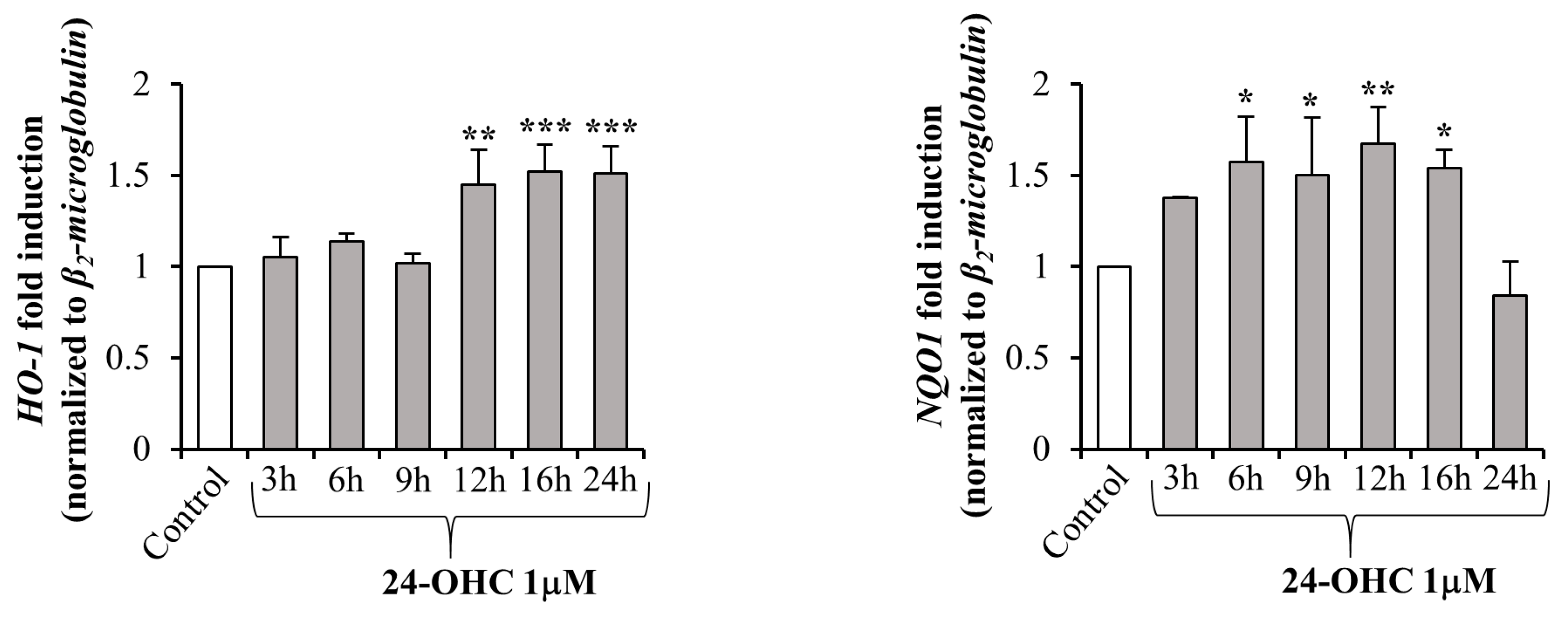
3.3. 24-OHC Promotes the Nuclear Translocation of Nrf2 and Activation of its Antioxidant Target Genes
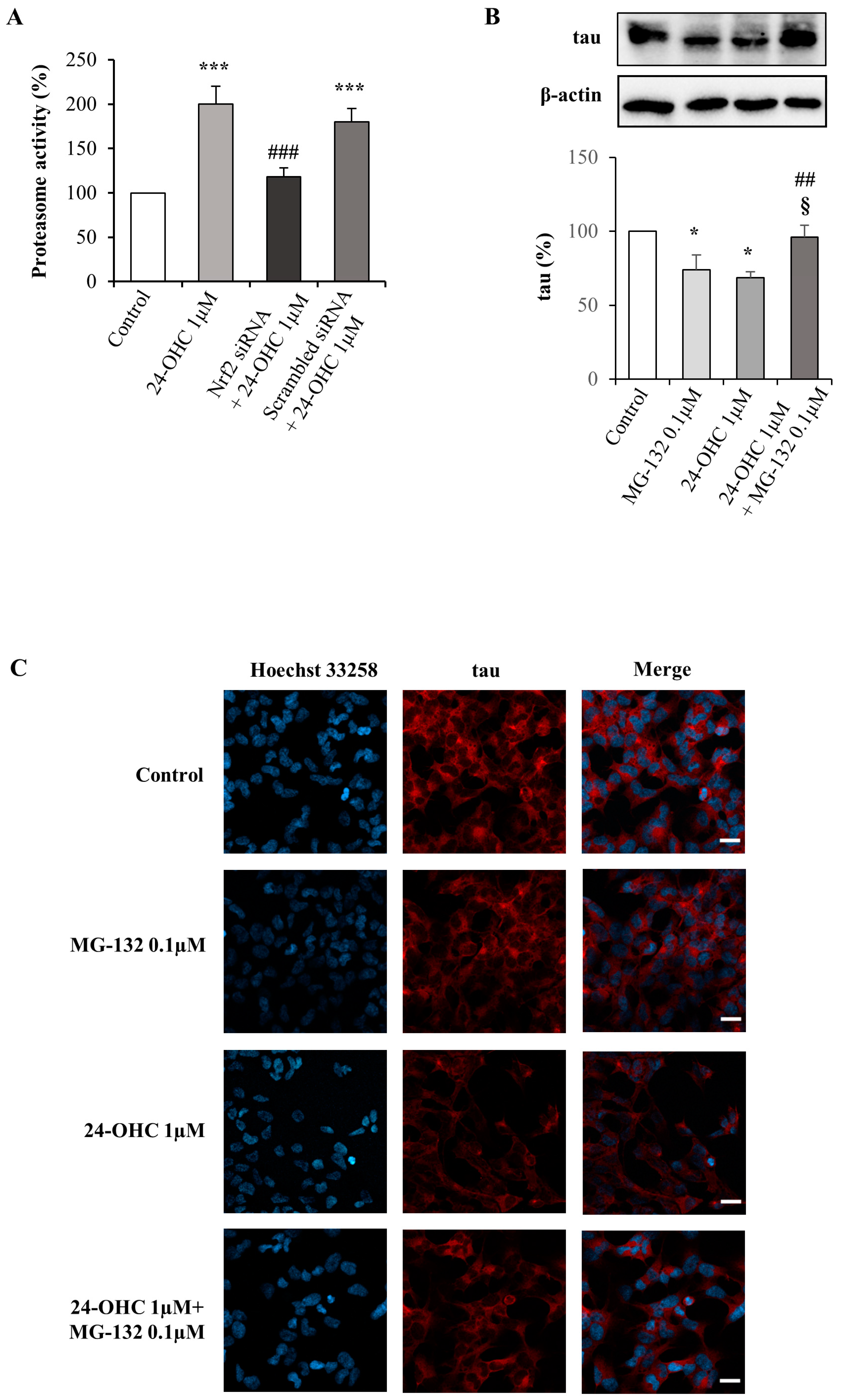
3.4. 24-OHC Induces Tau Protein Ubiquitination through Nrf2 Activation
3.5. Degradation of Tau by the Ubiquitin–Proteasome System
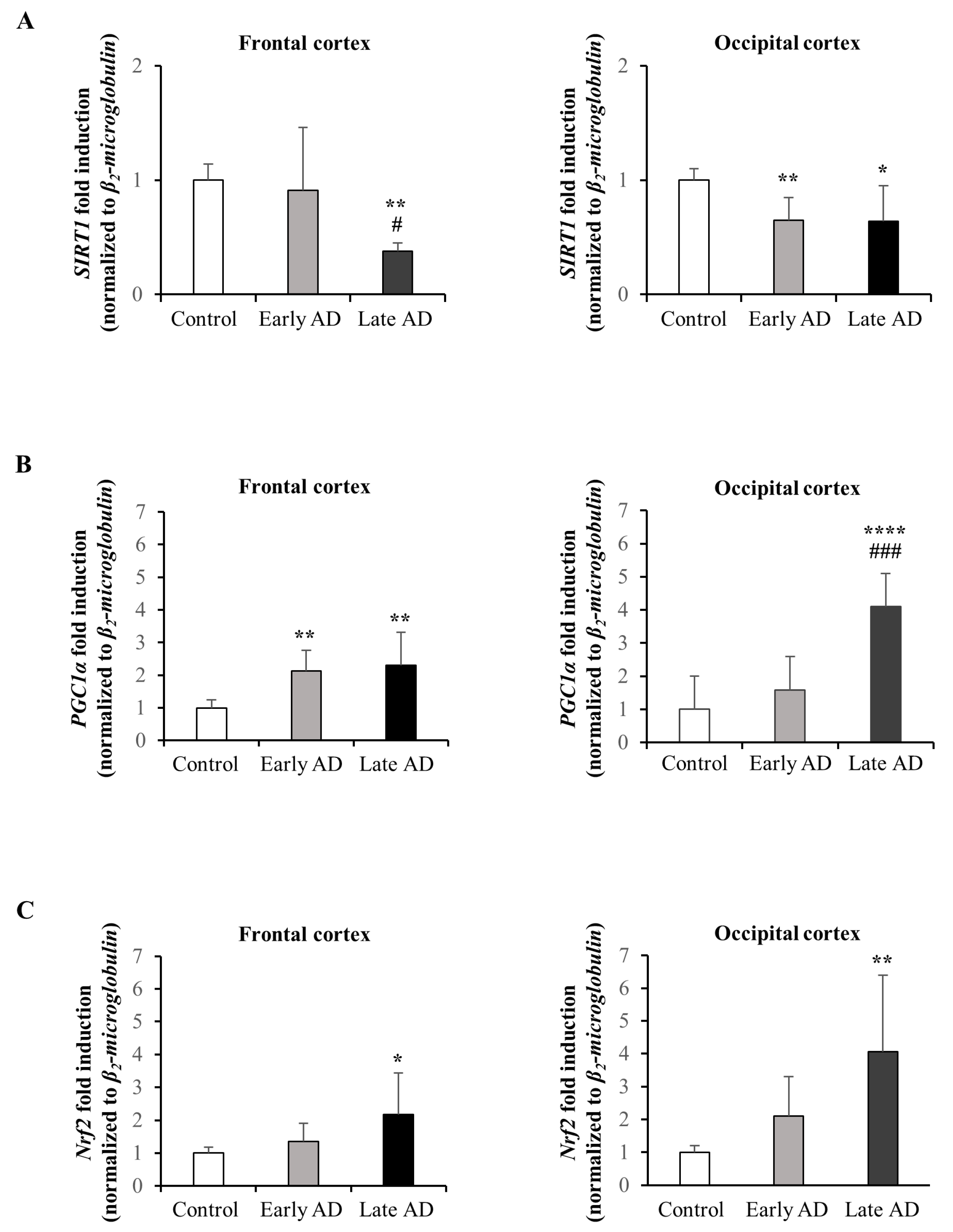
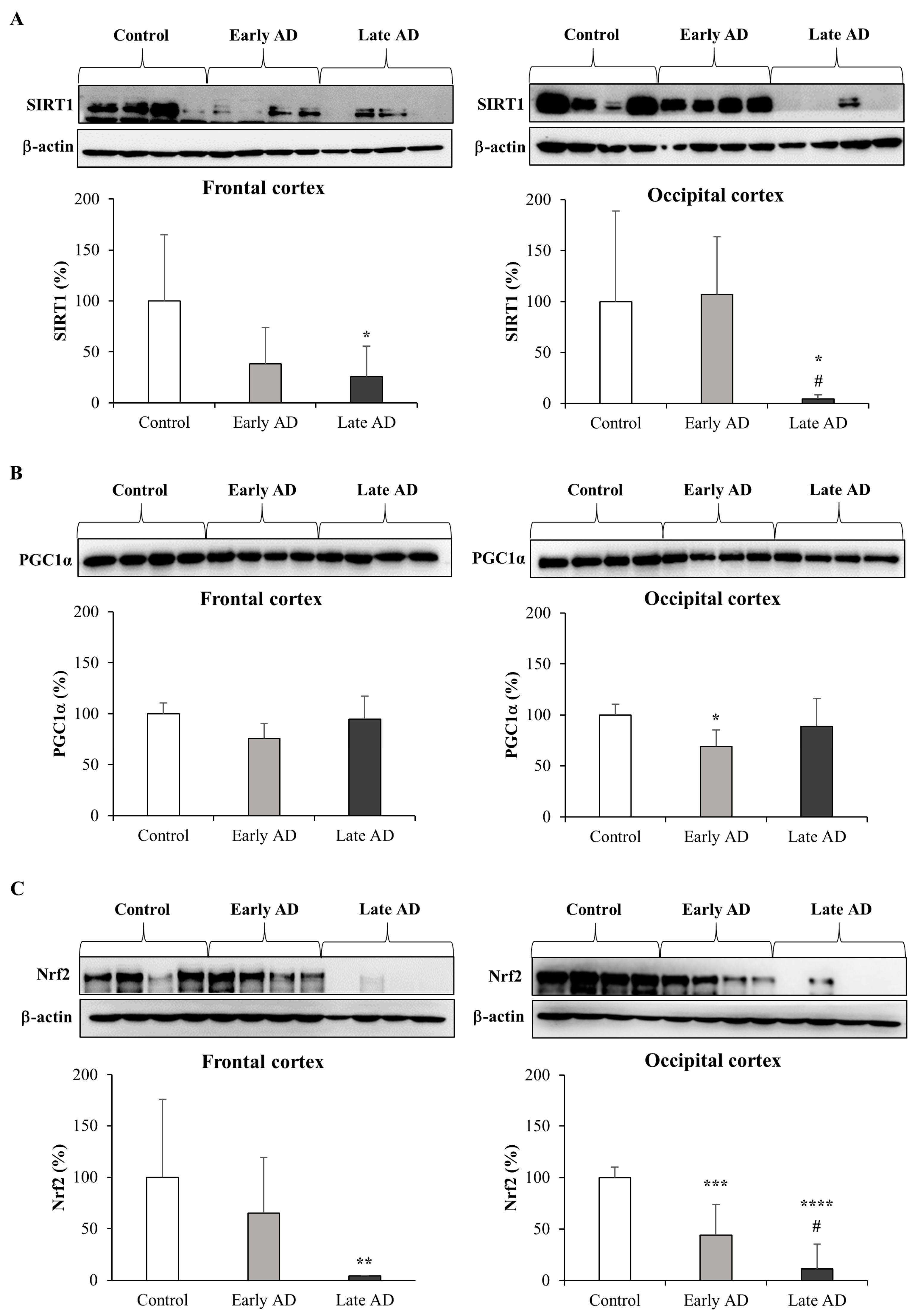
3.6. Expression and Synthesis of SIRT1, PGC1α, and Nrf2 in AD Brains
4. Discussion
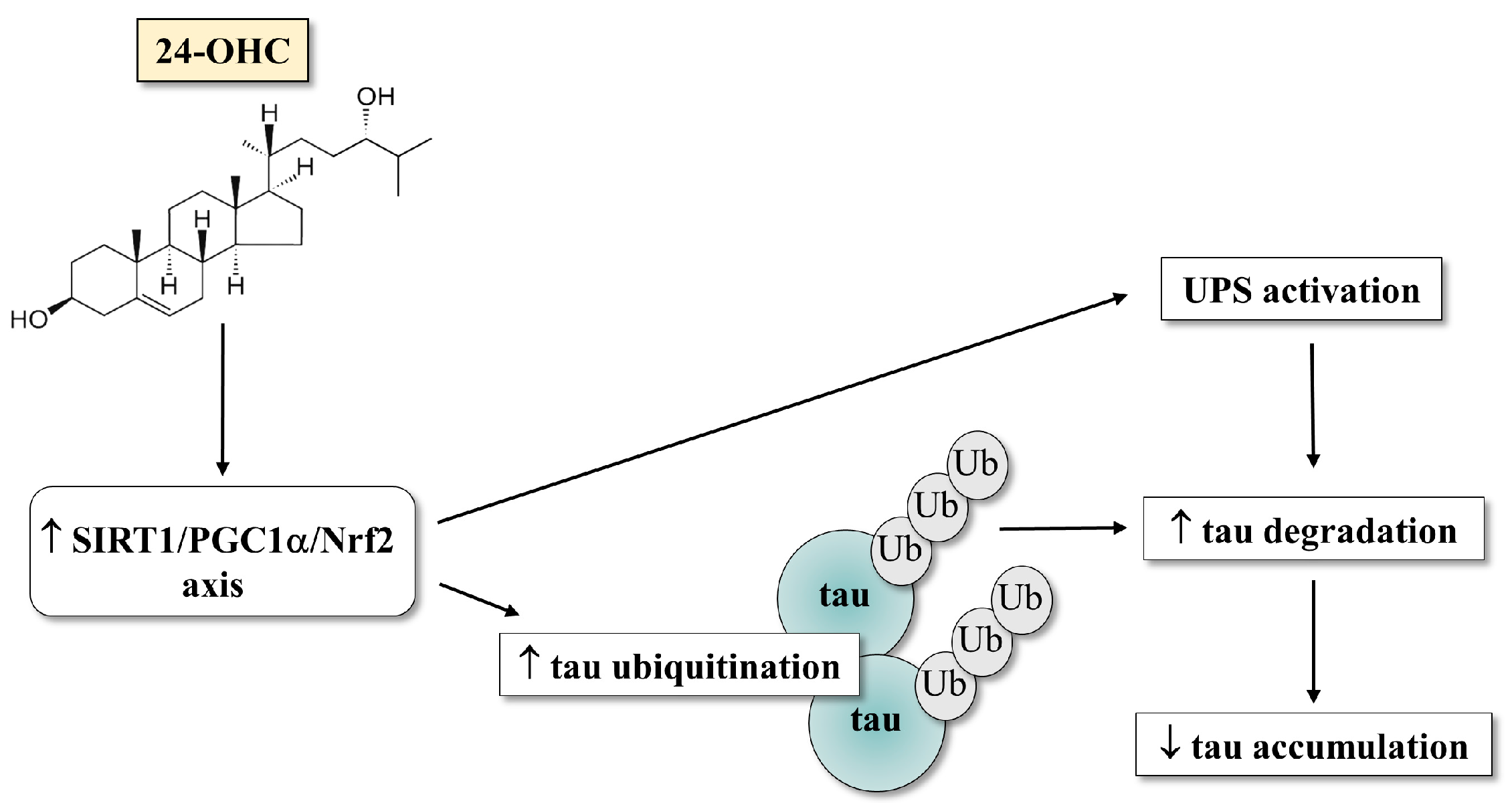
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemer, J.; Kins, S. Axonal transport and mitochondrial dysfunction in Alzheimer’s disease. Neurodegener. Dis. 2013, 12, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.G.; Li, L.; Müller, W.E.; Eckert, G.P. Cholesterol as a causative factor in Alzheimer’s disease: A debatable hypothesis. J. Neurochem. 2014, 129, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Cioffi, F.; Adam, R.H.I.; Broersen, K. Molecular Mechanisms and Genetics of Oxidative Stress in Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 981–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamba, P.; Staurenghi, E.; Testa, G.; Giannelli, S.; Sottero, B.; Leonarduzzi, G. A Crosstalk Between Brain Cholesterol Oxidation and Glucose Metabolism in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Sharma, S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J. Neurol. Sci. 2021, 421, 117253. [Google Scholar] [CrossRef]
- Ganguly, U.; Kaur, U.; Chakrabarti, S.S.; Sharma, P.; Agrawal, B.K.; Saso, L.; Chakrabarti, S. Oxidative Stress, Neuroinflammation, and NADPH Oxidase: Implications in the Pathogenesis and Treatment of Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 7086512. [Google Scholar] [CrossRef]
- Anwar, M.M. Oxidative stress-A direct bridge to central nervous system homeostatic dysfunction and Alzheimer’s disease. Cell Biochem. Funct. 2022, 40, 17–27. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Angeletti, M.; Keller, J.N.; Eleuteri, A.M. The fine-tuning of proteolytic pathways in Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 3433–3451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, X.; Zhao, Y.; Ponnusamy, M.; Liu, Y. The role of ubiquitin proteasomal system and autophagy-lysosome pathway in Alzheimer’s disease. Rev. Neurosci. 2017, 28, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Rahman, M.M.; Zaman, S.; Munira, M.S.; Uddin, M.S.; Rauf, A.; Banu, N.; Ashraf, M.G. Molecular Insight into the Crosstalk of UPS Components and Alzheimer’s Disease. Curr. Protein Pept. Sci. 2020, 21, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Pedraza-Chaverri, J.; Briones-Herrera, A.; Cruz-Ramos, E.; López-Canovas, L.; Zepeda-Cervantes, J. Protein degradation-associated mechanisms that are affected in Alzheimer’s disease. Mol. Cell. Biochem. 2022, 477, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhong, M.B.; Toro, C.A.; Zhang, L.; Cai, D. Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis. Neurosci. Lett. 2019, 703, 68–78. [Google Scholar] [CrossRef]
- Dar, K.B.; Bhat, A.H.; Amin, S.; Reshi, B.A.; Zargar, M.A.; Masood, A.; Ganie, S.A. Elucidating Critical Proteinopathic Mechanisms and Potential Drug Targets in Neurodegeneration. Cell. Mol. Neurobiol. 2020, 40, 313–345. [Google Scholar] [CrossRef]
- Tramutola, A.; Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease. Oxidative Med. Cell. Longev. 2016, 2016, 2756068. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Nemeroff, C.B.; Cooper, J.J.; Widge, A.; Rodriguez, C.; Carpenter, L.; McDonald, W.M. Amyloid and Tau in Alzheimer’s Disease: Biomarkers or Molecular Targets for Therapy? Are We Shooting the Messenger? Am. J. Psychiatry 2021, 178, 1014–1025. [Google Scholar] [CrossRef]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.D.; Hu, J.X.; Wu, J.R.; Zhou, B.; Huang, Y.P. Toxicities of amyloid-β and tau protein are reciprocally enhanced in the Drosophila model. Neural. Regen. Res. 2022, 17, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [Green Version]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2021, 11, 595532. [Google Scholar] [CrossRef]
- Boyarko, B.; Hook, V. Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neurodegeneration. Front. Neurosci. 2021, 15, 702788. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, J.; Sekiya, M.; Taniguchi, T.; Iijima, K.M.; Wang, R.; Ando, K. Global analysis of phosphorylation of tau by the checkpoint kinases Chk1 and Chk2 in vitro. J. Proteome Res. 2013, 12, 2654–2665. [Google Scholar] [CrossRef] [Green Version]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Testa, G.; Gargiulo, S.; Staurenghi, E.; Poli, G.; Leonarduzzi, G. Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front. Aging Neurosci. 2015, 7, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkhem, I.; Cedazo-Minguez, A.; Leoni, V.; Meaney, S. Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 2009, 30, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Masterman, T.; Patel, P.; Meaney, S.; Diczfalusy, U.; Björkhem, I. Side chain oxidized oxysterols in cerebrospinal fluid and the integrity of blood-brain and blood-cerebrospinal fluid barriers. J. Lipid Res. 2003, 44, 793–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, G.; Staurenghi, E.; Zerbinati, C.; Gargiulo, S.; Iuliano, L.; Giaccone, G.; Fantò, F.; Poli, G.; Leonarduzzi, G.; Gamba, P. Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol. 2016, 10, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Donmez, G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res. 2013, 23, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Testa, G.; Staurenghi, E.; Giannelli, S.; Gargiulo, S.; Guglielmotto, M.; Tabaton, M.; Tamagno, E.; Gamba, P.; Leonarduzzi, G. A silver lining for 24-hydroxycholesterol in Alzheimer’s disease: The involvement of the neuroprotective enzyme sirtuin 1. Redox Biol. 2018, 17, 423–431. [Google Scholar] [CrossRef]
- Jones, A.W.; Yao, Z.; Vicencio, J.M.; Karkucinska-Wieckowska, A.; Szabadkai, G. PGC-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signaling. Mitochondrion 2012, 12, 86–99. [Google Scholar] [CrossRef]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Nierenberg, A.A.; Ghaznavi, S.A.; Sande Mathias, I.; Ellard, K.K.; Janos, J.A.; Sylvia, L.G. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 α as a Novel Target for Bipolar Disorder and Other Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1α: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [Green Version]
- Uittenbogaard, M.; Chiaramello, A. Mitochondrial biogenesis: A therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. Curr. Pharm. Des. 2014, 20, 5574–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of Ab-deposition in the human brain and its relevance for the development of Alzheimers disease. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Giudetti, A.M.; Romano, A.; Lavecchia, A.M.; Gaetani, S. The Role of Brain Cholesterol and its Oxidized Products in Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 198–205. [Google Scholar] [CrossRef]
- Zarrouk, A.; Debbabi, M.; Bezine, M.; Karym, E.M.; Badreddine, A.; Rouaud, O.; Moreau, T.; Cherkaoui-Malki, M.; El Ayeb, M.; Nasser, B.; et al. Lipid Biomarkers in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 303–312. [Google Scholar] [CrossRef]
- Glöckner, F.; Meske, V.; Lütjohann, D.; Ohm, T.G. Dietary cholesterol and its effect on tau protein: A study in apolipoprotein E-deficient and P301L human tau mice. J. Neuropathol. Exp. Neurol. 2011, 70, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasanthi, J.R.; Larson, T.; Schommer, J.; Ghribi, O. Silencing GADD153/CHOP gene expression protects against Alzheimer’s disease-like pathology induced by 27-hydroxycholesterol in rabbit hippocampus. PLoS ONE 2011, 6, e26420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staurenghi, E.; Cerrato, V.; Gamba, P.; Testa, G.; Giannelli, S.; Leoni, V.; Caccia, C.; Buffo, A.; Noble, W.; Perez-Nievas, B.G.; et al. Oxysterols present in Alzheimer’s disease brain induce synaptotoxicity by activating astrocytes: A major role for lipocalin-2. Redox Biol. 2021, 39, 101837. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Rossin, D.; Poli, G.; Biasi, F.; Leonarduzzi, G. Implication of oxysterols in chronic inflammatory human diseases. Biochimie 2018, 153, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Gamba, P.; Giannelli, S.; Staurenghi, E.; Testa, G.; Sottero, B.; Biasi, F.; Poli, G.; Leonarduzzi, G. The Controversial Role of 24-S-Hydroxycholesterol in Alzheimer’s Disease. Antioxidants 2021, 10, 740. [Google Scholar] [CrossRef]
- Abildayeva, K.; Jansen, P.J.; Hirsch-Reinshagen, V.; Bloks, V.W.; Bakker, A.H.; Ramaekers, F.C.; de Vente, J.; Groen, A.K.; Wellington, C.L.; Kuipers, F.; et al. 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J. Biol. Chem. 2006, 281, 12799–12808. [Google Scholar] [CrossRef] [Green Version]
- Iuliano, S.; Ayton, J. Dietary intakes of expeditioners during prolonged sunlight deprivation in polar enviroments do not support bone health. Int. J. Circumpolar Health 2015, 74, 27965. [Google Scholar] [CrossRef]
- Okabe, A.; Urano, Y.; Itoh, S.; Suda, N.; Kotani, R.; Nishimura, Y.; Saito, Y.; Noguchi, N. Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol. 2013, 2, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Famer, D.; Meaney, S.; Mousavi, M.; Nordberg, A.; Björkhem, I.; Crisby, M. Regulation of α- and β-secretase activity by oxysterols: Cerebrosterol stimulates processing of APP via the α-secretase pathway. Biochem. Biophys. Res. Commun. 2007, 359, 46–50. [Google Scholar] [CrossRef]
- Prasanthi, J.R.; Huls, A.; Thomasson, S.; Thompson, A.; Schommer, E.; Ghribi, O. Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on β-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol. Neurodegener. 2009, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Schweinzer, C.; Kober, A.; Lang, I.; Etschmaier, K.; Scholler, M.; Kresse, A.; Sattler, W.; Panzenboeck, U. Processing of endogenous AβPP in blood-brain barrier endothelial cells is modulated by liver-X receptor agonists and altered cellular cholesterol homeostasis. J. Alzheimers Dis. 2011, 27, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pol, J.; Candela, P.; Boucau, M.C.; Fenart, L.; Gosselet, F. Oxysterols decrease apical-to-basolateral transport of Aβ peptides via an ABCB1-mediated process in an in vitro Blood-brain barrier model constituted of bovine brain capillary endothelial cells. Brain Res. 2013, 1517, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Ochiai, S.; Noguchi, N. Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013, 27, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Doherty, J.J.; Robichaud, A.J.; Belfort, G.M.; Chow, B.Y.; Hammond, R.S.; Crawford, D.C.; Linsenbardt, A.J.; Shu, H.-J.; Izumi, Y.; et al. The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J. Neurosci. 2013, 33, 17290–17300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsenbardt, A.J.; Taylor, A.; Emnett, C.M.; Doherty, J.J.; Krishnan, K.; Covey, D.F.; Paul, S.M.; Zorumski, C.F.; Mennerick, S. Different oxysterols have opposing actions at N-methyl-D-aspartate receptors. Neuropharmacology 2014, 85, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Burlot, M.A.; Braudeau, J.; Michaelsen-Preusse, K.; Potier, B.; Ayciriex, S.; Varin, J.; Gautier, B.; Djelti, F.; Audrain, M.; Dauphinot, L.; et al. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [Green Version]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxidative Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Cuadrado, A.; Rojo, A.I. Modulation of proteostasis by transcription factor NRF2 and impact in neurodegenerative diseases. Redox Biol. 2017, 11, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Park, H.M.; Kim, J.A.; Kwak, M.K. Protection against amyloid β cytotoxicity by sulforaphane: Role of the proteasome. Arch. Pharm. Res. 2009, 32, 109–115. [Google Scholar] [CrossRef]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.-M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.A.; Johnson, J.A. Nrf2—A therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, S.R.; Donepudi, A.C.; Xu, J.; Wei, W.; Cheng, Q.C.; Driscoll, M.V.; Johnson, D.A.; Johnson, J.A.; Li, X.; Slitt, A.L. Fasting induces nuclear factor E2-related factor 2 and ATP-binding Cassette transporters via protein kinase A and Sirtuin-1 in mouse and human. Antioxid. Redox Signal. 2014, 20, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, F.; Jia, X.; Yan, Q.; Zhang, X.; Mu, P. Amyloid-β (25-35) regulates neuronal damage and memory loss via SIRT1/Nrf2 in the cortex of mice. J. Chem. Neuroanat. 2021, 114, 101945. [Google Scholar] [CrossRef]
- Xue, F.; Huang, J.W.; Ding, P.Y.; Zang, H.G.; Kou, Z.J.; Li, T.; Fan, J.; Peng, Z.-W.; Yan, W.-J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef]
- da Cunha, M.S.B.; Arruda, S.F. Tucum-do-Cerrado (Bactris setosa Mart.) May Promote Anti-Aging Effect by Upregulating SIRT1-Nrf2 Pathway and Attenuating Oxidative Stress and Inflammation. Nutrients 2017, 9, 1243. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, Y.; Wei, X.; Fan, T. Neuroprotective effect of licochalcone A against oxygen-glucose deprivation/reperfusion in rat primary cortical neurons by attenuating oxidative stress injury and inflammatory response via the SIRT1/Nrf2 pathway. J. Cell. Biochem. 2018, 119, 3210–3219. [Google Scholar] [CrossRef]
- Lu, J.; Huang, Q.; Zhang, D.; Lan, T.; Zhang, Y.; Tang, X.; Xu, P.; Zhao, D.; Cong, D.; Zhao, D.; et al. The Protective Effect of DiDang Tang Against AlCl3-Induced Oxidative Stress and Apoptosis in PC12 Cells Through the Activation of SIRT1-Mediated Akt/Nrf2/HO-1 Pathway. Front. Pharmacol. 2020, 11, 466. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Shah, S.A.; Zaman, N.; Uddin, M.N.; Khan, W.; Ali, A.; Riaz, M.; Kamil, A. Vitamin D exerts neuroprotection via SIRT1/nrf-2/NF-kB signaling pathways against D-galactose-induced memory impairment in adult mice. Neurochem. Int. 2021, 142, 104893. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.J.; Bekdash, A.M.; Itani, H.A.; Borjac, J.M. Saffron extract attenuates neuroinflammation in rmTBI mouse model by suppressing NLRP3 inflammasome activation via SIRT1. PLoS ONE 2021, 16, e0257211. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Jo Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxidative Med. Cell. Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.V.; Dave, K.R.; Saul, I.; Perez-Pinzon, M.A. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke 2015, 46, 1626–1632. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Garg, S.; Mandelkow, E.M.; Mandelkow, E. Proteolytic processing of tau. Biochem. Soc. Trans. 2010, 38, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.H.; Reits, E.A.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell. Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef]
- Dickey, C.A.; Dunmore, J.; Lu, B.; Wang, J.W.; Lee, W.C.; Kamal, A.; Burrows, F.; Eckman, C.; Hutton, M.; Petrucelli, L. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006, 20, 753–755. [Google Scholar] [CrossRef]
- Dolan, P.J.; Johnson, G.V. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J. Biol. Chem. 2010, 285, 21978–21987. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Chesser, A.S.; Pritchard, S.M.; Johnson, G.V. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front. Neurol. 2013, 4, 122. [Google Scholar] [CrossRef] [Green Version]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Tao Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulic, B.; Pickhardt, M.; Schmidt, B.; Mandelkow, E.M.; Waldmann, H.; Mandelkow, E. Development of tau aggregation inhibitors for Alzheimer’s disease. Angew. Chem. Int. Ed. Eng. 2009, 48, 1740–1752. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.-Q.; et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Zhang, K.; Brodbeck, J.; Daub, A.C.; Sharma, P.; Finkbeiner, S.; Cui, B.; Mucke, L. Tau reduction prevents Aβ-induced defects in axonal transport. Science 2010, 330, 198. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Chu, T.T.; Gao, N.; Li, Q.Q.; Chen, P.G.; Yang, X.F.; Chen, Y.X.; Zhao, Y.-F.; Li, Y.-M. Specific Knockdown of Endogenous Tau Protein by Peptide-Directed Ubiquitin-Proteasome Degradation. Cell Chem. Biol. 2016, 23, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Liu, T.; Jiao, Q.; Ji, J.; Tao, M.; Liu, Y.; You, Q.; Jiang, Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem. 2018, 146, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.-H.; Kang, E.-B.; Oh, Y.-S.; Yang, D.-S.; Cho, J.-Y. Treadmill exercise decreases amyloid-β burden possibly via activation of SIRT-1 signaling in a mouse model of Alzheimer’s disease. Exp. Neurol. 2017, 288, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Michán, S.; Li, Y.; Chou, M.M.-H.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, M.I.; Milenkovic, I.; Regelsberger, G.; Kovacs, G.G. Distinct patterns of sirtuin expression during progression of Alzheimer’s disease. Neuromol. Med. 2014, 16, 405–414. [Google Scholar] [CrossRef]
- Cao, K.; Dong, Y.T.; Xiang, J.; Xu, Y.; Hong, W.; Song, H.; Guan, Z.Z. Reduced expression of SIRT1 and SOD-1 and the correlation between these levels in various regions of the brains of patients with Alzheimer’s disease. J. Clin. Pathol. 2018, 71, 1090–1099. [Google Scholar] [CrossRef]
- Mahady, L.; Nadeem, M.; Malek-Ahmadi, M.; Chen, K.; Perez, S.E.; Mufson, E.J. Frontal cortex epigenetic dysregulation during the progression of Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 115–131. [Google Scholar] [CrossRef]
- Lattanzio, F.; Carboni, L.; Carretta, D.; Rimondini, R.; Candeletti, S.; Romualdi, P. Human apolipoprotein E4 modulates the expression of Pin1, Sirtuin 1, and Presenilin 1 in brain regions of targeted replacement apoE mice. Neuroscience 2014, 256, 360–369. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef]
- Kanninen, K.; Heikkinen, R.; Malm, T.; Rolova, T.; Kuhmonen, S.; Leinonen, H.; Ylä-Herttuala, S.; Tanila, H.; Levonen, A.-L.; Koistinaho, M.; et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 16505–16510. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Sex | Age (Years) | Post-Mortem (h) | Braak Stage | Thal Stage |
|---|---|---|---|---|---|
| Control (n = 9) 1 2 3 4 5 6 7 8 9 | F F F F F M F M F | 61 61 60 68 58 75 72 70 69 | 36 36 18 48 24 40 43 39 20 | 0 0 0 0 0 0 0 0 0 | 0 0 0 0 0 0 0 0 0 |
| Early AD (n = 6) 10 11 12 13 14 15 | M M F M F M | 69 71 81 86 77 85 | 38 36 39 20 46 25 | I I I II II III | 1 1 1 1 1 3 |
| Late AD (n = 8) 16 17 18 19 20 21 22 23 | F F M M M F F F | 67 82 75 79 77 58 72 81 | 28 21 26 45 16 48 34 37 | V VI VI VI VI VI VI VI | 4 4 4 4 4 4 4 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, G.; Giannelli, S.; Sottero, B.; Staurenghi, E.; Giaccone, G.; Caroppo, P.; Gamba, P.; Leonarduzzi, G. 24-Hydroxycholesterol Induces Tau Proteasome-Dependent Degradation via the SIRT1/PGC1α/Nrf2 Pathway: A Potential Mechanism to Counteract Alzheimer’s Disease. Antioxidants 2023, 12, 631. https://doi.org/10.3390/antiox12030631
Testa G, Giannelli S, Sottero B, Staurenghi E, Giaccone G, Caroppo P, Gamba P, Leonarduzzi G. 24-Hydroxycholesterol Induces Tau Proteasome-Dependent Degradation via the SIRT1/PGC1α/Nrf2 Pathway: A Potential Mechanism to Counteract Alzheimer’s Disease. Antioxidants. 2023; 12(3):631. https://doi.org/10.3390/antiox12030631
Chicago/Turabian StyleTesta, Gabriella, Serena Giannelli, Barbara Sottero, Erica Staurenghi, Giorgio Giaccone, Paola Caroppo, Paola Gamba, and Gabriella Leonarduzzi. 2023. "24-Hydroxycholesterol Induces Tau Proteasome-Dependent Degradation via the SIRT1/PGC1α/Nrf2 Pathway: A Potential Mechanism to Counteract Alzheimer’s Disease" Antioxidants 12, no. 3: 631. https://doi.org/10.3390/antiox12030631