
Selective Enzymatic Esterification of Lignin-Derived Phenolics for the Synthesis of Lipophilic Antioxidants

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Enzymatic Esterification of DCA
2.3. Purification and Characterization of DCA-C8
- 4-(3-hydroxypropyl)-2-methoxyphenyl octanoate (DCA-C8)
- 1H-NMR (80 MHz, CDCl3): δ 6.79 (m, 3H, Ar); 5.47 (s, 1H, Ar-OH); 4.09 (t, J = 6.5 Hz), 2H, -CH2-COO-); 3.88 (s, 3H, -OCH3); 2.80–2.47 (m, 2H, -CH2-); 2.44–2.15 (m, 2H, -CH2-); 2.11–1.78 (m, 2H, -CH2-); 1.76–0.68 (m, 15H, -CH2-). Integration of the signals between 2.80 and 0.68 ppm results in 2 extra H (21 instead of 19), possibly due to the presence of traces of water in CDCl3 (NMR solvent) or traces of grease in the petroleum ether (purification solvent).
- IR (neat) ν: 3560–3188, 2958–2854, 1733, 1606, 1519, 1363, 1271, 1238, 1155, 1032 cm−1.
- HRMS in positive mode, [C18H28O4 + NH4]+ m/z 326.2324, [C18H28O4 + H]+ m/z 309.2058, HRMS in negative mode, [C18H28O4 − H]− m/z 307.19092.
2.4. UPLC Analysis
2.5. Antioxidant Activity Test
2.6. Lignin Depolymerization
2.7. Enzymatic Esterification of Lignin
2.8. Characterization of the Lignin Fractions
3. Results and Discussion
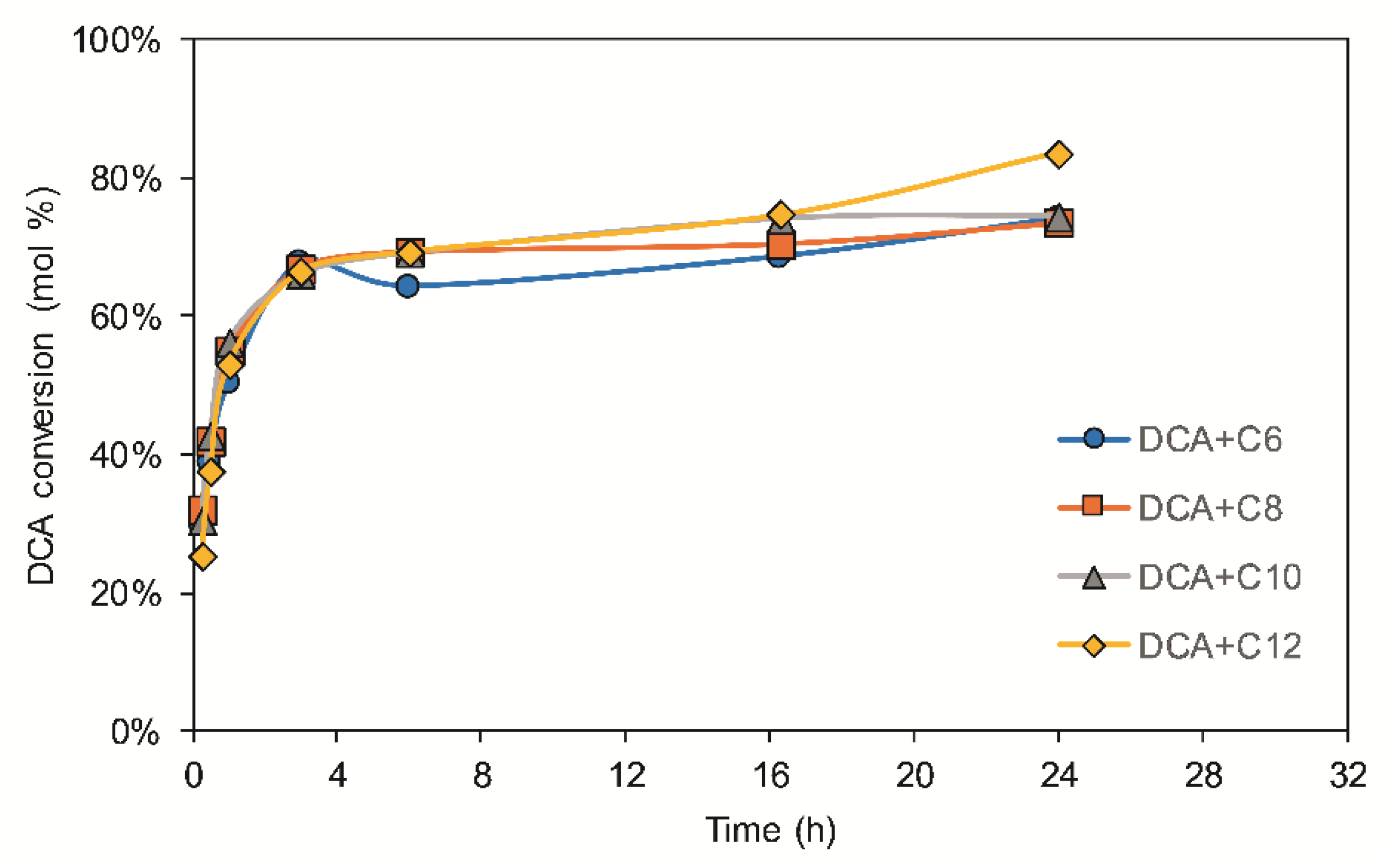
3.1. DCA Esterification with Different Fatty Acids
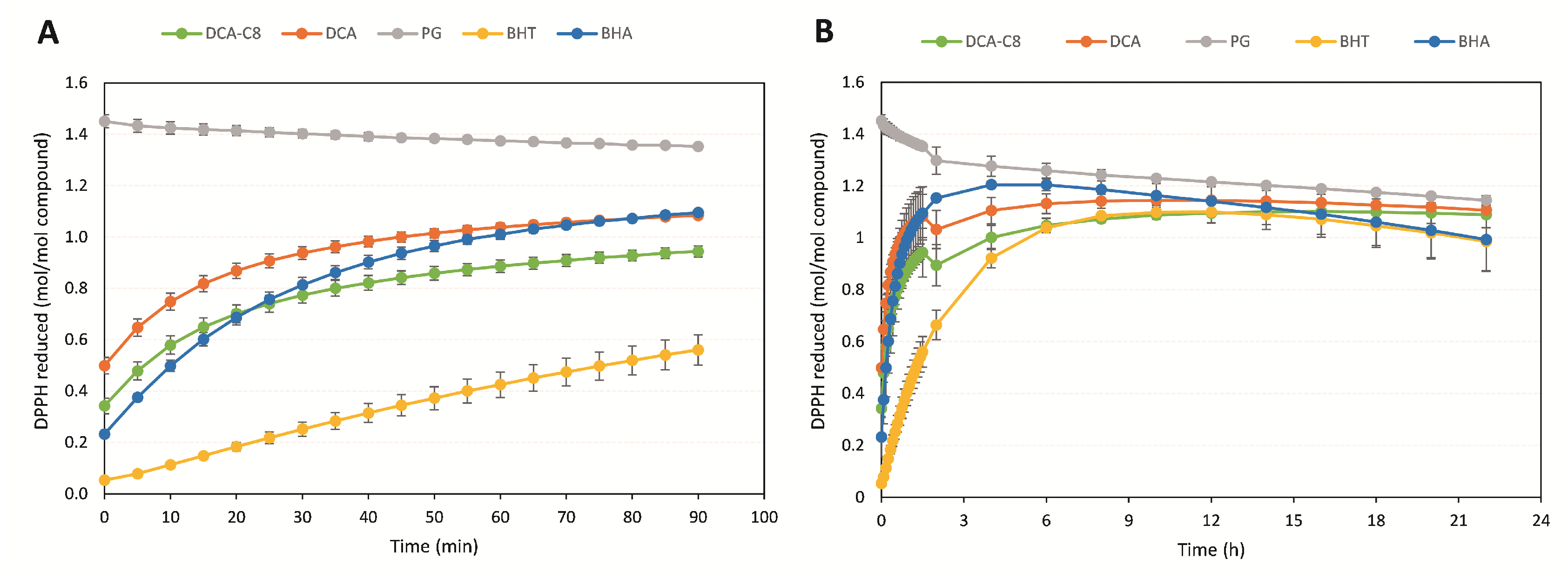
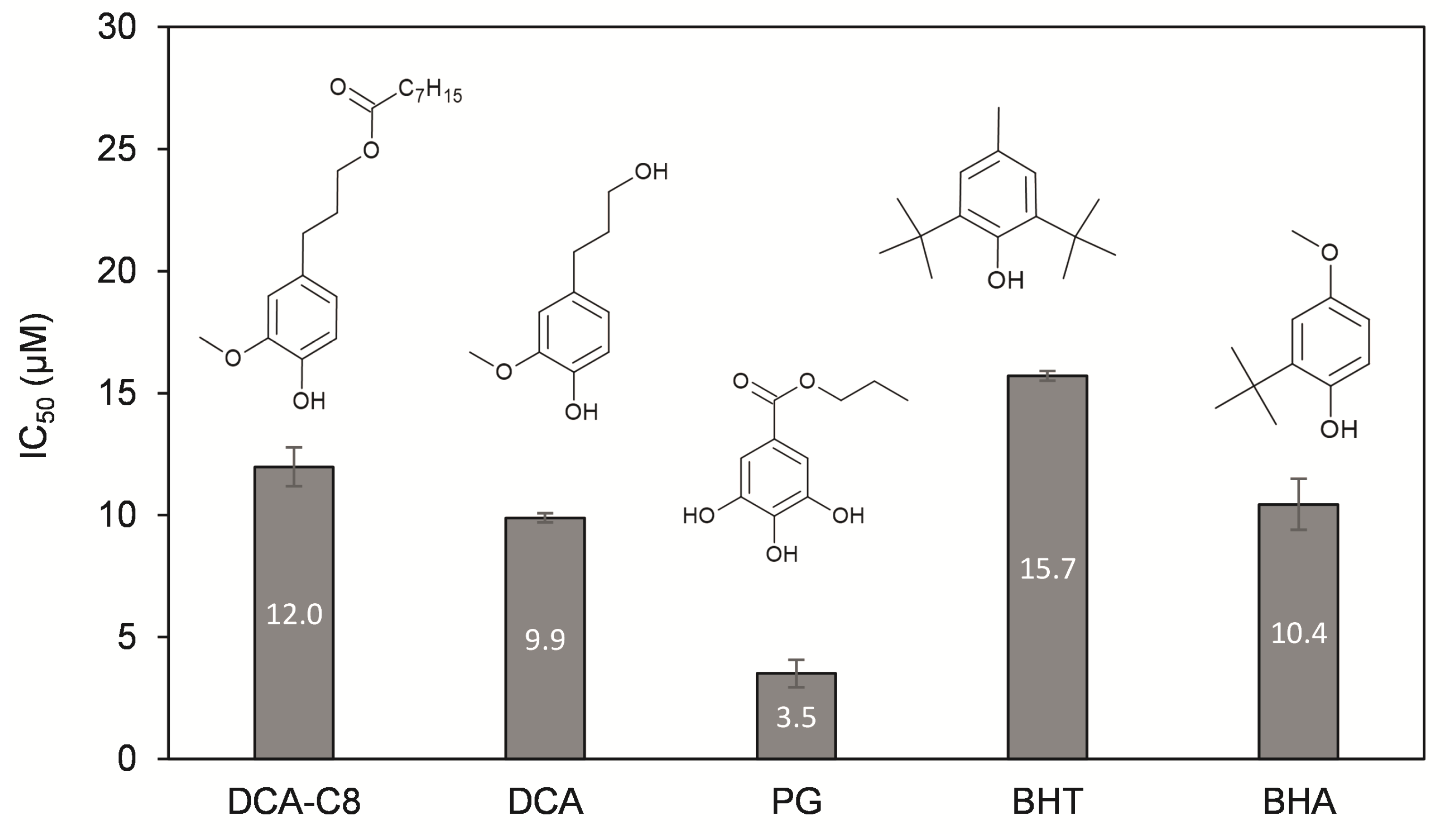
3.2. Antioxidant Activity of DCA-C8
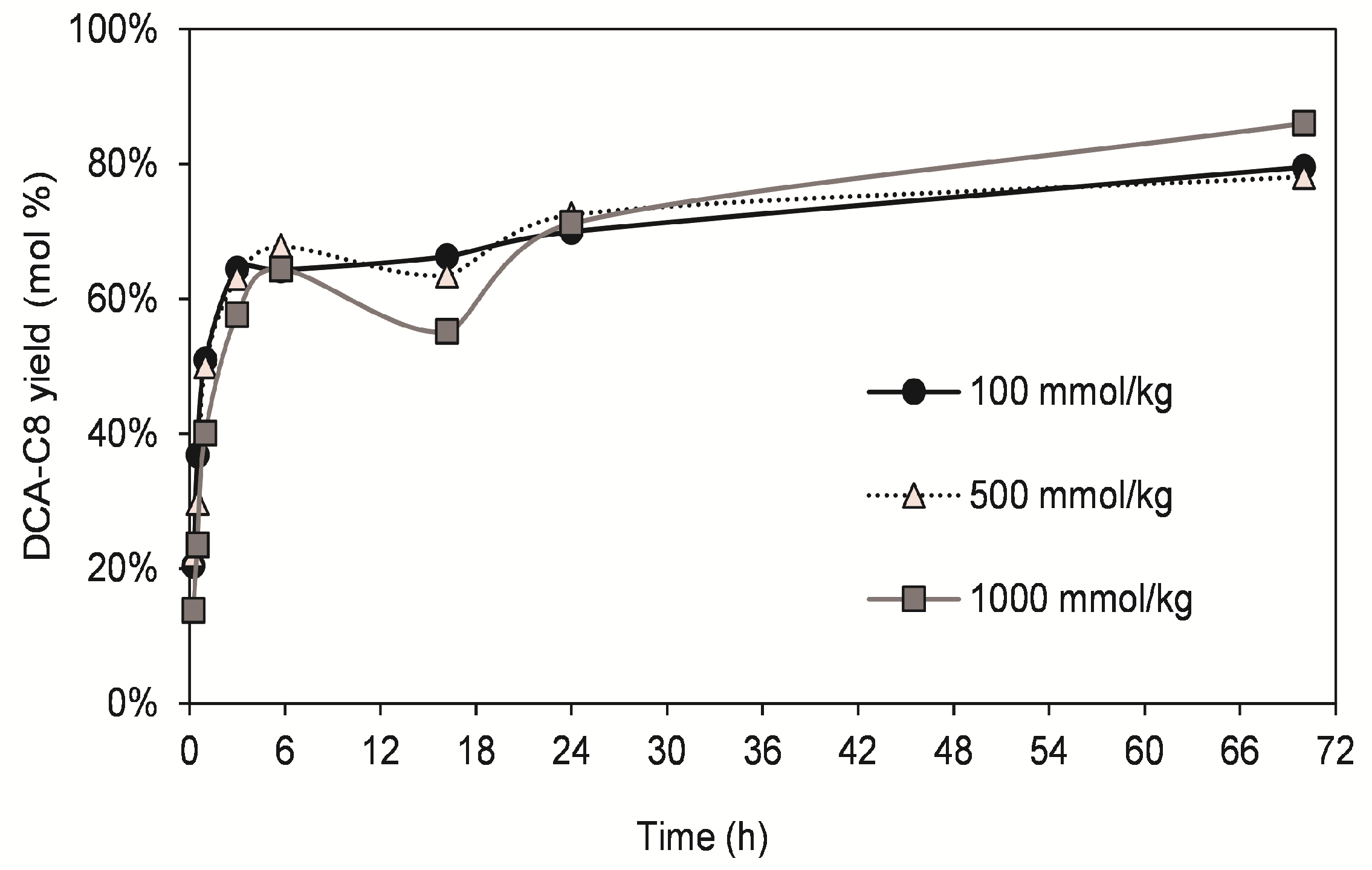
3.3. Optimization of DCA Esterification with Octanoic Acid
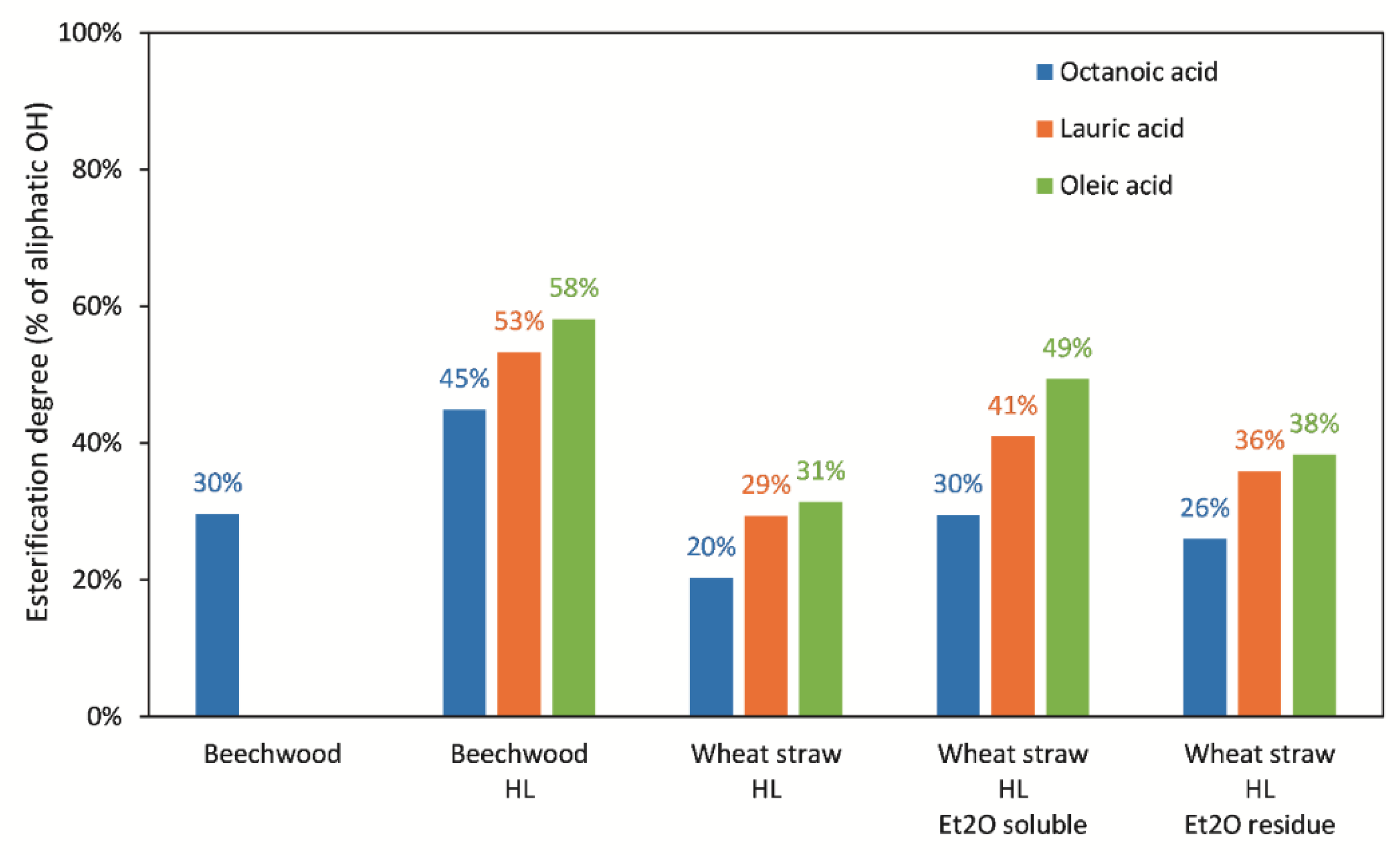
3.4. Enzymatic Esterification of Lignin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leopoldini, M.; Marino, T.; Russo, N.; Toscano, M. Antioxidant Properties of Phenolic Compounds: H-Atom versus Electron Transfer Mechanism. J. Phys. Chem. A 2004, 108, 4916–4922. [Google Scholar] [CrossRef]
- Polati, S.; Gosetti, F.; Gennaro, M.C. Preservatives in Cosmetics. Analytical Methods. Anal. Cosmet. Prod. 2007, 211–241. [Google Scholar] [CrossRef]
- Dizhbite, T.; Telysheva, G.; Jurkjane, V.; Viesturs, U. Characterization of the Radical Scavenging Activity of Lignins-Natural Antioxidants. Bioresour. Technol. 2004, 95, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Kabir, A.S.; Li, H.; Yuan, H.Z.; Kuboki, T.; Xu, C. Effects of De-Polymerized Lignin Content on Thermo-Oxidative and Thermal Stability of Polyethylene. J. Anal. Appl. Pyrolysis 2019, 140, 413–422. [Google Scholar] [CrossRef]
- Kabir, A.S.; Yuan, Z.; Kuboki, T.; Xu, C. De-Polymerization of Industrial Lignins to Improve the Thermo-Oxidative Stability of Polyolefins. Ind. Crops Prod. 2018, 120, 238–249. [Google Scholar] [CrossRef]
- Domenek, S.; Louaifi, A.; Guinault, A.; Baumberger, S. Potential of Lignins as Antioxidant Additive in Active Biodegradable Packaging Materials. J. Polym. Environ. 2013, 21, 692–701. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Odelius, K.; Hakkarainen, M. Microwave Hydrophobized Lignin with Antioxidant Activity for Fused Filament Fabrication. ACS Appl. Polym. Mater. 2021, 3, 3538–3548. [Google Scholar] [CrossRef]
- Ye, D.; Li, S.; Lu, X.; Zhang, X.; Rojas, O.J. Antioxidant and Thermal Stabilization of Polypropylene by Addition of Butylated Lignin at Low Loadings. ACS Sustain. Chem. Eng. 2016, 4, 5248–5257. [Google Scholar] [CrossRef]
- Jedrzejczyk, M.A.; Van den Bosch, S.; Van Aelst, J.; Van Aelst, K.; Kouris, P.D.; Moalin, M.; Haenen, G.R.M.M.; Boot, M.D.; Hensen, E.J.M.; Lagrain, B.; et al. Lignin-Based Additives for Improved Thermo-Oxidative Stability of Biolubricants. ACS Sustain. Chem. Eng. 2021, 9, 12548–12559. [Google Scholar] [CrossRef]
- Chandrasekaran, S.R.; Murali, D.; Marley, K.A.; Larson, R.A.; Doll, K.M.; Moser, B.R.; Scott, J.; Sharma, B.K. Antioxidants from Slow Pyrolysis Bio-Oil of Birch Wood: Application for Biodiesel and Biobased Lubricants. ACS Sustain. Chem. Eng. 2016, 4, 1414–1421. [Google Scholar] [CrossRef]
- Larson, R.A.; Sharma, B.K.; Marley, K.A.; Kunwar, B.; Murali, D.; Scott, J. Potential Antioxidants for Biodiesel from a Softwood Lignin Pyrolyzate. Ind. Crops Prod. 2017, 109, 476–482. [Google Scholar] [CrossRef]
- Widsten, P. Lignin-Based Sunscreens—State-of-the-Art, Prospects and Challenges. Cosmetics 2020, 7, 85. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Qin, L.; Ge, Y. Enhancing Antioxidant Performance of Lignin by Enzymatic Treatment with Laccase. ACS Sustain. Chem. Eng. 2018, 6, 2591–2595. [Google Scholar] [CrossRef]
- Renders, T.; Van den Bossche, G.; Vangeel, T.; Van Aelst, K.; Sels, B. Reductive Catalytic Fractionation: State of the Art of the Lignin-First Biorefinery. Curr. Opin. Biotechnol. 2019, 56, 193–201. [Google Scholar] [CrossRef]
- Schutyser, W.; Van Den Bosch, S.; Renders, T.; De Boe, T.; Koelewijn, S.F.; Dewaele, A.; Ennaert, T.; Verkinderen, O.; Goderis, B.; Courtin, C.M.; et al. Influence of Bio-Based Solvents on the Catalytic Reductive Fractionation of Birch Wood. Green Chem. 2015, 17, 5035–5045. [Google Scholar] [CrossRef]
- Van Den Bosch, S.; Schutyser, W.; Koelewijn, S.F.; Renders, T.; Courtin, C.M.; Sels, B.F. Tuning the Lignin Oil OH-Content with Ru and Pd Catalysts during Lignin Hydrogenolysis on Birch Wood. Chem. Commun. 2015, 51, 13158–13161. [Google Scholar] [CrossRef] [Green Version]
- Van Aelst, K.; Van Sinay, E.; Vangeel, T.; Cooreman, E.; Van Den Bossche, G.; Renders, T.; Van Aelst, J.; Van Den Bosch, S.; Sels, B.F. Reductive Catalytic Fractionation of Pine Wood: Elucidating and Quantifying the Molecular Structures in the Lignin Oil. Chem. Sci. 2020, 11, 11498–11508. [Google Scholar] [CrossRef]
- Weißbach, U.; Dabral, S.; Konnert, L.; Bolm, C.; Hernández, J.G. Selective Enzymatic Esterification of Lignin Model Compounds in the Ball Mill. Beilstein J. Org. Chem. 2017, 13, 1788–1795. [Google Scholar] [CrossRef] [Green Version]
- Torres de Pinedo, A.; Peñalver, P.; Pérez-Victoria, I.; Rondón, D.; Morales, J.C. Synthesis of New Phenolic Fatty Acid Esters and Their Evaluation as Lipophilic Antioxidants in an Oil Matrix. Food Chem. 2007, 105, 657–665. [Google Scholar] [CrossRef]
- Torres De Pinedo, A.; Peñalver, P.; Rondón, D.; Morales, J.C. Efficient Lipase-Catalyzed Synthesis of New Lipid Antioxidants Based on a Catechol Structure. Tetrahedron 2005, 61, 7654–7660. [Google Scholar] [CrossRef]
- Torres de Pinedo, A.; Peñalver, P.; Morales, J.C. Synthesis and Evaluation of New Phenolic-Based Antioxidants: Structure-Activity Relationship. Food Chem. 2007, 103, 55–61. [Google Scholar] [CrossRef]
- Sharma, O.P.; Bhat, T.K. DPPH Antioxidant Assay Revisited. Food Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Gracia-Vitoria, J.; Rubens, M.; Feghali, E.; Adriaensens, P.; Vanbroekhoven, K.; Vendamme, R. Low-Field Benchtop versus High-Field NMR for Routine 31P Analysis of Lignin, a Comparative Study. Ind. Crops Prod. 2022, 176, 114405. [Google Scholar] [CrossRef]
- Kirk, O.; Björkling, F.; Godtfredsen, S.E.; Larsen, T.O. Fatty Acid Specificity in Lipase-Catalyzed Synthesis of Glucoside Esters. Biocatal. Biotransformation 1992, 6, 127–134. [Google Scholar] [CrossRef]
- Tofani, D.; Balducci, V.; Gasperi, T.; Incerpi, S.; Gambacorta, A. Fatty Acid Hydroxytyrosyl Esters: Structure/Antioxidant Activity Relationship by Abts and in Cell-Culture DCF Assays. J. Agric. Food Chem. 2010, 58, 5292–5299. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A.; Cimpeanu, C.; Predoi, G. Antioxidant Capacity Determination in Plants and Plant-Derived Products: A Review. Oxid. Med. Cell. Longev. 2016, 2016, 9130976. [Google Scholar] [CrossRef] [Green Version]
- Widsten, P.; Liitiä, T.; Immonen, K.; Borrega, M.; Jääskeläinen, A.-S.; Wikberg, H.; Ohra-aho, T.; Tamminen, T. Potential of Lignin as Antioxidant for Thermoplastics and Other Materials. Lignin 2020, 1, 11–19. [Google Scholar]
- Barclay, L.R.C.; Xi, F.; Norris, J.Q. Antioxidant Properties of Phenolic Lignin Model Compounds. J. Wood Chem. Technol. 1997, 17, 73–90. [Google Scholar] [CrossRef]
- Jessop, P.G. Searching for Green Solvents. Green Chem. 2011, 13, 1391–1398. [Google Scholar] [CrossRef]
- Buisman, G.J.H.; Van Helteren, C.T.W.; Kramer, G.F.H.; Veldsink, J.W.; Derksen, J.T.P.; Cuperus, F.P. Enzymatic Esterifications of Functionalized Phenols for the Synthesis of Lipophilic Antioxidants. Biotechnol. Lett. 1998, 20, 131–136. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Prinsen, P.; Martínez, Á.T.; Ralph, J.; Gutiérrez, A. Structural Characterization of Wheat Straw Lignin as Revealed by Analytical Pyrolysis, 2D-NMR, and Reductive Cleavage Methods. J. Agric. Food Chem. 2012, 60, 5922–5935. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Crestini, C.; Ben, H.; Hao, N.; Pu, Y.; Ragauskas, A.J.; Argyropoulos, D.S. Determination of Hydroxyl Groups in Biorefinery Resources via Quantitative 31P NMR Spectroscopy. Nat. Protoc. 2019, 14, 2627–2647. [Google Scholar] [CrossRef]
- Hilgers, R.; Vincken, J.P.; Kabel, M.A. Facile Enzymatic Cγ-Acylation of Lignin Model Compounds. Catal. Commun. 2020, 136, 105919. [Google Scholar] [CrossRef]
- Hulin, L.; Husson, E.; Bonnet, J.P.; Stevanovic, T.; Sarazin, C. Enzymatic Transesterification of Kraft Lignin with Long Acyl Chains in Ionic Liquids. Molecules 2015, 20, 16334–16353. [Google Scholar] [CrossRef] [Green Version]
- Vibha, K.; Negi, S. Enzymatic Plasticising of Lignin and Styrene with Adipic Acid to Synthesize a Biopolymer with High Antioxidant and Thermostability. Polym. Degrad. Stab. 2020, 174, 109081. [Google Scholar] [CrossRef]
- Ugartondo, V.; Mitjans, M.; Vinardell, M.P. Comparative Antioxidant and Cytotoxic Effects of Lignins from Different Sources. Bioresour. Technol. 2008, 99, 6683–6687. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Reaction | DCA-C8 Yield (mol%) (Hours) |
|---|---|
| a In solvent + desiccants | |
| C8/DCA = 1.25 | 64.9% (3 h), 83.0% (24 h) |
| C8/DCA = 1.5 | 71.9% (3 h), 84.6% (24 h) |
| C8/DCA = 2 | 78.7% (3 h), 97.1% (24 h) |
| b Solvent-free + desiccants | |
| C8/DCA = 2.5 | 88.5% (3 h), 86.2% (24 h) |
| C8/DCA = 5 | 93.6% (3 h), 89.3% (24 h) |
| C8/DCA = 10 | 97.5% (3 h), 95.1% (24 h) |
| No lipase, C8/DCA = 2.5 | 20.1% (24 h) |
| No lipase, C8/DCA = 5 | 28.3% (24 h) |
| No lipase, C8/DCA = 10 | 33.6% (24 h) |
| c Solvent-free + vacuum | |
| C8/DCA = 10 | 90.3% (3 h), 89.5% (24 h) |
| No lipase, C8/DCA = 10 | 17.3% (24 h) |
| Aliphatic OH | Phenolic OH | COOH | Mw | Mn | Ɖ | |
|---|---|---|---|---|---|---|
| Lignin | (mmol/g) | (mmol/g) | (mmol/g) | (Da) | (Da) | Mw/Mn |
| Beechwood | 2.32 | 4.09 | 0.10 | 2210 | 1010 | 2.19 |
| Beechwood_HL | 2.32 | 5.01 | 0.09 | 1590 | 874 | 1.82 |
| Wheat straw | 2.31 | 1.51 | 0.71 | 3200 | 1050 | 3.0 |
| Wheat straw_HL | 1.94 | 2.87 | 0.31 | 1510 | 744 | 2.03 |
| Wheat straw_HL_Et2O_soluble | 1.41 | 2.28 | 0.23 | 593 | 438 | 1.35 |
| Wheat straw_HL_Et2O_residue | 2.31 | 2.93 | 0.20 | 1920 | 1000 | 1.92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Garcia, M.; Gracia-Vitoria, J.; Vanbroekhoven, K.; Dejonghe, W.; Satyawali, Y. Selective Enzymatic Esterification of Lignin-Derived Phenolics for the Synthesis of Lipophilic Antioxidants. Antioxidants 2023, 12, 657. https://doi.org/10.3390/antiox12030657
Martinez-Garcia M, Gracia-Vitoria J, Vanbroekhoven K, Dejonghe W, Satyawali Y. Selective Enzymatic Esterification of Lignin-Derived Phenolics for the Synthesis of Lipophilic Antioxidants. Antioxidants. 2023; 12(3):657. https://doi.org/10.3390/antiox12030657
Chicago/Turabian StyleMartinez-Garcia, Marta, Jaime Gracia-Vitoria, Karolien Vanbroekhoven, Winnie Dejonghe, and Yamini Satyawali. 2023. "Selective Enzymatic Esterification of Lignin-Derived Phenolics for the Synthesis of Lipophilic Antioxidants" Antioxidants 12, no. 3: 657. https://doi.org/10.3390/antiox12030657