
Auranofin Synergizes with the PARP Inhibitor Olaparib to Induce ROS-Mediated Cell Death in Mutant p53 Cancers

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Cell Lines
2.2. Murine Cell Line
2.3. Sulforhodamine B Cytotoxicity Assay
2.4. Western Blotting
2.5. Oxidative Stress Detection
2.6. 2D Spark Cyto Cytotoxicity Assay
2.7. IncuCyte ZOOM Analysis of Cell Death Pathways
2.8. Lipid Peroxidation
2.9. Immunofluorescent Labeling
2.10. Generation and Culturing of 344SQ Spheroids
2.11. 3D Multi-Spheroid Cytotoxicity Assay
2.12. Mice
2.13. Tumor Kinetics and Survival
2.14. RNA Sequencing of Mice Tumors
2.15. Statistics
3. Results
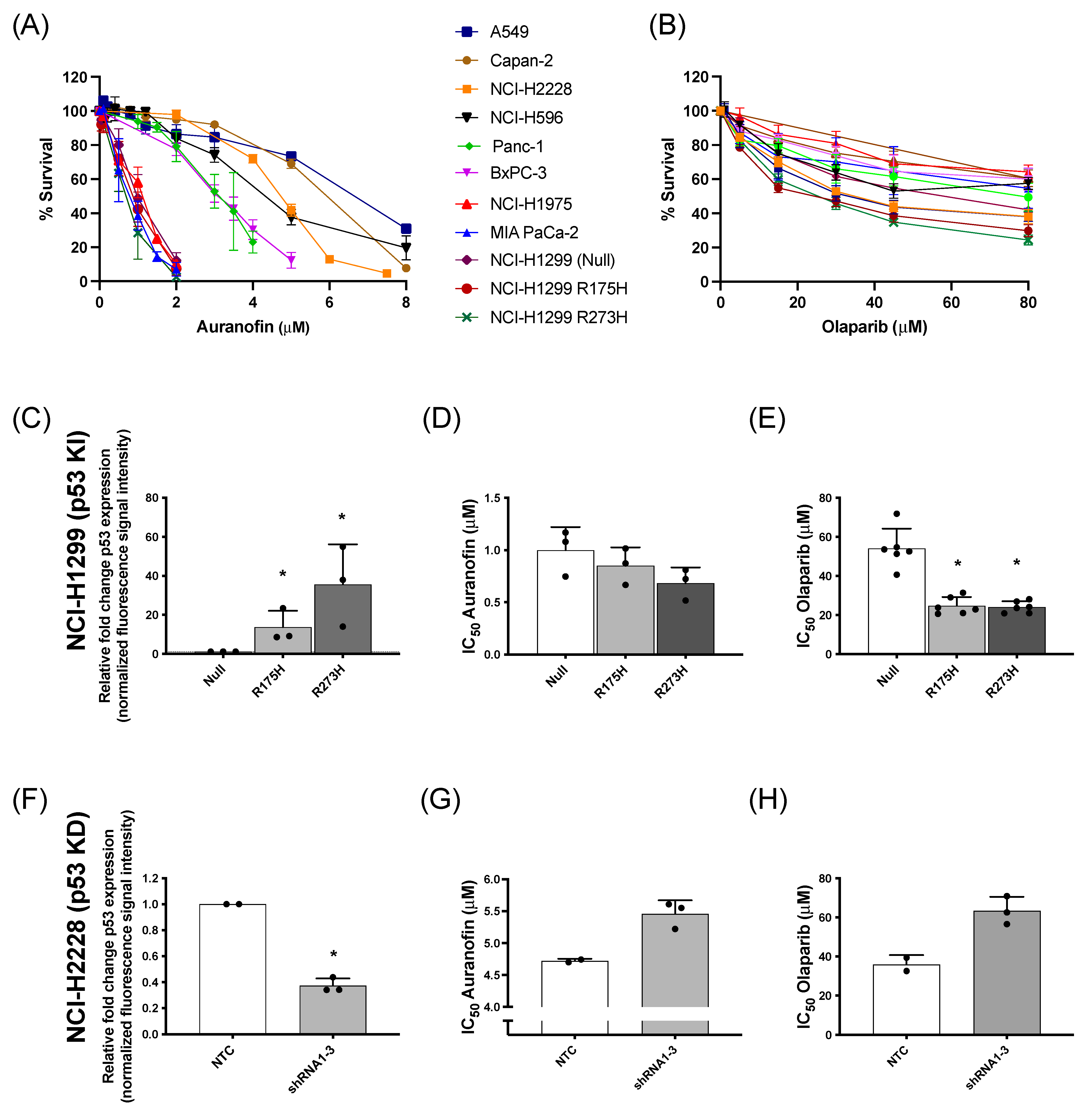
3.1. Effect of AF and Olaparib Monotherapy on Mutant p53 NSCLC and PDAC Cell Panel
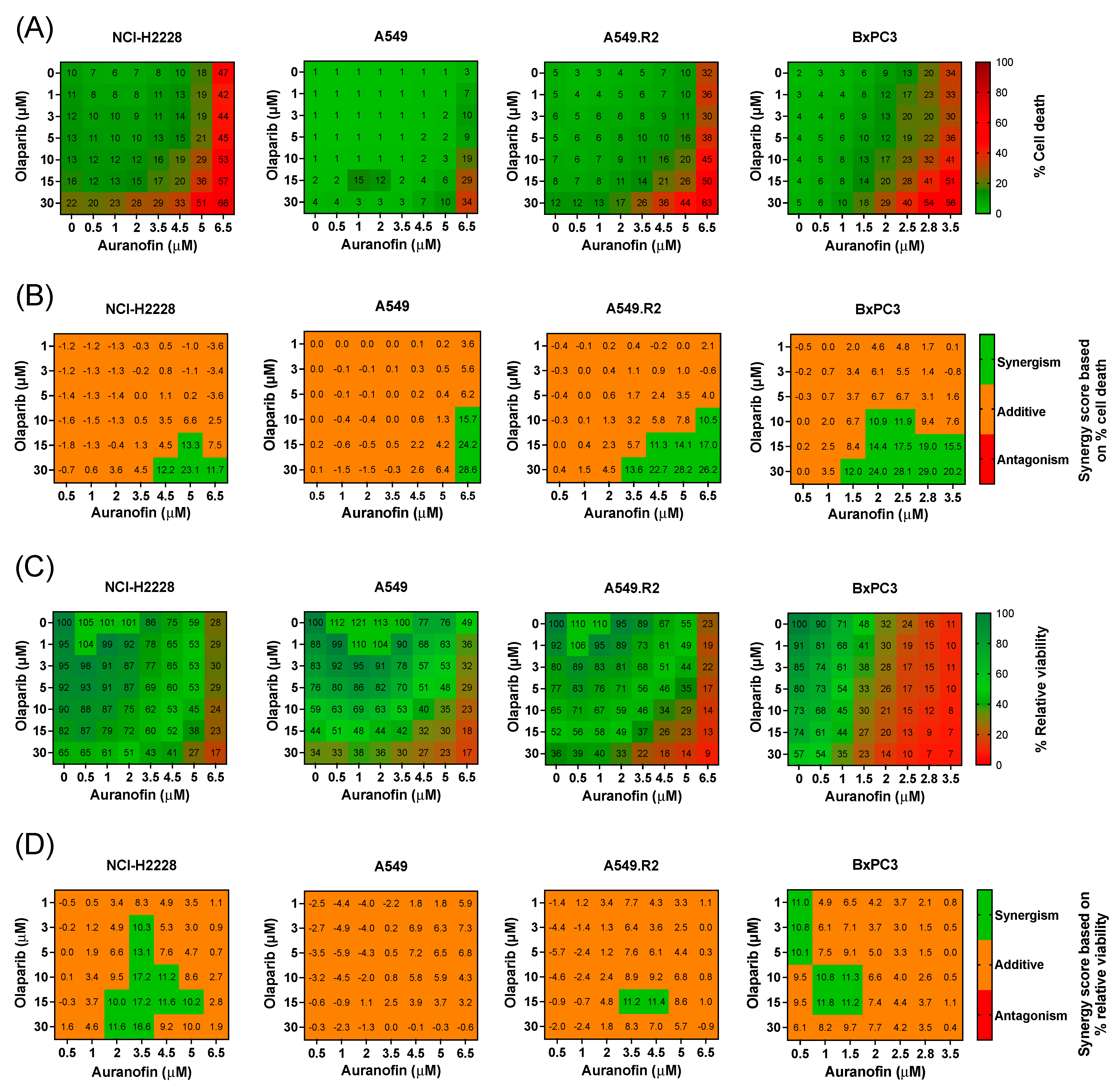
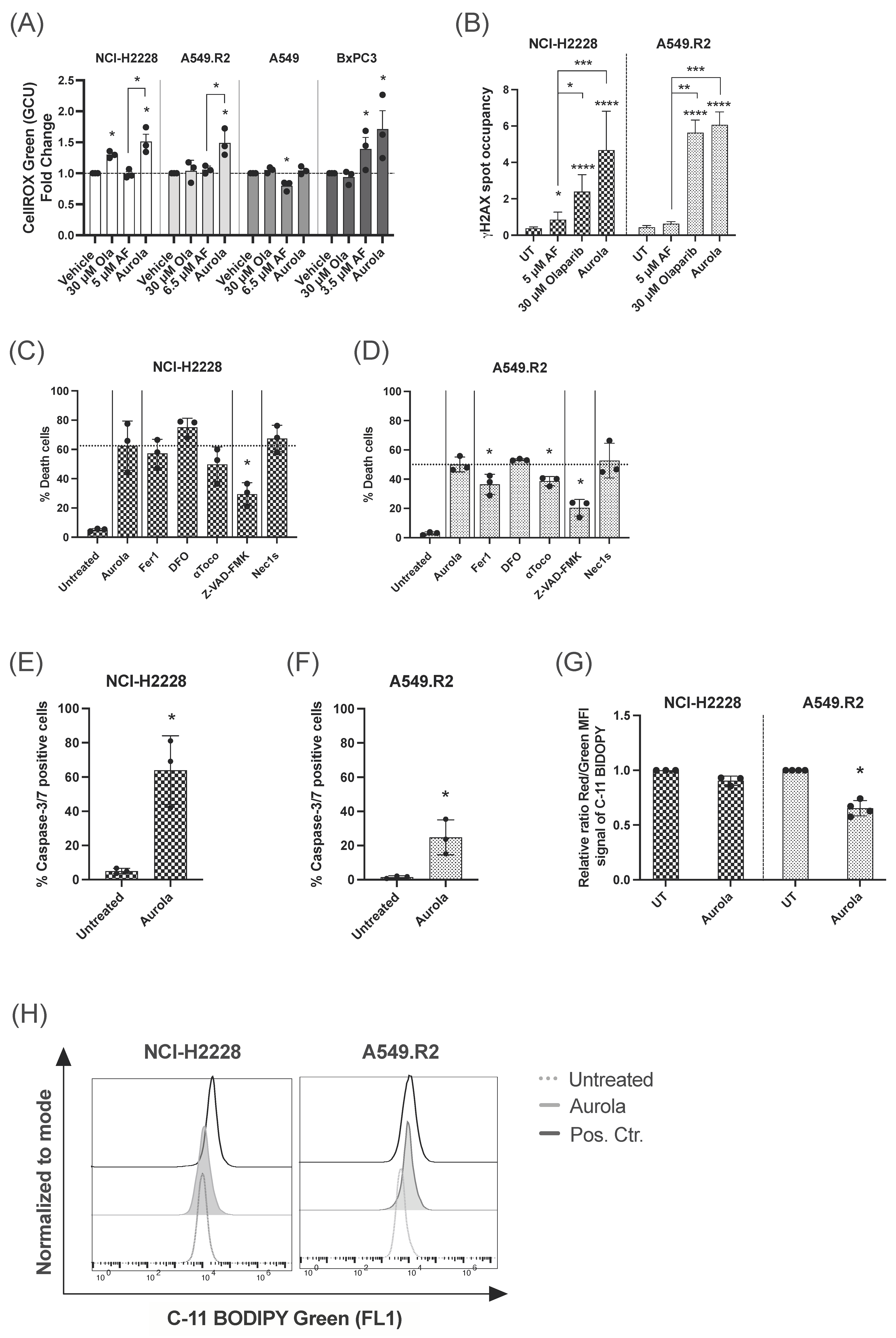
3.2. Aurola Treatment Leads to Synergistic ROS-Dependent Cytotoxicity in Mutant p53 NSCLC and PDAC Cell Lines with Low Mutant p53 Protein Levels
3.3. Aurola Treatment Results in Apoptotic and Ferroptotic Cell Death Characteristics in Mutant p53 NSCLC Cells
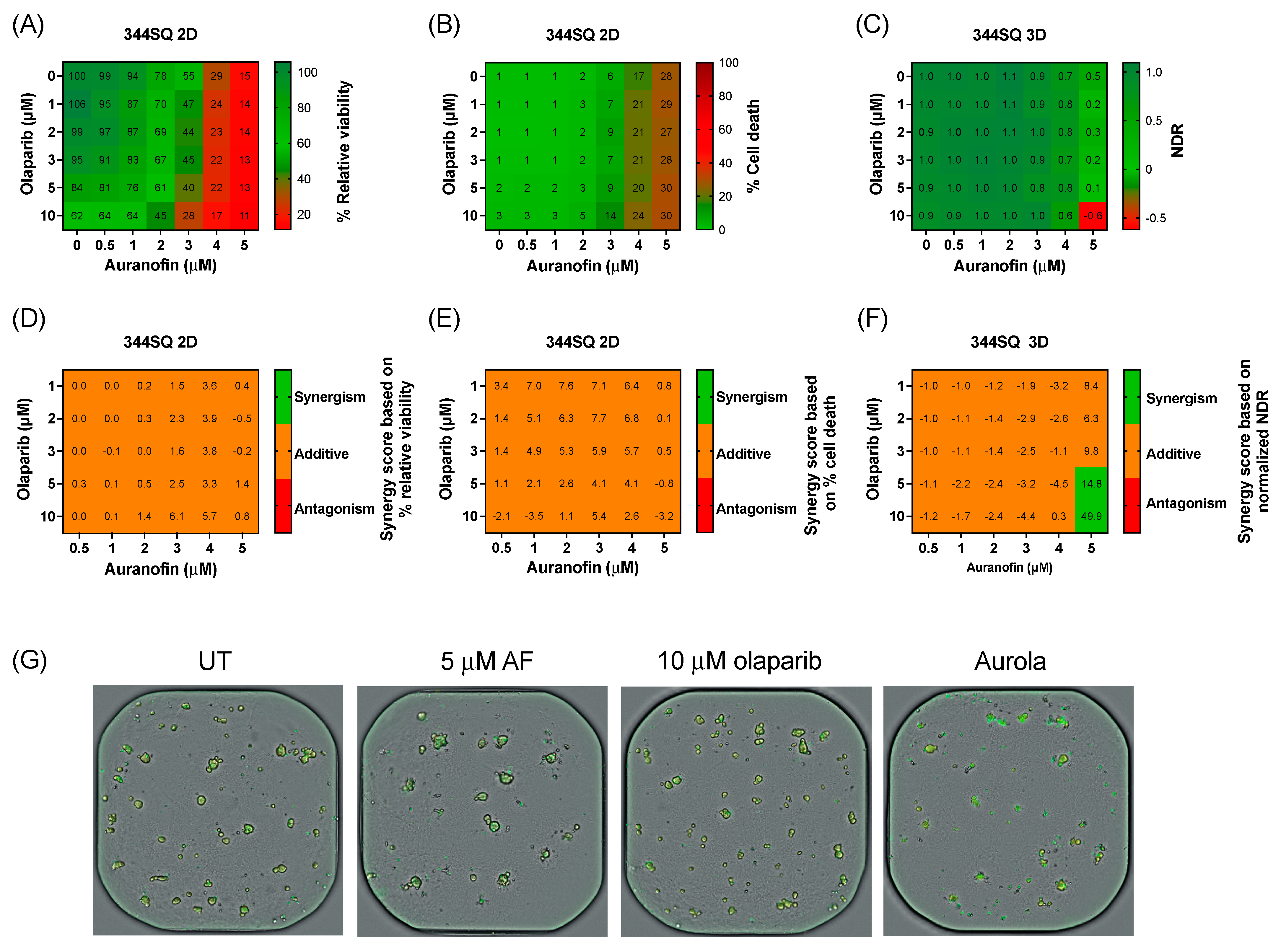
3.4. Clinically Relevant Concentrations of Olaparib Induce a Synergistic Effect in Combination with AF in 3D Cultures of the Murine Lung Adenocarcinoma Cell Line 344SQ
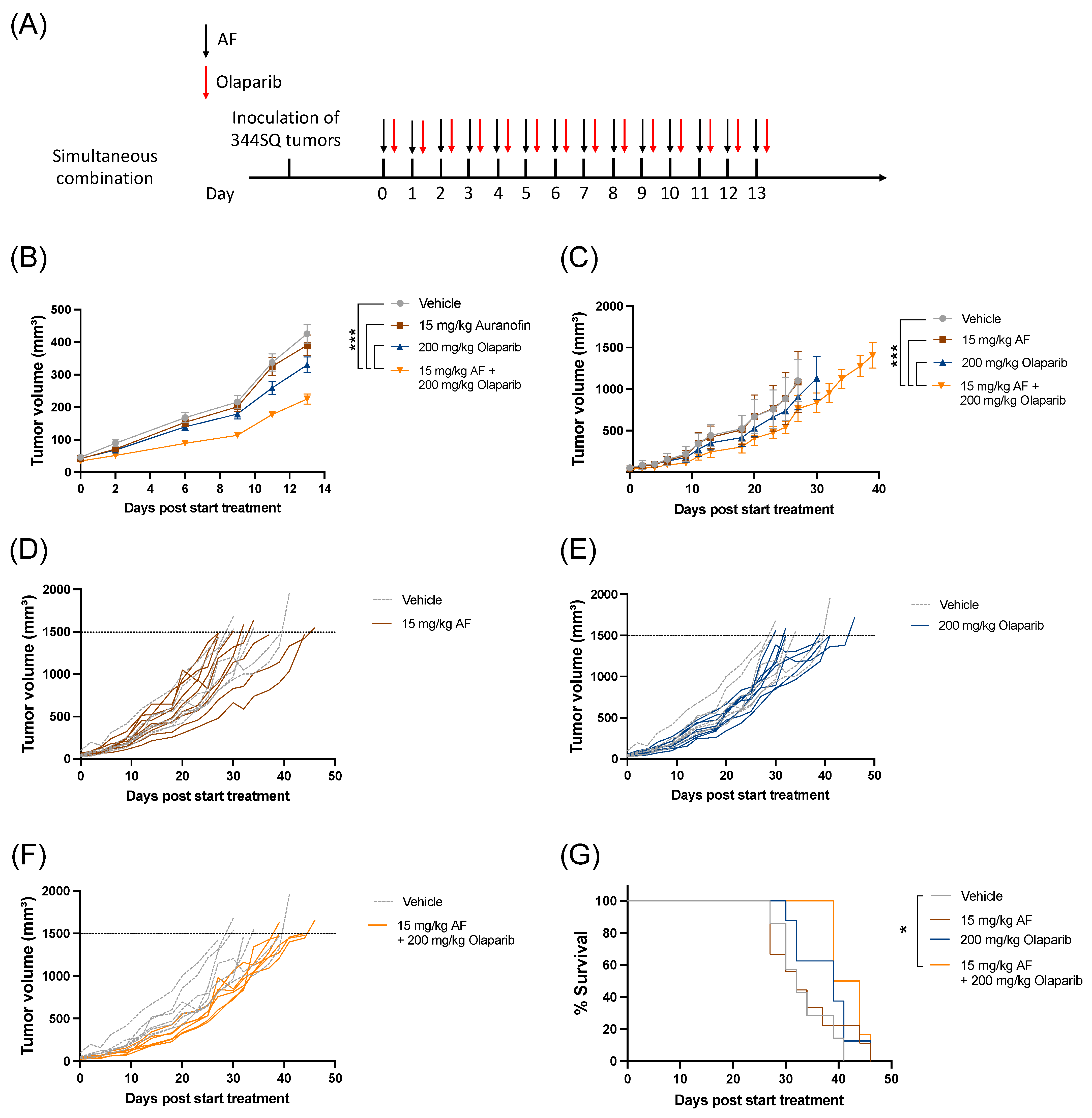
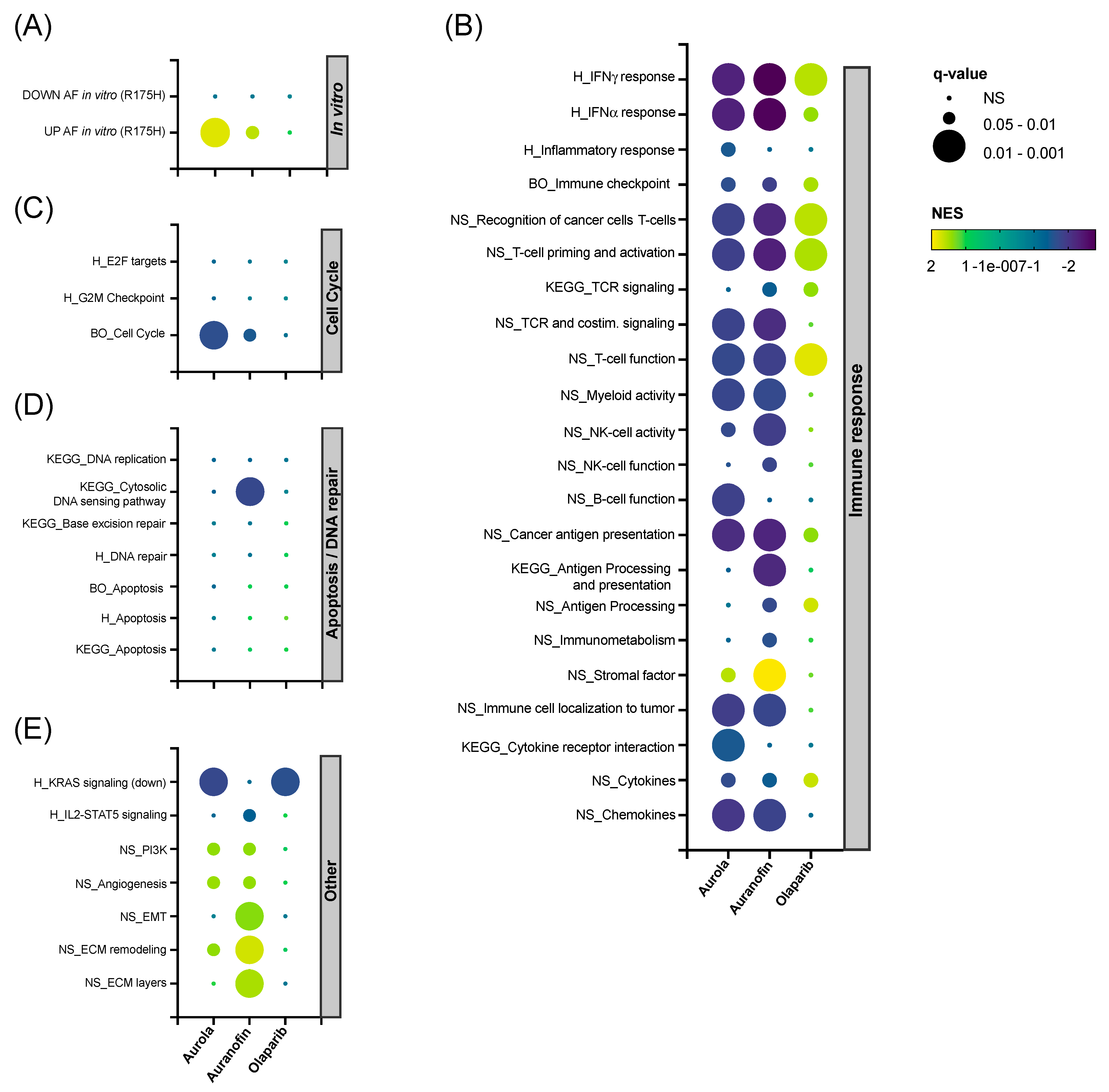
3.5. Aurola Delays Tumor Growth and Downregulates Immune-Related and Cell Cycle-Related Gene Sets in 344SQ Mice Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Uzunparmak, B.; Sahin, I.H. Pancreatic cancer microenvironment: A current dilemma. Clin. Transl. Med. 2019, 8, 2. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef]
- Deben, C.; Bossche, J.V.D.; Van Der Steen, N.; Lardon, F.; Wouters, A.; de Beeck, K.O.; Hermans, C.; Jacobs, J.; Peeters, M.; Van Camp, G.; et al. Deep sequencing of the TP53 gene reveals a potential risk allele for non–small cell lung cancer and supports the negative prognostic value of TP53 variants. Tumor Biol. 2017, 39, 1010428317694327. [Google Scholar] [CrossRef]
- Greulich, H. The Genomics of Lung Adenocarcinoma: Opportunities for Targeted Therapies. Genes Cancer 2010, 1, 1200–1210. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug. Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxidative Med. Cell. Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular Pathways: Reactive Oxygen Species Homeostasis in Cancer Cells and Implications for Cancer Therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R&D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Pantziarka, P.; Verbaanderd, C.; Sukhatme, V.; Capistrano, I.R.; Crispino, S.; Gyawali, B.; Rooman, I.; Van Nuffel, A.; Meheus, L.; Sukhatme, V.P.; et al. ReDO_DB: The repurposing drugs in oncology database. Ecancermedicalscience 2018, 12, 886. [Google Scholar] [CrossRef]
- Boullosa, L.F.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol. 2021, 42, 101949. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Hwang-Bo, H.; Jeong, J.-W.; Han, M.H.; Park, C.; Hong, S.-H.; Kim, G.-Y.; Moon, S.-K.; Cheong, J.; Kim, W.-J.; Yoo, Y.H.; et al. Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis in hepatocellular carcinoma Hep3B cells by generation of reactive oxygen species. Gen. Physiol. Biophys. 2017, 36, 117–128. [Google Scholar] [CrossRef]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Das Gupta, S.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin induces apoptosis by ROS-mediated ER stress and mitochondrial dysfunction and displayed synergistic lethality with piperlongumine in gastric cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef]
- Kim, I.S.; Jin, J.Y.; Lee, I.H.; Park, S.J. Auranofin induces apoptosis and when combined with retinoic acid enhances differentiation of acute promyelocytic leukaemia cells in vitro. Br. J. Pharmacol. 2004, 142, 749–755. [Google Scholar] [CrossRef]
- Karsa, M.; Kosciolek, A.; Bongers, A.; Mariana, A.; Failes, T.; Gifford, A.J.; Kees, U.R.; Cheung, L.C.; Kotecha, R.S.; Arndt, G.M.; et al. Exploiting the reactive oxygen species imbalance in high-risk paediatric acute lymphoblastic leukaemia through auranofin. Br. J. Cancer 2021, 125, 55–64. [Google Scholar] [CrossRef]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2015, 7, 3548–3558. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.; Song, L.; Qu, M.; Zou, Z. Synergistic lethality between PARP-trapping and alantolactone-induced oxidative DNA damage in homologous recombination-proficient cancer cells. Oncogene 2020, 39, 2905–2920. [Google Scholar] [CrossRef]
- Kim, D.; Nam, H.J. PARP Inhibitors: Clinical Limitations and Recent Attempts to Overcome Them. Int. J. Mol. Sci. 2022, 23, 8412. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Deben, C.; Lardon, F.; Wouters, A.; de Beeck, K.O.; Van den Bossche, J.; Jacobs, J.; Van Der Steen, N.; Peeters, M.; Rolfo, C.; Deschoolmeester, V.; et al. APR-246 (PRIMA-1 MET) strongly synergizes with AZD2281 (olaparib) induced PARP inhibition to induce apoptosis in non-small cell lung cancer cell lines. Cancer Lett. 2016, 375, 313–322. [Google Scholar] [CrossRef]
- Yin, Z.-X.; Hang, W.; Liu, G.; Wang, Y.-S.; Shen, X.-F.; Sun, Q.-H.; Li, D.-D.; Jian, Y.-P.; Zhang, Y.-H.; Quan, C.-S.; et al. PARP-1 inhibitors sensitize HNSCC cells to APR-246 by inactivation of thioredoxin reductase 1 (TrxR1) and promotion of ROS accumulation. Oncotarget 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Liu, Q.; Gheorghiu, L.; Drumm, M.; Clayman, R.; Eidelman, A.; Wszolek, M.F.; Olumi, A.; Feldman, A.; Wang, M.; Marcar, L.; et al. PARP-1 inhibition with or without ionizing radiation confers reactive oxygen species-mediated cytotoxicity preferentially to cancer cells with mutant TP53. Oncogene 2018, 37, 2793–2805. [Google Scholar] [CrossRef]
- Huang, X.; Motea, E.A.; Moore, Z.R.; Yao, J.; Dong, Y.; Chakrabarti, G.; Kilgore, J.A.; Silvers, M.A.; Patidar, P.L.; Cholka, A.; et al. Leveraging an NQO1 Bioactivatable Drug for Tumor-Selective Use of Poly(ADP-ribose) Polymerase Inhibitors. Cancer Cell 2016, 30, 940–952. [Google Scholar] [CrossRef]
- Marcar, L.; Bardhan, K.; Gheorghiu, L.; Dinkelborg, P.; Pfäffle, H.; Liu, Q.; Wang, M.; Piotrowska, Z.; Sequist, L.V.; Borgmann, K.; et al. Acquired Resistance of EGFR-Mutated Lung Cancer to Tyrosine Kinase Inhibitor Treatment Promotes PARP Inhibitor Sensitivity. Cell Rep. 2019, 27, 3422–3432.e4. [Google Scholar] [CrossRef]
- Blandino, G.; Levine, A.J.; Oren, M. Mutant p53 gain of function: Differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999, 18, 477–485. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Bossche, J.V.D.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers 2018, 10, 126. [Google Scholar] [CrossRef]
- Deben, C.; Boullosa, L.F.; Domen, A.; Wouters, A.; Cuypers, B.; Laukens, K.; Lardon, F.; Pauwels, P. Characterization of acquired nutlin-3 resistant non-small cell lung cancer cells. Cancer Drug Resist. 2021, 4, 233–243. [Google Scholar] [CrossRef]
- Pauwels, B.; Korst, A.E.C.; De Pooter, C.M.J.; Pattyn, G.G.O.; Lambrechts, H.A.J.; Baay, M.F.D.; Lardon, F.; Vermorken, J.B. Comparison of the sulforhodamine B assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother. Pharmacol. 2003, 51, 221–226. [Google Scholar] [CrossRef]
- Ianevski, A.; Giri, A.K.; Aittokallio, T. SynergyFinder 3.0: An interactive analysis and consensus interpretation of multi-drug synergies across multiple samples. Nucleic Acids Res. 2022, 50, W739–W743. [Google Scholar] [CrossRef]
- Fiji. Available online: https://fiji.sc (accessed on 12 December 2022).
- De Vos, W.H.; Van Neste, L.; Dieriks, B.; Joss, G.H.; Van Oostveldt, P. High content image cytometry in the context of subnuclear organization. Cytom. A 2010, 77, 64–75. [Google Scholar] [CrossRef]
- Schmidt, U.; Weigert, M.; Broaddus, C.; Myers, G. (Eds.) Cell Detection with Star-Convex Polygons. In Proceedings of the International Conference on Medical Image Computing and Computer-Assisted Intervention, Granada, Spain, 16–20 September 2018; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Deben, C.; De La Hoz, E.C.; Le Compte, M.; Van Schil, P.; Hendriks, J.M.; Lauwers, P.; Yogeswaran, S.K.; Lardon, F.; Pauwels, P.; Van Laere, S.; et al. OrBITS: Label-free and time-lapse monitoring of patient derived organoids for advanced drug screening. Cell. Oncol. 2022, 1–16. [Google Scholar] [CrossRef]
- Gupta, A.; Gautam, P.; Wennerberg, K.; Aittokallio, T. A normalized drug response metric improves accuracy and consistency of anticancer drug sensitivity quantification in cell-based screening. Commun. Biol. 2020, 3, 42. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Hermans, C.; Lau, H.W.; Merlin, C.; Marcq, E.; Takhsha, F.S.; Martinet, W.; De Meyer, G.R.; Lardon, F.; et al. Optimization of the Solvent and In Vivo Administration Route of Auranofin in a Syngeneic Non-Small Cell Lung Cancer and Glioblastoma Mouse Model. Pharmaceutics 2022, 14, 2761. [Google Scholar] [CrossRef]
- Akhmedov, M.; Martinelli, A.; Geiger, R.; Kwee, I. Omics Playground: A comprehensive self-service platform for visualization, analytics and exploration of Big Omics Data. NAR Genom. Bioinform. 2019, 2, lqz019. [Google Scholar] [CrossRef]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Fisher, S.A.; Peddle-McIntyre, C.J.; Burton, K.; Newton, R.U.; Marcq, E.; Lake, R.A.; Nowak, A.K. Voluntary exercise in mesothelioma: Effects on tumour growth and treatment response in a murine model. BMC Res. Notes 2020, 13, 435. [Google Scholar] [CrossRef]
- AbdulSalam, S.F.; Thowfeik, F.S.; Merino, E.J. Excessive Reactive Oxygen Species and Exotic DNA Lesions as an Exploitable Liability. Biochemistry 2016, 55, 5341–5352. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, e881. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef]
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef]
- Zweemer, R.P.; Shaw, P.A.; Verheijen, R.M.; Ryan, A.; Berchuck, A.; Ponder, B.A.; Risch, H.; McLaughlin, J.R.; Narod, S.A.; Menko, F.H.; et al. Accumulation of p53 protein is frequent in ovarian cancers associated with BRCA1 and BRCA2 germline mutations. J. Clin. Pathol. 1999, 52, 372–375. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, J.; Zhou, J.; Liu, H.; Xu, C. Olaparib induced senescence under P16 or P53 dependent manner in ovarian cancer. J. Gynecol. Oncol. 2019, 30, e26. [Google Scholar] [CrossRef]
- US National Library of Medicine. Available online: https://clinicaltrials.gov (accessed on 21 January 2022).
- Chang, M.; Wang, H.; Niu, J.; Song, Y.; Zou, Z. Alkannin-Induced Oxidative DNA Damage Synergizes with PARP Inhibition to Cause Cancer-Specific Cytotoxicity. Front. Pharmacol. 2020, 11, 610205. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Freire Boullosa, L.; Quatannens, D.; De Waele, J.; Merlin, C.; Lambrechts, H.; Lau, H.W.; Hermans, C.; Lin, A.; Lardon, F.; et al. Auranofin and Cold Atmospheric Plasma Synergize to Trigger Distinct Cell Death Mechanisms and Immunogenic Responses in Glioblastoma. Cells 2021, 10, 2936. [Google Scholar] [CrossRef] [PubMed]
- Abdalbari, F.H.; Telleria, C.M. The gold complex auranofin: New perspectives for cancer therapy. Discov. Oncol. 2021, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, X.-J.; Tian, W.; Jaramillo, M.C.; Lau, A.; Zhang, D.D. Poly(ADP-ribose) polymerase-1 modulates Nrf2-dependent transcription. Free Radic. Biol. Med. 2014, 67, 69–80. [Google Scholar] [CrossRef]
- Ma, X.; Dang, C.; Min, W.; Diao, Y.; Hui, W.; Wang, X.; Dai, Z.; Wang, X.; Kang, H. Downregulation of APE1 potentiates breast cancer cells to olaparib by inhibiting PARP-1 expression. Breast Cancer Res. Treat. 2019, 176, 109–117. [Google Scholar] [CrossRef]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive Oxygen Species (ROS) Regulates Different Types of Cell Death by Acting as a Rheostat. Oxidative Med. Cell. Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Metformin Affects Olaparib Sensitivity through Induction of Apoptosis in Epithelial Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 10557. [Google Scholar] [CrossRef]
- Gaymes, T.J.; Shall, S.; MacPherson, L.J.; Twine, N.A.; Lea, N.C.; Farzaneh, F.; Mufti, G.J. Inhibitors of poly ADP-ribose polymerase (PARP) induce apoptosis of myeloid leukemic cells: Potential for therapy of myeloid leukemia and myelodysplastic syndromes. Haematologica 2009, 94, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhou, F.; Jiang, F.; Lu, H.; Wang, J.; Cheng, C. PARP inhibitor reduces proliferation and increases apoptosis in breast cancer cells. Chin. J. Cancer Res. 2014, 26, 142–147. [Google Scholar] [CrossRef]
- Hong, T.; Lei, G.; Chen, X.; Li, H.; Zhang, X.; Wu, N.; Zhao, Y.; Zhang, Y.; Wang, J. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol. 2021, 42, 101928. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. The mechanism of cancer cell death by PARP inhibitors goes beyond DNA damage alone. Int. J. Cancer 2019, 145, 2594–2596. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, W. Which concentrations are optimal for in vitro testing? EXCLI J. 2020, 19, 1172–1183. [Google Scholar] [PubMed]
- Díez-Martínez, R.; García-Fernández, E.; Manzano, M.; Martínez, Á.; Domenech, M.; Vallet-Regí, M.; García, P. Auranofin-loaded nanoparticles as a new therapeutic tool to fight streptococcal infections. Sci. Rep. 2016, 6, 19525. [Google Scholar] [CrossRef]
- Awasthi, R.; Roseblade, A.; Hansbro, P.; Rathbone, M.J.; Dua, K.; Bebawy, M. Nanoparticles in Cancer Treatment: Opportunities and Obstacles. Curr. Drug Targets 2018, 19, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Chaffman, M.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Auranofin. A preliminary review of its pharmacological properties and therapeutic use in rheumatoid arthritis. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef]
- Kim, N.-H.; Lee, M.-Y.; Park, S.-J.; Choi, J.-S.; Oh, M.-K.; Kim, I.-S. Auranofin blocks interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunology 2007, 122, 607–614. [Google Scholar] [CrossRef]
- Jeon, K.-I.; Jeong, J.-Y.; Jue, D.-M. Thiol-Reactive Metal Compounds Inhibit NF-κB Activation by Blocking IκB Kinase. J. Immunol. 2000, 164, 5981. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.C.; Sinha, D.; Shih, Y.Y.; Mittal, D.; Makhale, A.; Bain, A.L.; Nanayakarra, D.; Tonissen, K.F.; Kalimutho, M.; et al. Therapeutic cooperation between auranofin, a thioredoxin reductase inhibitor and anti-PD-L1 antibody for treatment of triple-negative breast cancer. Int. J. Cancer 2020, 146, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Konstantinopoulos, P.A. PARP inhibition and immune modulation: Scientific rationale and perspectives for the treatment of gynecologic cancers. Ther. Adv. Med. Oncol. 2020, 12, 1758835920944116. [Google Scholar] [CrossRef] [PubMed]
- Staniszewska, A.D.; Armenia, J.; King, M.; Michaloglou, C.; Reddy, A.; Singh, M.; Martin, M.S.; Prickett, L.; Wilson, Z.; Proia, T.; et al. PARP inhibition is a modulator of anti-tumor immune response in BRCA-deficient tumors. Oncoimmunology 2022, 11, 2083755. [Google Scholar] [CrossRef]
- Hatem, E.; El Banna, N.; Heneman-Masurel, A.; Baïlle, D.; Vernis, L.; Riquier, S.; Golinelli-Cohen, M.-P.; Guittet, O.; Vallières, C.; Camadro, J.-M.; et al. Novel Insights into Redox-Based Mechanisms for Auranofin-Induced Rapid Cancer Cell Death. Cancers 2022, 14, 4864. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; Hermans, C.; Merlin, C.; Lau, H.W.; Marcq, E.; Verschuuren, M.; De Vos, W.H.; Lardon, F.; et al. Auranofin Synergizes with the PARP Inhibitor Olaparib to Induce ROS-Mediated Cell Death in Mutant p53 Cancers. Antioxidants 2023, 12, 667. https://doi.org/10.3390/antiox12030667
Freire Boullosa L, Van Loenhout J, Flieswasser T, Hermans C, Merlin C, Lau HW, Marcq E, Verschuuren M, De Vos WH, Lardon F, et al. Auranofin Synergizes with the PARP Inhibitor Olaparib to Induce ROS-Mediated Cell Death in Mutant p53 Cancers. Antioxidants. 2023; 12(3):667. https://doi.org/10.3390/antiox12030667
Chicago/Turabian StyleFreire Boullosa, Laurie, Jinthe Van Loenhout, Tal Flieswasser, Christophe Hermans, Céline Merlin, Ho Wa Lau, Elly Marcq, Marlies Verschuuren, Winnok H. De Vos, Filip Lardon, and et al. 2023. "Auranofin Synergizes with the PARP Inhibitor Olaparib to Induce ROS-Mediated Cell Death in Mutant p53 Cancers" Antioxidants 12, no. 3: 667. https://doi.org/10.3390/antiox12030667