
Dimethyl Itaconate Inhibits Melanogenesis in B16F10 Cells



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemicals, Antibodies, and Plasmids
2.2. Measurement of Intracellular Melanin
2.3. Trypan Blue Exclusion Assay
2.4. RNA Extraction and Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.5. Western Blot Analysis
2.6. Generation of Stable Cells by Lentiviral Transduction
2.7. Firefly Luciferase Assay
2.8. Measurement of α-Melanocyte Stimulating Hormone (α-MSH)
2.9. Knocking Down Mitf and Nrf2 mRNAs by Lentiviral Transduction
2.10. Statistics
3. Results
3.1. Dimethyl Itaconate (DMI) Inhibits the Production of Melanin in B16F10 and SK-MEL-3 Cells
3.2. DMI Inhibits TYR, TRP-1, and TRP-2 in B16F10 Cells
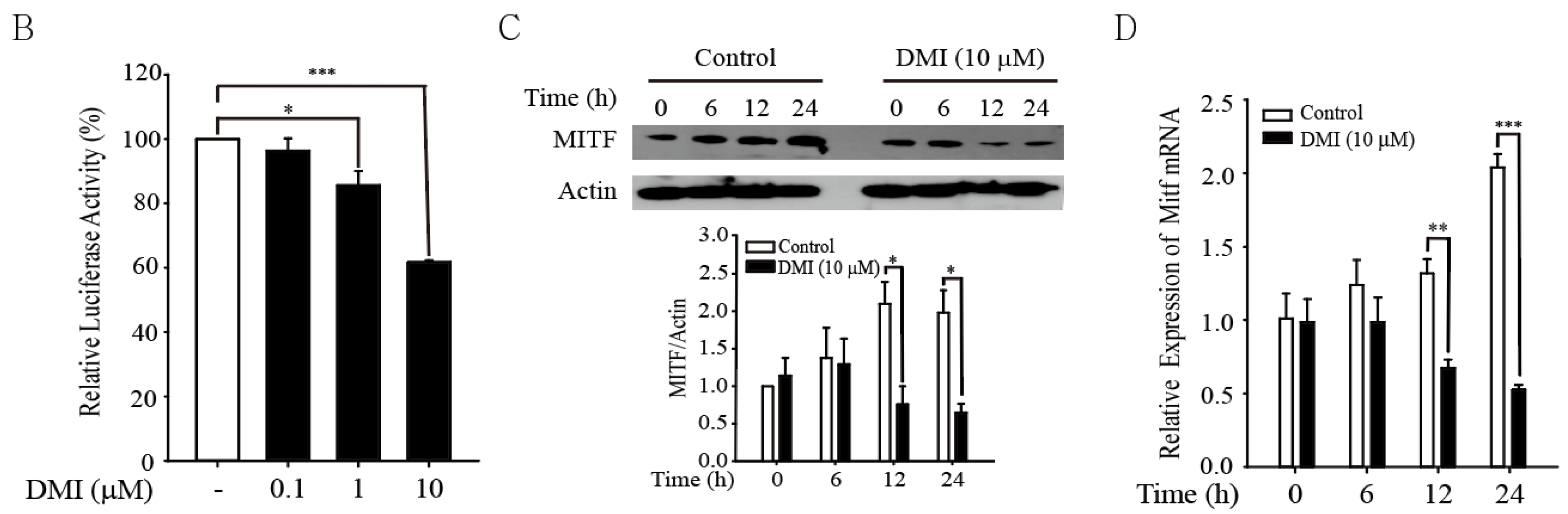
3.3. DMI Inhibits MITF in B16F10 Cells
3.4. DMI Inhibits the α-MSH/MC1R-ERK1/2-MITF Axis in B16F10 Cells
3.5. The α,β-Unsaturated Carbonyl Moiety in DMI Is Required to Inhibit Melanogenesis in B16F10 Cells
3.6. NRF2 Is Required for the Inhibition of Melanogenesis by DMI in B16F10 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moreiras, H.; Seabra, M.C.; Barral, D.C. Melanin Transfer in the Epidermis: The Pursuit of Skin Pigmentation Control Mechanisms. Int. J. Mol. Sci. 2021, 22, 4466. [Google Scholar] [CrossRef]
- Boissy, R.E. Melanosome transfer to and translocation in the keratinocyte. Exp. Dermatol. 2003, 12, 5–12. [Google Scholar] [CrossRef]
- Serre, C.; Busuttil, V.; Botto, J.M. Intrinsic and extrinsic regulation of human skin melanogenesis and pigmentation. Int. J. Cosmet. Sci. 2018, 40, 328–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.A.; Park, N.H.; Na, Y.J.; Lee, H.K.; Lee, J.H.; Kim, Y.J.; Lee, C.S. Coumestrol Down-Regulates Melanin Production in Melan-a Murine Melanocytes through Degradation of Tyrosinase. Biol. Pharm. Bull. 2017, 40, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Weng, Q.Y.; Fisher, D.E. UV signaling pathways within the skin. J. Investig. Dermatol. 2014, 134, 2080–2085. [Google Scholar] [CrossRef] [Green Version]
- Sander, M.; Sander, M.; Burbidge, T.; Beecker, J. The efficacy and safety of sunscreen use for the prevention of skin cancer. CMAJ 2020, 192, E1802–E1808. [Google Scholar] [CrossRef] [PubMed]
- Gillbro, J.M.; Olsson, M.J. The melanogenesis and mechanisms of skin-lightening agents—Existing and new approaches. Int. J. Cosmet. Sci. 2011, 33, 210–221. [Google Scholar] [CrossRef]
- Qian, W.; Liu, W.; Zhu, D.; Cao, Y.; Tang, A.; Gong, G.; Su, H. Natural skin-whitening compounds for the treatment of melanogenesis (Review). Exp. Ther. Med. 2020, 20, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; O’Neill, L.A.J. Krebs Cycle Reborn in Macrophage Immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Deng, M.; Scott, M.J.; Fu, G.; Loughran, P.A.; Lei, Z.; Li, S.; Sun, P.; Yang, C.; Li, W.; et al. Immune-Responsive Gene 1/Itaconate Activates Nuclear Factor Erythroid 2-Related Factor 2 in Hepatocytes to Protect against Liver Ischemia-Reperfusion Injury. Hepatology 2020, 72, 1394–1411. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Ren, J.; Gao, D.S.; Dai, Y.; Yu, L. The Emerging Application of Itaconate: Promising Molecular Targets and Therapeutic Opportunities. Front. Chem. 2021, 9, 669308. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Campanello, G.C.; Flicker, D.; Grabarek, Z.; Hu, J.; Luo, C.; Banerjee, R.; Mootha, V.K. The Human Knockout Gene CLYBL Connects Itaconate to Vitamin B12. Cell 2017, 171, 771–782.e11. [Google Scholar] [CrossRef]
- Okabe, M.; Lies, D.; Kanamasa, S.; Park, E.Y. Biotechnological production of itaconic acid and its biosynthesis in Aspergillus terreus. Appl. Microbiol. Biotechnol. 2009, 84, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordes, T.; Michelucci, A.; Hiller, K. Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu. Rev. Nutr. 2015, 35, 451–473. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Yang, J.Y.; Jeon, B.Y.; Yoon, Y.J.; Cho, S.N.; Kang, Y.H.; Ryu, D.H.; Hwang, G.S. (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J. Proteome Res. 2011, 10, 2238–2247. [Google Scholar] [CrossRef]
- Strelko, C.L.; Lu, W.; Dufort, F.J.; Seyfried, T.N.; Chiles, T.C.; Rabinowitz, J.D.; Roberts, M.F. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 2011, 133, 16386–16389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A Metabolite Regulates Inflammation Response and Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 5404780. [Google Scholar] [CrossRef] [PubMed]
- Luan, H.H.; Medzhitov, R. Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab. 2016, 24, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Mailar, K.; Yoo, O.K.; Choi, W.J.; Keum, Y.S. Marliolide inhibits skin carcinogenesis by activating NRF2/ARE to induce heme oxygenase-1. Eur. J. Med. Chem. 2018, 150, 113–126. [Google Scholar] [CrossRef]
- Hoang, H.N.; Hill, T.A.; Fairlie, D.P. Connecting Hydrophobic Surfaces in Cyclic Peptides Increases Membrane Permeability. Angew. Chem. Int. Ed. 2021, 60, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Koo, J.H.; Song, Y.G.; Kwon, K.B.; Lee, J.H.; Sohn, H.S.; Park, B.H.; Jhee, E.C.; Park, J.W. Stimulation of melanogenesis by scoparone in B16 melanoma cells. Acta Pharmacol. Sin. 2006, 27, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of Eumelanin and Pheomelanin and Its Pathophysiological Implications. Photochem. Photobiol. 2018, 94, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis--controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404. [Google Scholar] [CrossRef]
- Shibahara, S.; Takeda, K.; Yasumoto, K.; Udono, T.; Watanabe, K.; Saito, H.; Takahashi, K. Microphthalmia-associated transcription factor (MITF): Multiplicity in structure, function, and regulation. J. Investig. Dermatol. Symp. Proc. 2001, 6, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Pogenberg, V.; Ogmundsdottir, M.H.; Bergsteinsdottir, K.; Schepsky, A.; Phung, B.; Deineko, V.; Milewski, M.; Steingrimsson, E.; Wilmanns, M. Restricted leucine zipper dimerization and specificity of DNA recognition of the melanocyte master regulator MITF. Genes Dev. 2012, 26, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Yang, K.; Deng, F.; Xing, Y.; Li, Y.; Lian, X.; Yang, T. Wnt3a inhibits proliferation but promotes melanogenesis of melan-a cells. Int. J. Mol. Med. 2012, 30, 636–642. [Google Scholar] [CrossRef]
- Herraiz, C.; Garcia-Borron, J.C.; Jimenez-Cervantes, C.; Olivares, C. MC1R signaling. Intracellular partners and pathophysiological implications. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2448–2461. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Feng, Y.; Xu, M.; Yang, J.; Wang, Z.; Di, G. Four-octyl itaconate activates Keap1-Nrf2 signaling to protect neuronal cells from hydrogen peroxide. Cell Commun. Signal. 2018, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Diskin, C.; Zotta, A.; Corcoran, S.E.; Tyrrell, V.J.; Zaslona, Z.; O’Donnell, V.B.; O’Neill, L.A.J. 4-Octyl-Itaconate and Dimethyl Fumarate Inhibit COX2 Expression and Prostaglandin Production in Macrophages. J. Immunol. 2021, 207, 2561–2569. [Google Scholar] [CrossRef]
- Shin, J.M.; Kim, M.Y.; Sohn, K.C.; Jung, S.Y.; Lee, H.E.; Lim, J.W.; Kim, S.; Lee, Y.H.; Im, M.; Seo, Y.J.; et al. Nrf2 negatively regulates melanogenesis by modulating PI3K/Akt signaling. PLoS ONE 2014, 9, e96035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.; Chi, W.J.; Kim, S.Y. Dimethyl Itaconate Reduces alpha-MSH-Induced Pigmentation via Modulation of AKT and p38 MAPK Signaling Pathways in B16F10 Mouse Melanoma Cells. Molecules 2022, 27, 4183. [Google Scholar] [CrossRef]
- Flydal, M.I.; Martinez, A. Phenylalanine hydroxylase: Function, structure, and regulation. IUBMB Life 2013, 65, 341–349. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Arshad, L.; Jantan, I.; Bukhari, S.N.; Haque, M.A. Immunosuppressive Effects of Natural alpha, beta-Unsaturated Carbonyl-Based Compounds, and Their Analogs and Derivatives, on Immune Cells: A Review. Front. Pharmacol. 2017, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Eggler, A.L.; Mesecar, A.D.; van Breemen, R.B. Modification of keap1 cysteine residues by sulforaphane. Chem. Res. Toxicol. 2011, 24, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Ishitsuka, Y. NRF2 in the Epidermal Pigmentary System. Biomolecules 2022, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganjac, M.; Milkovic, L.; Sunjic, S.B.; Zarkovic, N. The NRF2, Thioredoxin, and Glutathione System in Tumorigenesis and Anticancer Therapies. Antioxidants 2020, 9, 1151. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Gene | Primer Sequence | |
|---|---|---|---|
| Mouse | NM_001289726.1 | Gapdh | Forward: 5′-GGAGAGTGTTTCCTCGTCCC-3′ Reverse: 5′-ACTGTGCCGTTGAATTTGCC-3′ |
| XM_036165907.1 | Mitf | Forward: 5′-AGCGTGTATTTTCCCCACAG-3′ Reverse: 5′-TAGCTCCTTAATGCGGTCGT-3′ | |
| NM_011661.5 | Tyr | Forward: 5′-GTCCACTCACAGGGATAGCAG-3′ Reverse: 5′-AGGTGCATTGGCTTCTGGGTA-3′ | |
| XM_006537781.2 | Trp-1 | Forward: 5′-GGACAGGAAAGCTTTGGGGA-3′ Reverse: 5′-GTCCTCCCGTTCCATTCAGG-3′ | |
| NM_010024.3 | Trp-2 | Forward: 5′-TACGTGATCACCACGCAACA-3′ Reverse: 5′-ACGTCACACTCGTTCTTCCC-3′ | |
| NM_008777.3 | Pah | Forward: 5′-GGGAACGGTGTTCAGGAC-3′ Reverse: 5′-GACAAGAGCCCAGCACCA-3′ | |
| NM_008513.3 | Lrp5 | Forward: 5′-ACACTC TCTGGGGACACAC-3′ Reverse: 5′-CCTCCAGGGGATCGTAGT-3′ | |
| NM_001122733.1 | c-Kit | Forward: 5′-TGTGAACCAACTTCGCCTGA-3′ Reverse: 5′-GCCTGGATTT GCTCTTTGTTG-3′ | |
| NM_008559.3 | Mc1r | Forward: 5′-TTCTAGCCATGCTGGCAC-3′ Reverse: 5′-CTGGCTGCGGAAAGCATA-3′ | |
| NM_010902.5 | Nrf2 | Forward: 5′-CACAGTGCTCCTATGCGTG-3′ Reverse: 5′-TCTGGGCGGCGACTTTATT-3′ |
| Gene | Primer Sequence (5′→3′) |
|---|---|
| shMitf | Forward: CCGGATGCTGGAAATGCTAGAATACCTCGAGGTATTCTAGCATTTCCAGCATTTTTTG Age I 21bp sense Loop 21bp antisense Terminal Reverse: AATTCAAAAAATGCTGGAAATGCTAGAATACCTCGAGGTATTCTAGCATTTCCAGCAT EcoRI Terminal 21bp sense Loop 21bp antisense |
| shNrf2 | Forward: CCGGCCCGAATTACAGTGTCTTAATCTCGAGATTAAGACACTGTAATTCGGGTTTTTG Age I 21bp sense Loop 21bp antisense Terminal Reverse: AATTCAAAAACCCGAATTACAGTGTCTTAATCTCGAGATTAAGACACTGTAATTCGGG EcoRI Terminal 21bp sense Loop 21bp antisense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, B.-Y.; Ngo, H.H.; Choi, W.J.; Keum, Y.-S. Dimethyl Itaconate Inhibits Melanogenesis in B16F10 Cells. Antioxidants 2023, 12, 692. https://doi.org/10.3390/antiox12030692
Yu B-Y, Ngo HH, Choi WJ, Keum Y-S. Dimethyl Itaconate Inhibits Melanogenesis in B16F10 Cells. Antioxidants. 2023; 12(3):692. https://doi.org/10.3390/antiox12030692
Chicago/Turabian StyleYu, Bo-Yeong, Hoang Hai Ngo, Won Jun Choi, and Young-Sam Keum. 2023. "Dimethyl Itaconate Inhibits Melanogenesis in B16F10 Cells" Antioxidants 12, no. 3: 692. https://doi.org/10.3390/antiox12030692
APA StyleYu, B.-Y., Ngo, H. H., Choi, W. J., & Keum, Y.-S. (2023). Dimethyl Itaconate Inhibits Melanogenesis in B16F10 Cells. Antioxidants, 12(3), 692. https://doi.org/10.3390/antiox12030692