
Clinical Relevance of lncRNA and Mitochondrial Targeted Antioxidants as Therapeutic Options in Regulating Oxidative Stress and Mitochondrial Function in Vascular Complications of Diabetes

Abstract
:1. Introduction
2. Implication of Oxidative Stress in Diabetes Related Complication
2.1. Oxidative Stress and Mitochondria
2.2. Mediators of Oxidative Stress and Their Role in the Development of Diabetic Complications
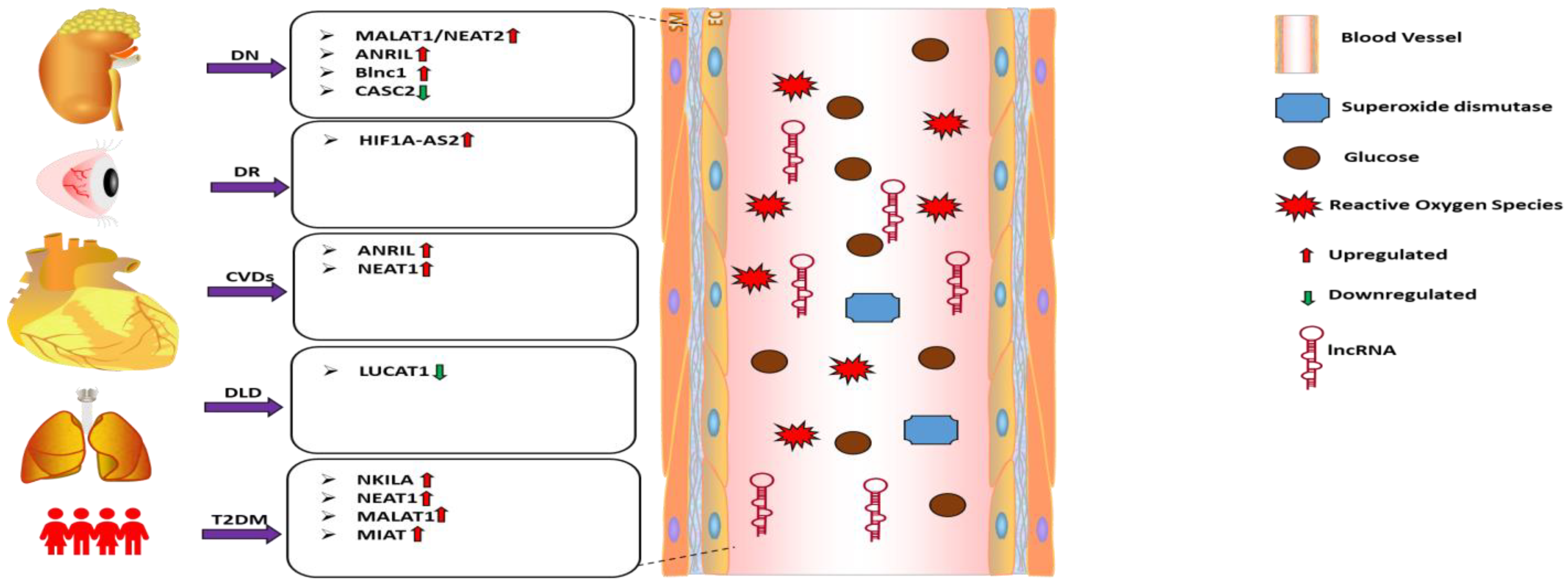
2.3. Circulating lncRNAs: Emerging Biomarkers of Oxidative Stress in Diabetes Complication
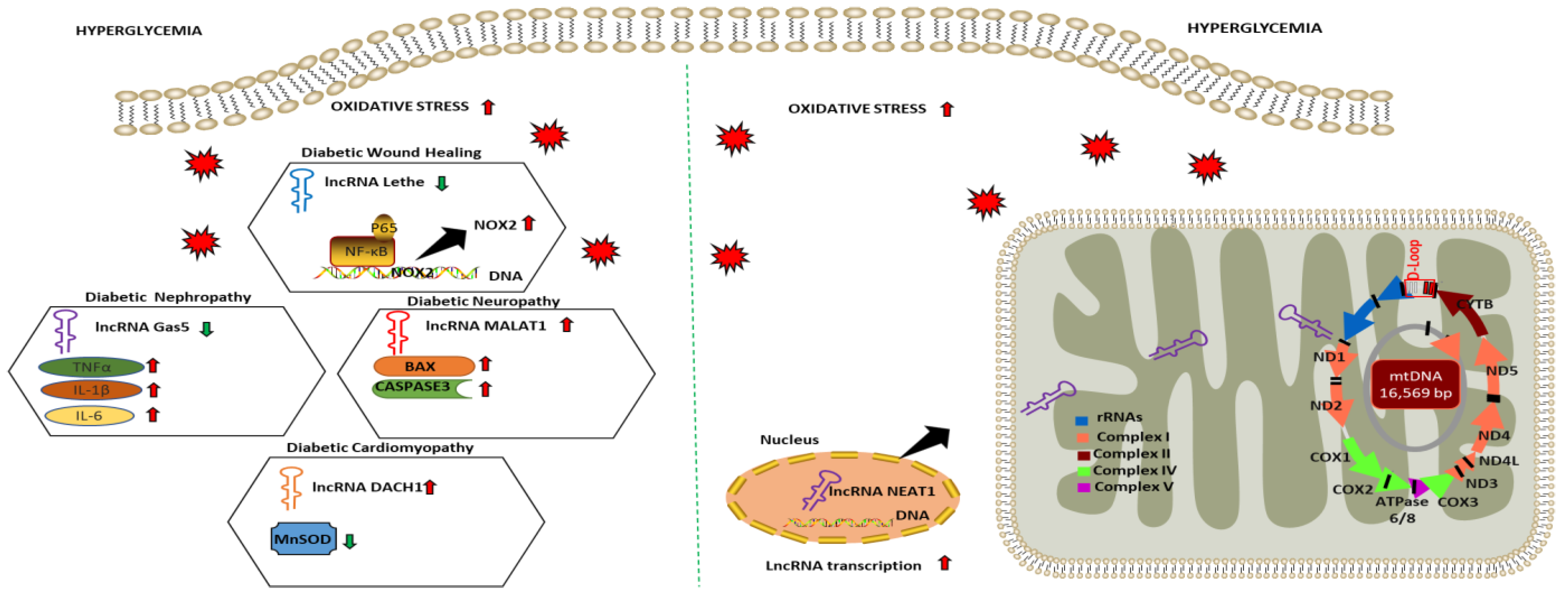
2.4. LncRNA as Regulators of Oxidative Stress and Mitochondrial Function in Diabetic Complications
2.5. Conventional Antioxidants and Their Application in Diabetic Complications: Success and Limitations
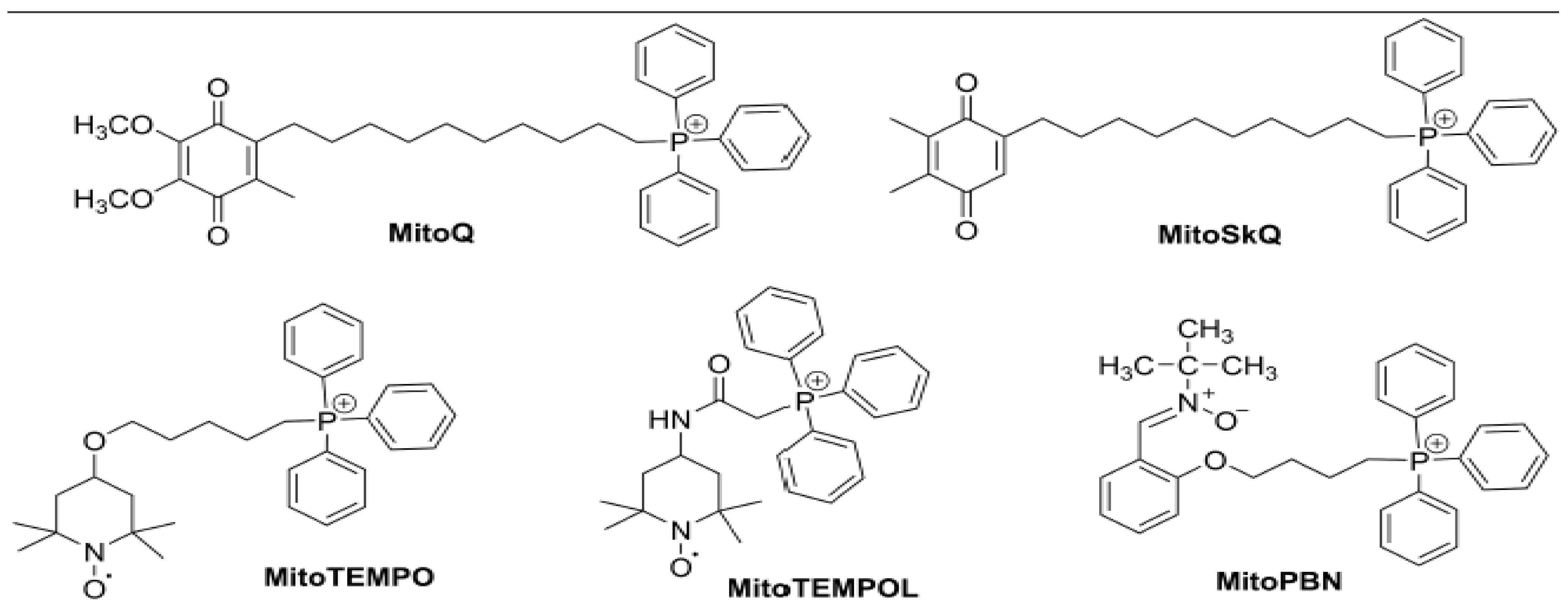
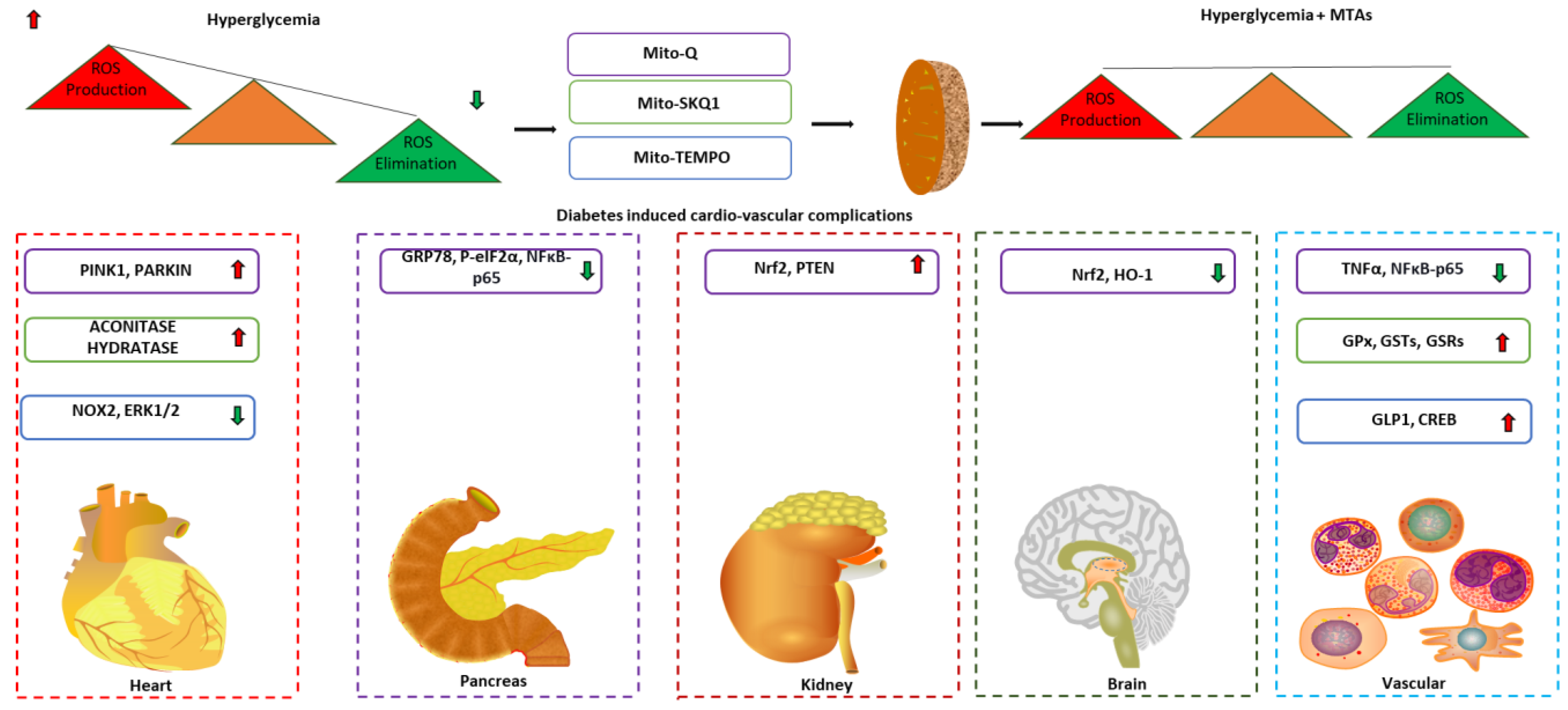
3. Mitochondrial-Targeted Antioxidants for Diabetic Complications
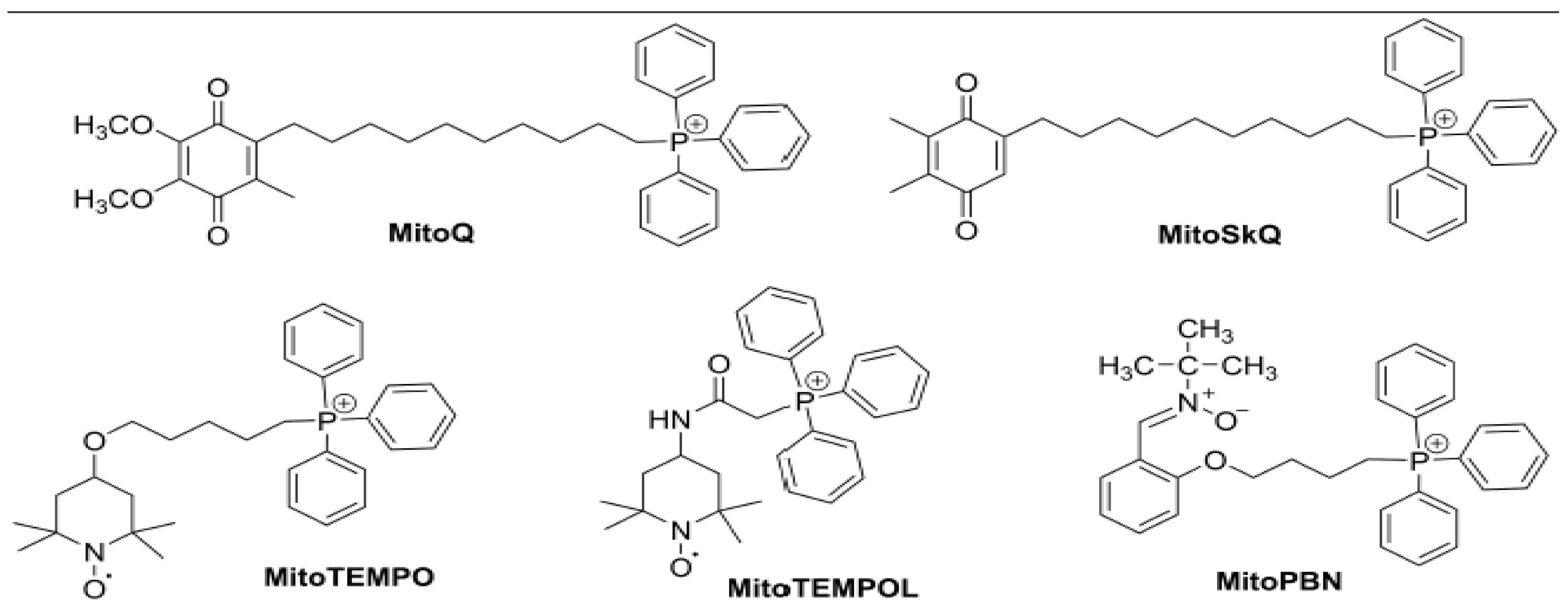
3.1. MitoQ
3.2. SKQ1
3.3. Mito-Tempo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondria Targeted Antioxidants | Experimental Models | Dosage | Effect/Mechanism | Limitation | References |
|---|---|---|---|---|---|
| MitoQ | Mice model of diabetes kidney disease | 5 mg/kg × bodyweight; intraperitoneal administration | Reverts tubular injury by ameliorating mtROS and mitochondrial fragmentation via activating NRF2 and PINK1. | [157] | |
| Mice model of diabetic kidney disease | 0.6 mg/kg × bodyweight; intragastric gavage | Mitigates mitochondrial dysfunction and conferred renal protection. | Contrasting effect on AER and ACR as response to therapies. | [158] | |
| Pancreatic β cell line INS-1E model of HG | 0.5 µmol/L in culture medium | Protects the β cell via preventing ROS production and decreasing endoplasmic reticulum stress and NFκB-p65 activation under high glucose. | [159] | ||
| Mice model of peripheral neuropathy | 0.93 g/kg × bodyweight; diet administration | Increases motor and sensory nerve conduction velocity cornea sensitivity and thermal nociception improving peripheral neuropathy. | [160] | ||
| Brain microvascular endothelial cells (BMECs) model of HG | 50 µmol/L in culture medium | Attenuates mitochondrial ROS production, cytoskeletal damage and apoptosis in BMEC via activating Nrf2/HO-1 pathway. | [161] | ||
| Rat model of myocardial ischemia reperfusion injury | 2.8 mg/kg × bodyweight; tail vein administration | Confers cardio protection by promoting mitophagy via modulating PINK1/Parkin pathway. | [162] | ||
| Leukocytes of T2D patients | 0.5 µmol/L in culture medium | Decreases ROS production, leukocyte endothelium interaction, TNFα in the leukocytes of T2D patients via regulating NF-κB pathway. | Small cohort size and lack of control group with similar BMI as T2D patients | [163] | |
| SKQ1 | Rat model of protamine sulfate induced hyperglycemia | 1250 nmol/kg × bodyweight; Intraperitoneal administration | Decreases ROS production, free radical oxidation and restored total antioxidant activity and mitigates hyperglycemic stress. | [165,166] | |
| Rat model of streptozotocin induced hyperglycemia | 1250 nmol/kg × bodyweight; Intraperitoneal administration | Decreases free radical production and oxidation restoring the antioxidant activity of catalase and SOD to the direction of control to mitigate hyperglycemic stress. | [167] | ||
| Mice model of diabetic dermal wound healing | 250 nmol/kg × bodyweight; Oral administration | Increases mitochondrial biogenesis, normalizes inflammation enhancing wound healing | [168] | ||
| Mito-Tempo | Mice model of diabetic cardiomyopathy | 0.7 mg/kg × bodyweight; Intraperitoneal administration | Decreases mitochondrial reactive oxygen species generation decreasing apoptosis and myocardial hypertrophy via regulation ERK1/2 pathway. | [171] | |
| Rat model of diabetic induced vascular constriction | 20 mg/kg × bodyweight; Intraperitoneal administration | Attenuates abnormal vascular tone and hypertension via modulation of GLP-1/CREB/adiponectin pathway. | [172] | ||
| Arterioles and mononuclear cells of T2D patients | 1 mmol/L in culture medium | Improves endothelial function and reduces mitochondrial superoxide levels. | Small study size, medication effects influencing mitochondrial function of mononuclear cells were not excluded | [173] | |
| Visceral adipose tissue of T2D patients | 10 µmol/L in culture medium | Restores the activity of mitochondrial complex II and insulin sensitivity. | Small sample size, inherent variability in complex II activity between subjects and markers of mitochondrial functions were not measured | [174] |
3.4. Mito-PBN
3.5. Szeto-Schiller (SS) Peptides
4. Challenges and Prospects of Mitochondrial Targets Antioxidants
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANRIL | Antisense non-coding RNA in the INK4 locus |
| BAX | Bcl-2-associated X protein |
| Blnc1 | Brown Fat lncRNA 1 |
| CVD | Cardiovascular disease |
| CASC2 | Cancer susceptibility candidate 2 |
| CREB | Cyclic AMP response element binding protein |
| CVDs | Cardiovascular diseases |
| DAG | Diacylglycerol |
| DCM | Diabetic cardiomyopathy |
| DLD | Diabetes lung disease |
| DN | Diabetic nephropathy |
| DR | Diabetic retinopathy |
| DRP1 | Dynamin related protein 1 |
| ERK1/2 | Extracellular signal-regulated protein kinase ½ |
| ETC | Electron transport chain |
| FoxP1 | Forehead box protein 1 |
| GLP-1 | Glucagon-like peptide 1 |
| GPx | Glutathione peroxidases |
| GRP78 | Glucose related protein 78 GRP78 |
| GSRs | Glutathione disulfide reductase |
| GSTs | Glutathione S transferase |
| HF1A-AS2 | Hypoxia inducible factor 1 alpha-antisense RNA 2 |
| HO1 | Heme oxygenase 1 |
| ICAM1 | Intracellular adhesion molecule 1 |
| IHD | Ischemic heart disease |
| iNOS | Inducible nitric oxide synthase |
| IRS2 | Insulin stimulated receptor substrate 2 |
| lncRNA | Long non-coding RNA |
| LUCAT1 | Lung cancer associated transcript 1 |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript |
| MCP1 | Monocyte chemoattractant protein 1 |
| MDA | Malonaldehyde |
| Mfn2 | Mitofusin2 |
| MIAT | Myocardial infarction-associated transcript. |
| MnSOD | Manganese superoxide dismutase |
| NEAT1 | Nuclear paraspeckle assembly transcript 1 |
| NF-κB | Nuclear factor-kappa B |
| NKILA | NF-κB-interacting long noncoding RNA |
| NO | Nitric oxide |
| NOX2 | NAPDH oxidase 2 |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| OPA1 | Optic atrophy protein 1 |
| OXPHOS | Oxidative phosphorylation |
| PI3K | Phosphatidylinositol 3 kinase |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TNFα | Tumor necrotic factor α |
| TPR | Translocated promoter region |
| VHD | Valvular heart disease |
References
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElSayed, N.A.; Aleppo, G.; Aroda, V.R.; Bannuru, R.R.; Brown, F.M.; Bruemmer, D.; Collins, B.S.; Hilliard, M.E.; Isaacs, D.; Johnson, E.L.; et al. 2. Classification and Diagnosis of Diabetes: Standards of Care in Diabetes—2023. Diabetes Care 2023, 46 (Suppl. 1), S19–S40. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [Green Version]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Prim. 2015, 1, 15019. [Google Scholar] [CrossRef]
- Nowotny, K.; Jung, T.; Hohn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fucho, R.; Casals, N.; Serra, D.; Herrero, L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J. 2017, 31, 1263–1272. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, M.; Cocco, T.; Lorusso, M. Ceramide interaction with the respiratory chain of heart mitochondria. Biochemistry 2000, 39, 6660–6668. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N. Ceramide-induced apoptosis in renal tubular cells: A role of mitochondria and sphingosine-1-phoshate. Int. J. Mol. Sci. 2015, 16, 5076–5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, J.G.; Xia, Z.K.; Tong, M.L.; Guo, X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Rodriguez, E.; Anderson, C.A.; Thayne, K.; Chitwood, W.R.; Kypson, A.P. Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H118–H124. [Google Scholar] [CrossRef] [Green Version]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-kappaB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Field, B.C.; Gordillo, R.; Scherer, P.E. The Role of Ceramides in Diabetes and Cardiovascular Disease Regulation of Ceramides by Adipokines. Front. Endocrinol. 2020, 11, 569250. [Google Scholar] [CrossRef] [PubMed]
- Beaudoin, M.S.; Perry, C.G.; Arkell, A.M.; Chabowski, A.; Simpson, J.A.; Wright, D.C.; Holloway, G.P. Impairments in mitochondrial palmitoyl-CoA respiratory kinetics that precede development of diabetic cardiomyopathy are prevented by resveratrol in ZDF rats. J. Physiol. 2014, 592, 2519–2533. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Oxidative stress and sarcomeric proteins. Circ. Res. 2013, 112, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Pan, N.H.; Chen, Y.J. Oxidative stress and inflammation modulate peroxisome proliferator-activated receptors with regional discrepancy in diabetic heart. Eur. J. Clin. Investig. 2010, 40, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Huang, J.H.; Hsiao, F.C.; Chen, Y.J. Peroxisome proliferator-activated receptors modulate cardiac dysfunction in diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2013, 100, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Burkart, E.M.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Gierasch, C.M.; Shoghi, K.; Welch, M.J.; Kelly, D.P. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J. Clin. Investig. 2007, 117, 3930–3939. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Okere, I.C.; Chandler, M.P.; McElfresh, T.A.; Rennison, J.H.; Sharov, V.; Sabbah, H.N.; Tserng, K.Y.; Hoit, B.D.; Ernsberger, P.; Young, M.E.; et al. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H38–H44. [Google Scholar] [CrossRef]
- Yu, M.; Shan, X.; Liu, Y.; Zhu, J.; Cao, Q.; Yang, F.; Liu, Y.; Wang, G.; Zhao, X. RNA-Seq analysis and functional characterization revealed lncRNA NONRATT007560.2 regulated cardiomyocytes oxidative stress and apoptosis induced by high glucose. J. Cell. Biochem. 2019, 120, 18278–18287. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, L.; He, X.; Ye, Z.; Li, L.; Liu, Z.; Zhou, S. Atorvastatin Regulates MALAT1/miR-200c/NRF2 Activity to Protect Against Podocyte Pyroptosis Induced by High Glucose. Diabetes Metab. Syndr. Obes. 2021, 14, 1631–1645. [Google Scholar] [CrossRef]
- Durackova, Z. Some current insights into oxidative stress. Physiol. Res. 2010, 59, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Pizzinat, N.; Copin, N.; Vindis, C.; Parini, A.; Cambon, C. Reactive oxygen species production by monoamine oxidases in intact cells. Naunyn-Schmiedebergs Arch. Pharmacol. 1999, 359, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Baudry, N.; Laemmel, E.; Vicaut, E. In vivo reactive oxygen species production induced by ischemia in muscle arterioles of mice: Involvement of xanthine oxidase and mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H821–H828. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena-Silva, R.A.; Miller, J.D.; Chu, Y.; Heistad, D.D. Serotonin produces monoamine oxidase-dependent oxidative stress in human heart valves. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1354–H1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [Green Version]
- Haynes, C.M.; Titus, E.A.; Cooper, A.A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 2004, 15, 767–776. [Google Scholar] [CrossRef]
- Wu, R.F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef] [PubMed]
- Abdal Dayem, A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Politi, P.M.; Sinha, B.K.; Myers, C.E. Adriamycin-induced free radical formation in the perfused rat heart: Implications for cardiotoxicity. Cancer Res. 1988, 48, 4766–4769. [Google Scholar] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Oldford, C.; Kuksal, N.; Gill, R.; Young, A.; Mailloux, R.J. Estimation of the hydrogen peroxide producing capacities of liver and cardiac mitochondria isolated from C57BL/6N and C57BL/6J mice. Free Radic. Biol. Med. 2019, 135, 15–27. [Google Scholar] [CrossRef]
- Aon, M.A.; Stanley, B.A.; Sivakumaran, V.; Kembro, J.M.; O’Rourke, B.; Paolocci, N.; Cortassa, S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J. Gen. Physiol. 2012, 139, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3663–3668. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira-Llopis, S.; Banuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Nan, J.; Hu, H.; Sun, Y.; Zhu, L.; Wang, Y.; Zhong, Z.; Zhao, J.; Zhang, N.; Wang, Y.; Wang, Y.; et al. TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB-Dependent Activation of OPA1 Expression. Circ. Res. 2017, 121, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, S.M.; Widlansky, M.E.; Chen, K.; Xu, G.; Holbrook, M.; Tabit, C.E.; Hamburg, N.M.; Frame, A.A.; Caiano, T.L.; Kluge, M.A.; et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation 2011, 124, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Perseghin, G.; Scifo, P.; De Cobelli, F.; Pagliato, E.; Battezzati, A.; Arcelloni, C.; Vanzulli, A.; Testolin, G.; Pozza, G.; Del Maschio, A.; et al. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 1999, 48, 1600–1606. [Google Scholar] [CrossRef]
- Summers, S.A. Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 2006, 45, 42–72. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Jornayvaz, F.R.; Birkenfeld, A.L.; Jurczak, M.J.; Kanda, S.; Guigni, B.A.; Jiang, D.C.; Zhang, D.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc. Natl. Acad. Sci. USA 2011, 108, 5748–5752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowska, E.; Blachnio-Zabielska, A. The Role of Ceramides in Insulin Resistance. Front. Endocrinol. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as mediator of NF-kB pathway activation in neuroinflammation and oxidative stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- El-Mesallamy, H.O.; Hamdy, N.M.; Ezzat, O.A.; Reda, A.M. Levels of soluble advanced glycation end product-receptors and other soluble serum markers as indicators of diabetic neuropathy in the foot. J. Investig. Med. 2011, 59, 1233–1238. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H491–H498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic Res. 2008, 40, 10–15. [Google Scholar] [CrossRef]
- Gao, X.; Belmadani, S.; Picchi, A.; Xu, X.; Potter, B.J.; Tewari-Singh, N.; Capobianco, S.; Chilian, W.M.; Zhang, C. Tumor necrosis factor-alpha induces endothelial dysfunction in Lepr(db) mice. Circulation 2007, 115, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Harja, E.; Bu, D.X.; Hudson, B.I.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Al-Aubaidy, H.A.; Jelinek, H.F. 8-Hydroxy-2-deoxy-guanosine identifies oxidative DNA damage in a rural prediabetes cohort. Redox Rep. 2010, 15, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.W.; Yao, Q.H.; Weng, Q.F.; Su, B.L.; Zhang, X.; Xiong, J.H. Study of urinary 8-hydroxydeoxyguanosine as a biomarker of oxidative DNA damage in diabetic nephropathy patients. J. Pharm. Biomed. Anal. 2004, 36, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Jiang, R.; Zhang, Q.; Wang, R.; Yang, C.; Ma, J.; Du, H. Increased 8-hydroxy-2′-deoxyguanosine in leukocyte DNA from patients with type 2 diabetes and microangiopathy. J. Int. Med. Res. 2016, 44, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.B.; Cui, N.H.; Liu, X.; Liu, X. Mitochondrial 8-hydroxy-2′-deoxyguanosine and coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2020, 19, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinonen, J.; Lehtimaki, T.; Toyokuni, S.; Okada, K.; Tanaka, T.; Hiai, H.; Ochi, H.; Laippala, P.; Rantalaiho, V.; Wirta, O.; et al. New biomarker evidence of oxidative DNA damage in patients with non-insulin-dependent diabetes mellitus. FEBS Lett. 1997, 417, 150–152. [Google Scholar] [CrossRef] [Green Version]
- Kaefer, M.; De Carvalho, J.A.; Piva, S.J.; da Silva, D.B.; Becker, A.M.; Sangoi, M.B.; Almeida, T.C.; Hermes, C.L.; Coelho, A.C.; Tonello, R.; et al. Plasma malondialdehyde levels and risk factors for the development of chronic complications in type 2 diabetic patients on insulin therapy. Clin. Lab. 2012, 58, 973–978. [Google Scholar]
- Mahreen, R.; Mohsin, M.; Nasreen, Z.; Siraj, M.; Ishaq, M. Significantly increased levels of serum malonaldehyde in type 2 diabetics with myocardial infarction. Int. J. Diabetes Dev. Ctries 2010, 30, 49–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulere, L.; Lagarde, M.; Soulage, C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, D.E.; Atalay, M.; Niskanen, L.; Uusitupa, M.; Hanninen, O.; Sen, C.K. Increased resting and exercise-induced oxidative stress in young IDDM men. Diabetes Care 1996, 19, 569–574. [Google Scholar] [CrossRef]
- Griesmacher, A.; Kindhauser, M.; Andert, S.E.; Schreiner, W.; Toma, C.; Knoebl, P.; Pietschmann, P.; Prager, R.; Schnack, C.; Schernthaner, G.; et al. Enhanced serum levels of thiobarbituric-acid-reactive substances in diabetes mellitus. Am. J. Med. 1995, 98, 469–475. [Google Scholar] [CrossRef]
- Tavares, A.M.; Silva, J.H.; Bensusan, C.O.; Ferreira, A.C.F.; Matos, L.P.L.; KLA, E.S.; Cardoso-Weide, L.C.; Taboada, G.F. Altered superoxide dismutase-1 activity and intercellular adhesion molecule 1 (ICAM-1) levels in patients with type 2 diabetes mellitus. PLoS ONE 2019, 14, e0216256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Sakamoto, T.; Komatsu, K.; Fujishima, H.; Morii, T.; Narita, T.; Takahashi, T.; Yamada, Y. Reduction of circulating superoxide dismutase activity in type 2 diabetic patients with microalbuminuria and its modulation by telmisartan therapy. Hypertens. Res. 2011, 34, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Gawlik, K.; Naskalski, J.W.; Fedak, D.; Pawlica-Gosiewska, D.; Grudzien, U.; Dumnicka, P.; Malecki, M.T.; Solnica, B. Markers of Antioxidant Defense in Patients with Type 2 Diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 2352361. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, K.; Tanaka, M.; Iwata, H.; Nishihara, R.; Ishihara, K.; Wang, D.H.; Ogino, K.; Taniuchi, K.; Masuoka, N. Low catalase activity in blood is associated with the diabetes caused by alloxan. Clin. Chim. Acta 2009, 407, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P.; Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viereck, J.; Thum, T. Circulating Noncoding RNAs as Biomarkers of Cardiovascular Disease and Injury. Circ. Res. 2017, 120, 381–399. [Google Scholar] [CrossRef] [Green Version]
- Pant, T.; Juric, M.; Bosnjak, Z.J.; Dhanasekaran, A. Recent Insight on the Non-coding RNAs in Mesenchymal Stem Cell-Derived Exosomes: Regulatory and Therapeutic Role in Regenerative Medicine and Tissue Engineering. Front. Cardiovasc. Med. 2021, 8, 737512. [Google Scholar] [CrossRef]
- Pant, T.; Dhanasekaran, A.; Fang, J.; Bai, X.; Bosnjak, Z.J.; Liang, M.; Ge, Z.D. Current status and strategies of long noncoding RNA research for diabetic cardiomyopathy. BMC Cardiovasc. Disord. 2018, 18, 197. [Google Scholar] [CrossRef] [Green Version]
- Pant, T.; DiStefano, J.K.; Logan, S.; Bosnjak, Z.J. Emerging Role of Long Noncoding RNAs in Perioperative Neurocognitive Disorders and Anesthetic-Induced Developmental Neurotoxicity. Anesth. Analg. 2021, 132, 1614–1625. [Google Scholar] [CrossRef]
- de Gonzalo-Calvo, D.; Kenneweg, F.; Bang, C.; Toro, R.; van der Meer, R.W.; Rijzewijk, L.J.; Smit, J.W.; Lamb, H.J.; Llorente-Cortes, V.; Thum, T. Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci. Rep. 2016, 6, 37354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, T.; Dhanasekaran, A.; Zhao, M.; Thorp, E.B.; Forbess, J.M.; Bosnjak, Z.J.; Benjamin, I.J.; Ge, Z.D. Identification and analysis of circulating long non-coding RNAs with high significance in diabetic cardiomyopathy. Sci. Rep. 2021, 11, 2571. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.J.; Yang, D.W.; Ou, L.N.; Guo, X.R.; Wu, B.L. Circulating Expression Level of LncRNA Malat1 in Diabetic Kidney Disease Patients and Its Clinical Significance. J. Diabetes Res. 2020, 2020, 4729019. [Google Scholar] [CrossRef]
- Zhang, X.L.; Zhu, H.Q.; Zhang, Y.; Zhang, C.Y.; Jiao, J.S.; Xing, X.Y. LncRNA CASC2 regulates high glucose-induced proliferation, extracellular matrix accumulation and oxidative stress of human mesangial cells via miR-133b/FOXP1 axis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Atef, M.M.; Shafik, N.M.; Hafez, Y.M.; Watany, M.M.; Selim, A.; Shafik, H.M.; Safwat El-Deeb, O. The evolving role of long noncoding RNA HIF1A-AS2 in diabetic retinopathy: A cross-link axis between hypoxia, oxidative stress and angiogenesis via MAPK/VEGF-dependent pathway. Redox Rep. 2022, 27, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Jiang, J. LncRNA ANRIL Silencing Alleviates High Glucose-Induced Inflammation, Oxidative Stress, and Apoptosis via Upregulation of MME in Podocytes. Inflammation 2020, 43, 2147–2155. [Google Scholar] [CrossRef]
- Feng, X.; Zhao, J.; Ding, J.; Shen, X.; Zhou, J.; Xu, Z. LncRNA Blnc1 expression and its effect on renal fibrosis in diabetic nephropathy. Am. J. Transl. Res. 2019, 11, 5664–5672. [Google Scholar]
- Zhu, Y.; Dai, L.; Yu, X.; Chen, X.; Li, Z.; Sun, Y.; Liang, Y.; Wu, B.; Wang, Q.; Wang, X. Circulating expression and clinical significance of LncRNA ANRIL in diabetic kidney disease. Mol. Biol. Rep. 2022, 49, 10521–10529. [Google Scholar] [CrossRef]
- Li, P.; Zhang, N.; Ping, F.; Gao, Y.; Cao, L. lncRNA SCAL1 inhibits inducible nitric oxide synthase in lung cells under high-glucose conditions. Exp. Ther. Med. 2019, 18, 1831–1836. [Google Scholar] [CrossRef] [Green Version]
- Karam, R.A.; Amer, M.M.; Zidan, H.E. Long Noncoding RNA NEAT1 Expression and Its Target miR-124 in Diabetic Ischemic Stroke Patients. Genet. Test. Mol. Biomark. 2022, 26, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Alfaifi, M.; Ali Beg, M.M.; Alshahrani, M.Y.; Ahmad, I.; Alkhathami, A.G.; Joshi, P.C.; Alshehri, O.M.; Alamri, A.M.; Verma, A.K. Circulating long non-coding RNAs NKILA, NEAT1, MALAT1, and MIAT expression and their association in type 2 diabetes mellitus. BMJ Open Diabetes Res. Care 2021, 9, e001821. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Kurland, C.G.; Andersson, S.G. Origin and evolution of the mitochondrial proteome. Microbiol. Mol. Biol. Rev. 2000, 64, 786–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.W. Mosaic nature of the mitochondrial proteome: Implications for the origin and evolution of mitochondria. Proc. Natl. Acad. Sci. USA 2015, 112, 10133–10138. [Google Scholar] [CrossRef] [Green Version]
- Bridges, M.C.; Daulagala, A.C.; Kourtidis, A. LNCcation: lncRNA localization and function. J. Cell. Biol. 2021, 220, e202009045. [Google Scholar] [CrossRef]
- Lin, Y.H. Crosstalk of lncRNA and Cellular Metabolism and Their Regulatory Mechanism in Cancer. Int. J. Mol. Sci. 2020, 21, 2947. [Google Scholar] [CrossRef] [PubMed]
- Rackham, O.; Shearwood, A.M.; Mercer, T.R.; Davies, S.M.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yoshitomi, T.; Hu, J.F.; Cui, J. Long noncoding RNAs coordinate functions between mitochondria and the nucleus. Epigenetics Chromatin 2017, 10, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, W.; Hoffman, A.R.; Cui, J.; Hu, J.F. The Nucleus/Mitochondria-Shuttling LncRNAs Function as New Epigenetic Regulators of Mitophagy in Cancer. Front. Cell Dev. Biol. 2021, 9, 699621. [Google Scholar] [CrossRef]
- Zgheib, C.; Hodges, M.M.; Hu, J.; Liechty, K.W.; Xu, J. Long non-coding RNA Lethe regulates hyperglycemia-induced reactive oxygen species production in macrophages. PLoS ONE 2017, 12, e0177453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Wu, W.; Tang, A.; Luo, N.; Tan, Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes Metab. Syndr. Obes. 2019, 12, 2609–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, F.; Han, Y.; Fu, J.; Wang, N.; Jia, Y.; Wang, K.; Ge, J. LncRNA MALAT1 induced by hyperglycemia promotes microvascular endothelial cell apoptosis through activation of the miR-7641/TPR axis to exacerbate neurologic damage caused by cerebral small vessel disease. Ann. Transl. Med. 2021, 9, 1762. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Geng, J.; Li, X.; Wan, J.; Liu, J.; Zhou, Z.; Liu, X. Long Noncoding RNA LINC01619 Regulates MicroRNA-27a/Forkhead Box Protein O1 and Endoplasmic Reticulum Stress-Mediated Podocyte Injury in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 29, 355–376. [Google Scholar] [CrossRef] [PubMed]
- Pant, T.; Dhanasekaran, A.; Bai, X.; Zhao, M.; Thorp, E.B.; Forbess, J.M.; Bosnjak, Z.J.; Ge, Z.D. Genome-wide differential expression profiling of lncRNAs and mRNAs associated with early diabetic cardiomyopathy. Sci. Rep. 2019, 9, 15345. [Google Scholar] [CrossRef] [Green Version]
- Pant, T.; Mishra, M.K.; Bai, X.; Ge, Z.D.; Bosnjak, Z.J.; Dhanasekaran, A. Microarray analysis of long non-coding RNA and mRNA expression profiles in diabetic cardiomyopathy using human induced pluripotent stem cell-derived cardiomyocytes. Diabetes Vasc. Dis. Res. 2019, 16, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Li, D.; Dong, X.; Zhang, X.; Liu, J.; Peng, L.; Meng, B.; Hua, Q.; Pei, X.; Zhao, L.; et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci. China Life Sci. 2022, 65, 1198–1212. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Nuclear Genome-Encoded Long Noncoding RNAs and Mitochondrial Damage in Diabetic Retinopathy. Cells 2021, 10, 3271. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Khan, A. Antioxidants and diabetes. Indian J. Endocrinol. Metab. 2012, 16, S267–S271. [Google Scholar] [CrossRef]
- Giugliano, D.; Ceriello, A.; Paolisso, G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996, 19, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M. The antioxidants of human extracellular fluids. Arch. Biochem. Biophys. 1990, 280, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Feskens, E.J.; Virtanen, S.M.; Rasanen, L.; Tuomilehto, J.; Stengard, J.; Pekkanen, J.; Nissinen, A.; Kromhout, D. Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study. Diabetes Care 1995, 18, 1104–1112. [Google Scholar] [CrossRef]
- Mason, S.A.; Keske, M.A.; Wadley, G.D. Effects of Vitamin C Supplementation on Glycemic Control and Cardiovascular Risk Factors in People with Type 2 Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis of Randomized Controlled Trials. Diabetes Care 2021, 44, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Saklayen, M.G.; Yap, J.; Vallyathan, V. Effect of month-long treatment with oral N-acetylcysteine on the oxidative stress and proteinuria in patients with diabetic nephropathy: A pilot study. J. Investig. Med. 2010, 58, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Darko, D.; Dornhorst, A.; Kelly, F.J.; Ritter, J.M.; Chowienczyk, P.J. Lack of effect of oral vitamin C on blood pressure, oxidative stress and endothelial function in Type II diabetes. Clin. Sci. 2002, 103, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Karne, R.J.; Hall, G.; Campia, U.; Panza, J.A.; Cannon, R.O., 3rd; Wang, Y.; Katz, A.; Levine, M.; Quon, M.J. High-dose oral vitamin C partially replenishes vitamin C levels in patients with Type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H137–H145. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Lu, X.Z.; Shen, M.Z.; Xing, C.Y.; Ma, J.; Duan, Y.Y.; Yuan, L.J. N-Acetyl Cysteine improves the diabetic cardiac function: Possible role of fibrosis inhibition. BMC Cardiovasc. Disord. 2015, 15, 84. [Google Scholar] [CrossRef] [Green Version]
- Argaev Frenkel, L.; Rozenfeld, H.; Rozenberg, K.; Sampson, S.R.; Rosenzweig, T. N-Acetyl-l-Cysteine Supplement in Early Life or Adulthood Reduces Progression of Diabetes in Nonobese Diabetic Mice. Curr. Dev. Nutr. 2019, 3, nzy097. [Google Scholar] [CrossRef] [Green Version]
- Dludla, P.V.; Orlando, P.; Silvestri, S.; Mazibuko-Mbeje, S.E.; Johnson, R.; Marcheggiani, F.; Cirilli, I.; Muller, C.J.F.; Louw, J.; Obonye, N.; et al. N-Acetyl cysteine ameliorates hyperglycemia-induced cardiomyocyte toxicity by improving mitochondrial energetics and enhancing endogenous Coenzyme Q(9/10) levels. Toxicol. Rep. 2019, 6, 1240–1245. [Google Scholar] [CrossRef]
- Montonen, J.; Knekt, P.; Jarvinen, R.; Reunanen, A. Dietary antioxidant intake and risk of type 2 diabetes. Diabetes Care 2004, 27, 362–366. [Google Scholar] [CrossRef]
- Khatami, P.G.; Soleimani, A.; Sharifi, N.; Aghadavod, E.; Asemi, Z. The effects of high-dose vitamin E supplementation on biomarkers of kidney injury, inflammation, and oxidative stress in patients with diabetic nephropathy: A randomized, double-blind, placebo-controlled trial. J. Clin. Lipidol. 2016, 10, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Leonard, S.; Traber, M.G.; Jialal, I. Gamma-tocopherol supplementation alone and in combination with alpha-tocopherol alters biomarkers of oxidative stress and inflammation in subjects with metabolic syndrome. Free Radic. Biol. Med. 2008, 44, 1203–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriello, A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care 2003, 26, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchioli, R.; Schweiger, C.; Levantesi, G.; Tavazzi, L.; Valagussa, F. Antioxidant vitamins and prevention of cardiovascular disease: Epidemiological and clinical trial data. Lipids 2001, 36, S53–S63. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C.; et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes: Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Sotler, R.; Poljsak, B.; Dahmane, R.; Jukic, T.; Pavan Jukic, D.; Rotim, C.; Trebse, P.; Starc, A. Prooxidant Activities of Antioxidants and Their Impact on Health. Acta Clin. Croat. 2019, 58, 726–736. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Matzinger, M.; Fischhuber, K.; Heiss, E.H. Activation of Nrf2 signaling by natural products-can it alleviate diabetes? Biotechnol. Adv. 2018, 36, 1738–1767. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Lund, L.G. Cellular reducing equivalents and oxidative stress. Free Radic. Biol. Med. 1994, 17, 65–75. [Google Scholar] [CrossRef]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S. Mitochondrial medicine: Pharmacological targeting of mitochondria in disease. Br. J. Pharmacol. 2007, 151, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Coulter, C.V.; Murphy, M.P. Selective targeting of an antioxidant to mitochondria. Eur. J. Biochem. 1999, 263, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Ward, M.S.; Flemming, N.B.; Gallo, L.A.; Fotheringham, A.K.; McCarthy, D.A.; Zhuang, A.; Tang, P.H.; Borg, D.J.; Shaw, H.; Harvie, B.; et al. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 2017, 7, 15190. [Google Scholar] [CrossRef] [Green Version]
- Escribano-Lopez, I.; Banuls, C.; Diaz-Morales, N.; Iannantuoni, F.; Rovira-Llopis, S.; Gomis, R.; Rocha, M.; Hernandez-Mijares, A.; Murphy, M.P.; Victor, V.M. The Mitochondria-Targeted Antioxidant MitoQ Modulates Mitochondrial Function and Endoplasmic Reticulum Stress in Pancreatic beta Cells Exposed to Hyperglycaemia. Cell. Physiol. Biochem. 2019, 52, 186–197. [Google Scholar] [CrossRef]
- Fink, B.; Coppey, L.; Davidson, E.; Shevalye, H.; Obrosov, A.; Chheda, P.R.; Kerns, R.; Sivitz, W.; Yorek, M. Effect of mitoquinone (Mito-Q) on neuropathic endpoints in an obese and type 2 diabetic rat model. Free Radic. Res. 2020, 54, 311–318. [Google Scholar] [CrossRef]
- Yang, M.Y.; Fan, Z.; Zhang, Z.; Fan, J. MitoQ protects against high glucose-induced brain microvascular endothelial cells injury via the Nrf2/HO-1 pathway. J. Pharmacol. Sci. 2021, 145, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Leng, Y.; Lei, S.; Qiu, Z.; Ming, H.; Zhang, Y.; Zhang, A.; Wu, Y.; Xia, Z. The mitochondria-targeted antioxidant MitoQ ameliorates myocardial ischemia-reperfusion injury by enhancing PINK1/Parkin-mediated mitophagy in type 2 diabetic rats. Cell Stress Chaperones 2022, 27, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Lopez, I.; Diaz-Morales, N.; Rovira-Llopis, S.; de Maranon, A.M.; Orden, S.; Alvarez, A.; Banuls, C.; Rocha, M.; Murphy, M.P.; Hernandez-Mijares, A.; et al. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016, 10, 200–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izyumov, D.S.; Domnina, L.V.; Nepryakhina, O.K.; Avetisyan, A.V.; Golyshev, S.A.; Ivanova, O.Y.; Korotetskaya, M.V.; Lyamzaev, K.G.; Pletjushkina, O.Y.; Popova, E.N.; et al. Mitochondria as source of reactive oxygen species under oxidative stress. Study with novel mitochondria-targeted antioxidants--the “Skulachev-ion” derivatives. Biochemistry 2010, 75, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Voronkova, Y.G.; Popova, T.N.; Agarkov, A.A.; Skulachev, M.V. Influence of 10-(6′-Plastoquinonyl)decyltriphenylphosphonium (SkQ1) on Oxidative Status in Rats with Protamine Sulfate-Induced Hyperglycemia. Biochemistry 2015, 80, 1606–1613. [Google Scholar] [CrossRef]
- Voronkova, Y.G.; Popova, T.N.; Agarkov, A.A.; Zinovkin, R.A. Effect of SkQ1 on Activity of the Glutathione System and NADPH-Generating Enzymes in an Experimental Model of Hyperglycemia. Biochemistry 2015, 80, 1614–1621. [Google Scholar] [CrossRef]
- Agarkov, A.A.; Popova, T.N.; Boltysheva, Y.G. Influence of 10-(6-plastoquinonyl) decyltriphenylphosphonium on free-radical homeostasis in the heart and blood serum of rats with streptozotocin-induced hyperglycemia. World J. Diabetes 2019, 10, 546–559. [Google Scholar] [CrossRef]
- Demyanenko, I.A.; Zakharova, V.V.; Ilyinskaya, O.P.; Vasilieva, T.V.; Fedorov, A.V.; Manskikh, V.N.; Zinovkin, R.A.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P.; et al. Mitochondria-Targeted Antioxidant SkQ1 Improves Dermal Wound Healing in Genetically Diabetic Mice. Oxid. Med. Cell. Longev. 2017, 2017, 6408278. [Google Scholar] [CrossRef] [Green Version]
- Dvoretskaya, Y.; Glanz, V.; Gryaznova, M.; Syromyatnikov, M.; Popov, V. Mitochondrial Antioxidant SkQ1 Has a Beneficial Effect in Experimental Diabetes as Based on the Analysis of Expression of microRNAs and mRNAs for the Oxidative Metabolism Regulators. Antioxidants 2021, 10, 1749. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Lu, W.; Qin, X.; Luo, Q.; Zhou, W. Downregulation of the GLP-1/CREB/adiponectin pathway is partially responsible for diabetes-induced dysregulated vascular tone and VSMC dysfunction. Biomed. Pharmacother. 2020, 127, 110218. [Google Scholar] [CrossRef]
- Kizhakekuttu, T.J.; Wang, J.; Dharmashankar, K.; Ying, R.; Gutterman, D.D.; Vita, J.A.; Widlansky, M.E. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2531–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, D.T.M.; Sverdlov, A.L.; Karki, S.; Macartney-Coxson, D.; Stubbs, R.S.; Farb, M.G.; Carmine, B.; Hess, D.T.; Colucci, W.S.; Gokce, N. Oxidative modifications of mitochondrial complex II are associated with insulin resistance of visceral fat in obesity. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E168–E177. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Liao, L.; Jiang, L.; Zhang, C.; Gao, H.; Qiao, L.; Liu, S.; Shi, D. Liver-targeted Nano-MitoPBN normalizes glucose metabolism by improving mitochondrial redox balance. Biomaterials 2019, 222, 119457. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.W.; Robinson, M.; Li, R.; Aldhowayan, H.; Geetha, T.; Babu, J.R. Mitochondrial dysfunction and beneficial effects of mitochondria-targeted small peptide SS-31 in Diabetes Mellitus and Alzheimer’s disease. Pharmacol. Res. 2021, 171, 105783. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, J.S.; Nunes, S.; Rolo, A.P.; Reis, F.; Palmeira, C.M. Therapeutic Options Targeting Oxidative Stress, Mitochondrial Dysfunction and Inflammation to Hinder the Progression of Vascular Complications of Diabetes. Front. Physiol. 2018, 9, 1857. [Google Scholar] [CrossRef]
- Kaludercic, N.; Di Lisa, F. Mitochondrial ROS Formation in the Pathogenesis of Diabetic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]

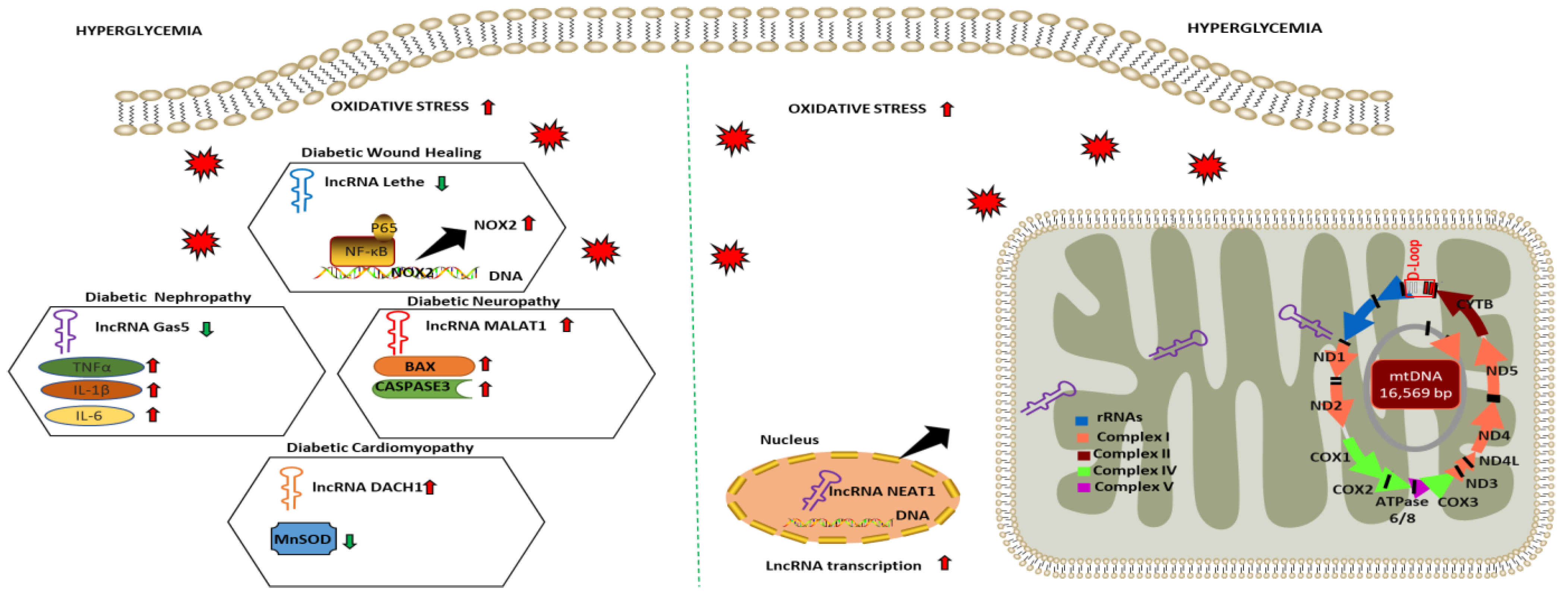

| Diabetic Complications | lncRNA | Study Design/Sample Size | Biological Fluid or Cells | lncRNA Expression Level | Interacting Factors/Relevant Pathways | Oxidative Stress Markers | References |
|---|---|---|---|---|---|---|---|
| Diabetic Nephropathy | MALAT1 | Diabetic Nephropathy patients (n = 47) | Serum | Upregulated | SOD | [103] | |
| Diabetic Nephropathy | CASC2 | Diabetic Nephropathy patients (n = 27) | Serum and HG-treated mesangial cells | Downregulated | miR-133b/FOXP1 | SOD MDA | [104] |
| Diabetic Retinopathy | HF1A-AS2 | Diabetic Retinopathy patients (n = 60) | Serum | Upregulated | MAPK/VEGF | ONOO− NO MDA | [105] |
| Diabetic Kidney disease | ANRIL | Diabetic Kidney disease patients (n = 22) | Serum and HG-treated podocytes | Upregulated | [107] | ||
| Diabetic Nephropathy | Blnc1 | Diabetic Nephropathy patients (n = 30) | Serum and HG-treated HK2 cells | Upregulated | NRF2/HO-1 NF-κB | [108] | |
| Diabetic Kidney disease | ANRIL | Diabetic Kidney disease patients (n = 21) | PBMC | Upregulated | [109] | ||
| Diabetic Lung Disease | SCAL1 | Diabetic Lung disease patients (n = 56) | Serum and HG-treated lung cells | Downregulated | NO iNOS | [110] | |
| Diabetic Ischemic Stroke | NEAT1 | Diabetic Ischemic stroke patients (n = 22) | plasma | Upregulated | miR-124 | [111] |
| Diabetic Complications | lncRNA | Experimental Model | Expression Level | Function | Oxidative Stress/Cell Viability Markers | References |
|---|---|---|---|---|---|---|
| Diabetic Wound Healing | Lethe | HG-treated RAW264.7 | Downregulated | Increases NOX 2 expression and ROS production via modulating NF-κB signaling | Intracellular ROS | [121] |
| Diabetic Nephropathy | Gas5 | HG-treated HK2 | Downregulated | Increases oxidative stress and pyroptosis via modulating miR-452-5p | MDA SOD | [122] |
| Diabetic Neuropathy | MALAT1 | HG-treated BMEC | Upregulated | Promotes cellular apoptosis via upregulating miR-7641/TPR expression | BAX CASPASE3 | [123] |
| Diabetic Nephropathy | LIN01619 | HG-treated podocytes cells | Downregulated | Augments ER stress via modulating miR-27a/FOXO1 | Intracellular ROS | [124] |
| Diabetic Cardiomyopathy | DACH1 | HG-treated cardiomyocytes | Upregulated | Increases mitochondrial-derived ROS, mitochondrial dysfunction and cellular apoptosis via increasing SIRT3 degradation | MnSOD | [127] |
| Diabetic Retinopathy | MALAT1 NEAT1 | HG-treated HREC | Downregulated | Dysregulates mitochondrial homeostasis by damaging mitochondrial structure and genome integrity | mtROS | [128] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pant, T.; Uche, N.; Juric, M.; Bosnjak, Z.J. Clinical Relevance of lncRNA and Mitochondrial Targeted Antioxidants as Therapeutic Options in Regulating Oxidative Stress and Mitochondrial Function in Vascular Complications of Diabetes. Antioxidants 2023, 12, 898. https://doi.org/10.3390/antiox12040898
Pant T, Uche N, Juric M, Bosnjak ZJ. Clinical Relevance of lncRNA and Mitochondrial Targeted Antioxidants as Therapeutic Options in Regulating Oxidative Stress and Mitochondrial Function in Vascular Complications of Diabetes. Antioxidants. 2023; 12(4):898. https://doi.org/10.3390/antiox12040898
Chicago/Turabian StylePant, Tarun, Nnamdi Uche, Matea Juric, and Zeljko J. Bosnjak. 2023. "Clinical Relevance of lncRNA and Mitochondrial Targeted Antioxidants as Therapeutic Options in Regulating Oxidative Stress and Mitochondrial Function in Vascular Complications of Diabetes" Antioxidants 12, no. 4: 898. https://doi.org/10.3390/antiox12040898
APA StylePant, T., Uche, N., Juric, M., & Bosnjak, Z. J. (2023). Clinical Relevance of lncRNA and Mitochondrial Targeted Antioxidants as Therapeutic Options in Regulating Oxidative Stress and Mitochondrial Function in Vascular Complications of Diabetes. Antioxidants, 12(4), 898. https://doi.org/10.3390/antiox12040898