
The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
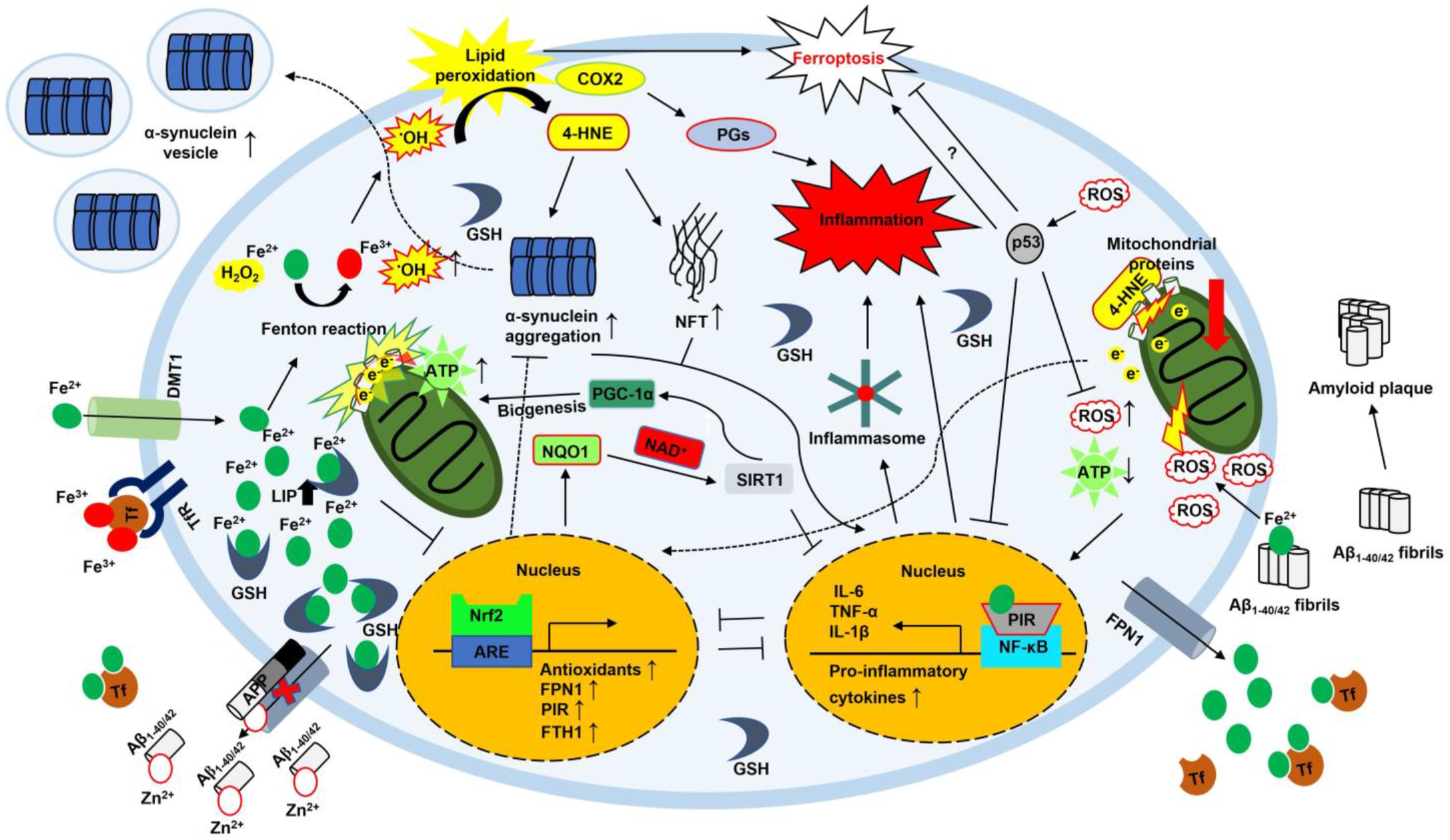
2. Intracellular Iron Homeostasis
2.1. Hepcidin
2.2. NCOA4
2.3. PCBPs
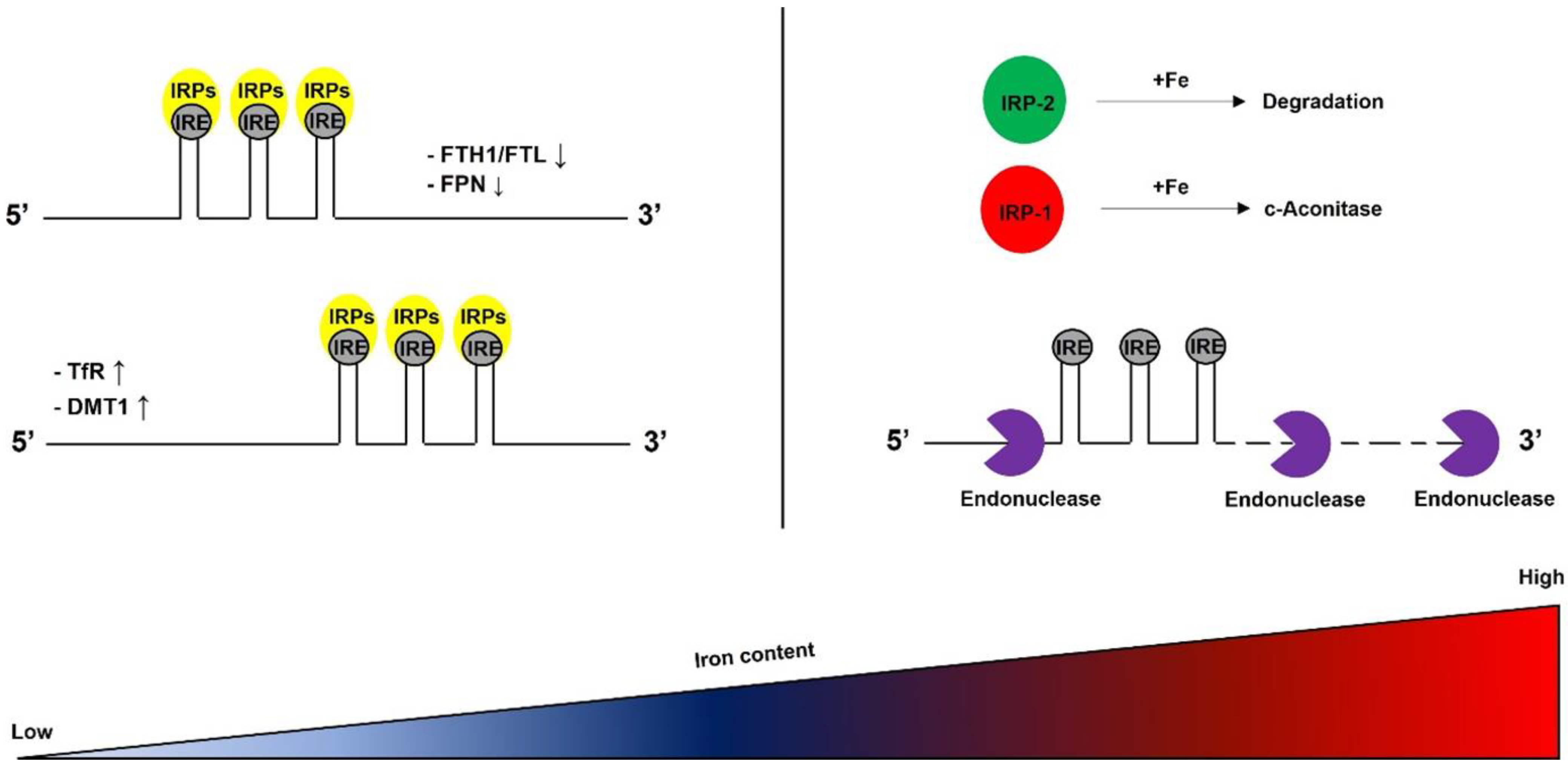
2.4. IRP/IRE System
2.5. DMT1
2.6. Ferritin
2.7. Ferroportin
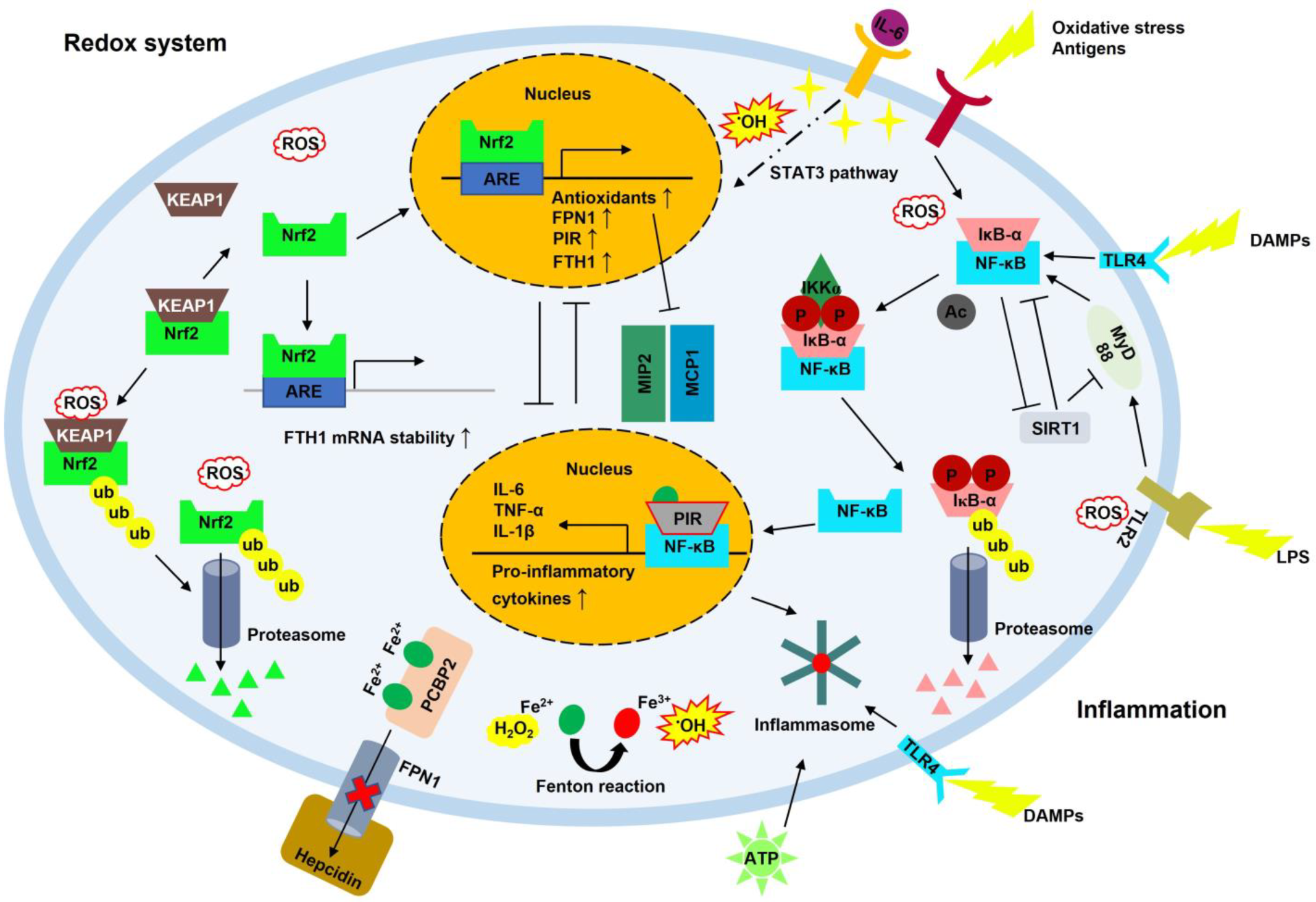
2.8. Neuroinflammation
2.9. NF-κB
2.10. SIRT1
2.11. Inflammasome
2.12. NRF2
3. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-syn | alpha-synuclein |
| Aβ | amyloid-beta |
| AD | Alzheimer’s disease |
| ALOX15 | arachidonate 15-lipoxygenase |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| ARE | antioxidant response element |
| ASC | apoptosis-associated speck-like protein containing a CARD |
| ATP | adenosine triphosphate |
| Bach1 | BTB and CNC homology 1 |
| BECN1 | beclin 1 |
| BMP | bone morphogenetic protein |
| CNS | central nervous system |
| COX2 | cyclooxygenase-2 |
| CRB | CREB-binding protein |
| DAMP | damage-associated molecular pattern |
| DcytB | duodenal cytochrome B |
| DMT1 | divalent metal transporter 1 |
| e− | electron |
| EMT | epithelial–mesenchymal transition |
| Fe2+ | ferrous iron |
| Fe3+ | ferric iron |
| FPN1 | ferroportin 1 |
| FTH1 | ferritin heavy chain |
| FTL | ferritin light chain |
| GSH | glutathione |
| 4-HNE | 4-hydroxy-2,3-trans-nonenal |
| HEPH | hephaestin |
| HERC2 | HECT domain and RCC1-like domain 2 |
| HMOX-1 | heme oxygenase |
| H2O2 | hydrogen peroxide |
| HO-1 | heme oxygenase-1 |
| IκB-α | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha |
| IKKα | IκB kinase alpha |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| IRE | iron-responsive element |
| IRP | iron-regulatory protein |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| LIP | labile iron pool |
| LPS | lipopolysaccharide |
| MCP1 | monocyte chemoattractant protein 1 |
| MIP2 | macrophage inflammatory protein 2 |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MyD88 | myeloid differentiation primary response protein 88 |
| NAD+ | nicotinamide adenine dinucleotide |
| NCOA4 | nuclear receptor coactivator 4 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFT | neurofibrillary tangle |
| NIK | NF-κB-inducing kinase |
| NLRP | nucleotide-binding domain and leucine-rich repeat and pyrin domain-containing protein |
| NOX2 | nicotinamide adenine dinucleotide phosphate oxidase 2 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| •OH | hydroxyl radical |
| OS | oxidative stress |
| P | phosphorylation |
| P2X7R | P2X purinergic receptor 7 |
| p53 | tumor protein P53 |
| PCBP | poly(rC)-binding protein |
| PD | Parkinson’s disease |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PIR | pirin |
| PPAR | peroxisome proliferator-activated receptor |
| PGs | prostaglandins |
| PUFA | polyunsaturated fatty acid |
| ROS | reactive oxygen species |
| SIRT1 | silent information regulator factor 2-related enzyme 1 |
| STAT3 | signal transducer and activator of transcription 3 |
| Steap3 | six-transmembrane epithelial antigen of prostate family member 3 |
| SMAD | suppressor of mothers against the decapentaplegic |
| Tf | transferrin |
| TfR | transferrin receptor |
| TLR | Toll-like receptor |
| TNF-α | tumor necrosis factor-α |
| TNFR | tumor necrosis factor receptor |
| TOM20 | translocase of the outer membrane 20 |
| Ub | ubiquitin |
| UPS | ubiquitin–proteosome system |
| UTR | untranslated region |
| Zn2+ | zinc ion |
References
- Drayer, B.; Burger, P.; Darwin, R.; Riederer, S.; Herfkens, R.; Johnson, G.A. MRI of brain iron. AJR Am. J. Roentgenol. 1986, 147, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Tishler, T.A.; Lu, P.H.; Villablanca, P.; Altshuler, L.L.; Carter, M.; Huang, D.; Edwards, N.; Mintz, J. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol. Aging 2007, 28, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Kernan, K.F.; Carcillo, J.A. Hyperferritinemia and inflammation. Int. Immunol. 2017, 29, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Wolozin, B.; Golts, N. Iron and Parkinson’s disease. Neuroscientist 2002, 8, 22–32. [Google Scholar] [CrossRef]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef]
- Meyer, E.; Kurian, M.A.; Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation: Genetic Diversity and Pathophysiological Mechanisms. Annu. Rev. Genomics Hum. Genet. 2015, 16, 257–279. [Google Scholar] [CrossRef]
- Reinert, A.; Morawski, M.; Seeger, J.; Arendt, T.; Reinert, T. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. 2019, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the human substantia nigra: An update. Neurotox. Res. 2014, 25, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.; Manceau, H.; Karim, Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse. Med. 2017, 46, e272–e278. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.A.; Huebers, H. Perspectives in iron metabolism. N. Engl. J. Med. 1982, 306, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Crichton, R. Iron Metabolism-From Molecular Mechanisms to Clinical Consequences; Wiley and Sons, Ltd.: Chichester, UK, 2009. [Google Scholar]
- Theil, E.C. Iron, ferritin, and nutrition. Annu. Rev. Nutr. 2004, 24, 327–343. [Google Scholar] [CrossRef] [Green Version]
- Macara, I.G.; Hoy, T.G.; Harrison, P.M. The formation of ferritin from apoferritin. Kinetics and mechanism of iron uptake. Biochem. J. 1972, 126, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Mancias, J.D.; Pontano Vaites, L.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [Green Version]
- Philpott, C.C.; Ryu, M.S.; Frey, A.; Patel, S. Cytosolic iron chaperones: Proteins delivering iron cofactors in the cytosol of mammalian cells. J. Biol. Chem. 2017, 292, 12764–12771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Hider, R.H. Iron and redox cycling. Do’s and don’ts. Free. Radic. Biol. Med. 2019, 133, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron: The cancer connection. Mol. Asp. Med. 2020, 75, 100860. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlgren-Beckendorf, J.A.; Reising, A.M.; Schander, M.A.; Herdler, J.W.; Johnson, J.A. Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: Role of the antioxidant/electrophile responsive element. Glia 1999, 25, 131–142. [Google Scholar] [CrossRef]
- Kennedy, K.A.; Sandiford, S.D.; Skerjanc, I.S.; Li, S.S. Reactive oxygen species and the neuronal fate. Cell. Mol. Life Sci. 2012, 69, 215–221. [Google Scholar] [CrossRef]
- Vieira, H.L.; Alves, P.M.; Vercelli, A. Modulation of neuronal stem cell differentiation by hypoxia and reactive oxygen species. Prog. Neurobiol. 2011, 93, 444–455. [Google Scholar] [CrossRef]
- Wilson, C.; Munoz-Palma, E.; Gonzalez-Billault, C. From birth to death: A role for reactive oxygen species in neuronal development. Semin. Cell Dev. Biol. 2018, 80, 43–49. [Google Scholar] [CrossRef]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Schulz, J.B.; Dringen, R. Glutathione release from cultured brain cells: Multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J. Neurosci. Res. 2002, 69, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-kappa B system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef]
- Komatsu, M.; Kageyama, S.; Ichimura, Y. p62/SQSTM1/A170: Physiology and pathology. Pharmacol. Res. 2012, 66, 457–462. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S369–S379. [Google Scholar] [CrossRef] [Green Version]
- Gow, A.J.; Duran, D.; Malcolm, S.; Ischiropoulos, H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996, 385, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Smith, M.A.; Avila, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.; Nunomura, A.; Honda, K.; Moreira, P.I.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005, 38, 746–754. [Google Scholar] [CrossRef]
- Bae, E.J.; Ho, D.H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.J.; Kim, K.P.; Lee, S.J. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid. Redox Signal. 2013, 18, 770–783. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, L.C. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics 2015, 7, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Roh, J.L. Poly(rC)-binding protein 1 represses ferritinophagy-mediated ferroptosis in head and neck cancer. Redox Biol. 2022, 51, 102276. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [Green Version]
- Hawula, Z.J.; Wallace, D.F.; Subramaniam, V.N.; Rishi, G. Therapeutic Advances in Regulating the Hepcidin/Ferroportin Axis. Pharmaceuticals 2019, 12, 170. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T.; Nemeth, E. Iron sequestration and anemia of inflammation. Semin. Hematol. 2009, 46, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Rivera, S.; Gabayan, V.; Keller, C.; Taudorf, S.; Pedersen, B.K.; Ganz, T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J. Clin. Investig. 2004, 113, 1271–1276. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Wincup, C.; Sawford, N.; Rahman, A. Pathological mechanisms of abnormal iron metabolism and mitochondrial dysfunction in systemic lupus erythematosus. Expert Rev. Clin. Immunol. 2021, 17, 957–967. [Google Scholar] [CrossRef]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129685. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Jadhav, S. The ins and outs of iron: Escorting iron through the mammalian cytosol. Free Radic. Biol. Med. 2019, 133, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Protchenko, O.; Shakoury-Elizeh, M.; Baratz, E.; Jadhav, S.; Philpott, C.C. The iron chaperone and nucleic acid-binding activities of poly(rC)-binding protein 1 are separable and independently essential. Proc. Natl. Acad. Sci. USA 2021, 118, e2104666118. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Shin, D.; Roh, J.L. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Ghanem, L.R.; Kromer, A.; Silverman, I.M.; Chatterji, P.; Traxler, E.; Penzo-Mendez, A.; Weiss, M.J.; Stanger, B.Z.; Liebhaber, S.A. The Poly(C) Binding Protein Pcbp2 and Its Retrotransposed Derivative Pcbp1 Are Independently Essential to Mouse Development. Mol. Cell. Biol. 2016, 36, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshinaga, M.; Nakatsuka, Y.; Vandenbon, A.; Ori, D.; Uehata, T.; Tsujimura, T.; Suzuki, Y.; Mino, T.; Takeuchi, O. Regnase-1 Maintains Iron Homeostasis via the Degradation of Transferrin Receptor 1 and Prolyl-Hydroxylase-Domain-Containing Protein 3 mRNAs. Cell Rep. 2017, 19, 1614–1630. [Google Scholar] [CrossRef] [Green Version]
- Leibold, E.A.; Munro, H.N. Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5’ untranslated region of ferritin heavy- and light-subunit mRNAs. Proc. Natl. Acad. Sci. USA 1988, 85, 2171–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, M.; Yoshimori, T.; Yamaguchi, K.; Yoshida, T.; Kishi, F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 2000, 275, 22220–22228. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.L.; Gelbart, T.; West, C.; Halloran, C.; Beutler, E. The human Nramp2 gene: Characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol. Dis. 1998, 24, 199–215. [Google Scholar] [CrossRef]
- Van Weert, A.W.; Dunn, K.W.; Geuze, H.J.; Maxfield, F.R.; Stoorvogel, W. Transport from late endosomes to lysosomes, but not sorting of integral membrane proteins in endosomes, depends on the vacuolar proton pump. J. Cell Biol. 1995, 130, 821–834. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Garrick, M.D.; Yang, F.; Dailey, L.A.; Piantadosi, C.A.; Ghio, A.J. TNF, IFN-gamma, and endotoxin increase expression of DMT1 in bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L24–L33. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N.Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Hartmann, A.; Troadec, J.D.; Hunot, S.; Kikly, K.; Faucheux, B.A.; Mouatt-Prigent, A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J. Neurosci. 2001, 21, 2247–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.D.; Tan, E.K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef] [Green Version]
- Hintze, K.J.; Katoh, Y.; Igarashi, K.; Theil, E.C. Bach1 repression of ferritin and thioredoxin reductase1 is heme-sensitive in cells and in vitro and coordinates expression with heme oxygenase1, beta-globin, and NADP(H) quinone (oxido) reductase1. J. Biol. Chem. 2007, 282, 34365–34371. [Google Scholar] [CrossRef] [Green Version]
- Jian, N.; Dowle, M.; Horniblow, R.D.; Tselepis, C.; Palmer, R.E. Morphology of the ferritin iron core by aberration corrected scanning transmission electron microscopy. Nanotechnology 2016, 27, 46LT02. [Google Scholar] [CrossRef]
- Mehlenbacher, M.; Poli, M.; Arosio, P.; Santambrogio, P.; Levi, S.; Chasteen, N.D.; Bou-Abdallah, F. Iron Oxidation and Core Formation in Recombinant Heteropolymeric Human Ferritins. Biochemistry 2017, 56, 3900–3912. [Google Scholar] [CrossRef]
- Bou-Abdallah, F.; Zhao, G.; Biasiotto, G.; Poli, M.; Arosio, P.; Chasteen, N.D. Facilitated diffusion of iron(II) and dioxygen substrates into human H-chain ferritin. A fluorescence and absorbance study employing the ferroxidase center substitution Y34W. J. Am. Chem. Soc. 2008, 130, 17801–17811. [Google Scholar] [CrossRef] [Green Version]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Nairz, M.; Weiss, G. Iron in infection and immunity. Mol. Aspects Med. 2020, 75, 100864. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.; Ganz, T.; Goodnough, L.T. Anemia of inflammation. Blood 2019, 133, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’Cruz, D.P.; Shoenfeld, Y. The hyperferritinemic syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibille, J.C.; Kondo, H.; Aisen, P. Interactions between isolated hepatocytes and Kupffer cells in iron metabolism: A possible role for ferritin as an iron carrier protein. Hepatology 1988, 8, 296–301. [Google Scholar] [CrossRef]
- Leimberg, M.J.; Prus, E.; Konijn, A.M.; Fibach, E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J. Cell. Biochem. 2008, 103, 1211–1218. [Google Scholar] [CrossRef]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [Green Version]
- Coffman, L.G.; Parsonage, D.; D’Agostino, R., Jr.; Torti, F.M.; Torti, S.V. Regulatory effects of ferritin on angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Knovich, M.A.; Coffman, L.G.; Torti, F.M.; Torti, S.V. Serum ferritin: Past, present and future. Biochim. Biophys. Acta 2010, 1800, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Ruddell, R.G.; Hoang-Le, D.; Barwood, J.M.; Rutherford, P.S.; Piva, T.J.; Watters, D.J.; Santambrogio, P.; Arosio, P.; Ramm, G.A. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatology 2009, 49, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Matzner, Y.; Hershko, C.; Polliack, A.; Konijn, A.M.; Izak, G. Suppressive effect of ferritin on in vitro lymphocyte function. Br. J. Haematol. 1979, 42, 345–353. [Google Scholar] [CrossRef]
- Broxmeyer, H.E.; Williams, D.E.; Geissler, K.; Hangoc, G.; Cooper, S.; Bicknell, D.C.; Levi, S.; Arosio, P. Suppressive effects in vivo of purified recombinant human H-subunit (acidic) ferritin on murine myelopoiesis. Blood 1989, 73, 74–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Luo, C.; Mines, M.; Zhang, J.; Fan, G.H. Chemokine CXCL12 induces binding of ferritin heavy chain to the chemokine receptor CXCR4, alters CXCR4 signaling, and induces phosphorylation and nuclear translocation of ferritin heavy chain. J. Biol. Chem. 2006, 281, 37616–37627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquita, G.; Silva, T.; Gomes, A.C.; Oliveira, P.F.; Alves, M.G.; Fernandes, R.; Almeida, A.A.; Moreira, A.C.; Gomes, M.S. H-Ferritin is essential for macrophages’ capacity to store or detoxify exogenously added iron. Sci. Rep. 2020, 10, 3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalcati, S.; Invernizzi, P.; Arosio, P.; Cairo, G. New functions for an iron storage protein: The role of ferritin in immunity and autoimmunity. J. Autoimmun. 2008, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Zandman-Goddard, G.; Shoenfeld, Y. Ferritin in autoimmune diseases. Autoimmun. Rev. 2007, 6, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.B.; Yang, F.; Haile, D.J. Functional consequences of ferroportin 1 mutations. Blood Cells Mol. Dis. 2005, 35, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Dielschneider, R.F.; Henson, E.S.; Xiao, W.; Choquette, T.R.; Blankstein, A.R.; Chen, Y.; Gibson, S.B. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS ONE 2017, 12, e0182921. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Abe, N.; Nishihara, T.; Yorozuya, T.; Tanaka, J. Microglia and Macrophages in the Pathological Central and Peripheral Nervous Systems. Cells 2020, 9, 2132. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet. Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, A.; Radhakrishnan, A.; Cabrero, I.A.; Cotsonis, G.; Cohen, C. COX-2 expression in invasive breast cancer: Correlation with prognostic parameters and outcome. Appl. Immunohistochem. Mol. Morphol. 2007, 15, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, S.K.; Zelic, M.; Han, Y.; Teeple, E.; Chen, L.; Sadeghi, M.; Shankara, S.; Guo, L.; Li, C.; Pontarelli, F.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef]
- Martin, M.; Sun, M.; Motolani, A.; Lu, T. The Pivotal Player: Components of NF-kappaB Pathway as Promising Biomarkers in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 7429. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Pra, I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-kappaB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2019, 326, 62–74. [Google Scholar] [CrossRef]
- Mattson, M.P.; Camandola, S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasstrom, T.; Fagerqvist, T.; Barbu, M.; Karlsson, M.; Nikolajeff, F.; Kasrayan, A.; Ekberg, M.; Lannfelt, L.; Ingelsson, M.; Bergstrom, J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of alpha-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 2011, 50, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. (Lond.) 2013, 124, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Deng, C.X.; Mostoslavsky, R. Recent progress in the biology and physiology of sirtuins. Nature 2009, 460, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Kim, M.S.; Roh, J.L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020, 37, 101697. [Google Scholar] [CrossRef]
- Hao, C.; Zhu, P.X.; Yang, X.; Han, Z.P.; Jiang, J.H.; Zong, C.; Zhang, X.G.; Liu, W.T.; Zhao, Q.D.; Fan, T.T.; et al. Overexpression of SIRT1 promotes metastasis through epithelial-mesenchymal transition in hepatocellular carcinoma. BMC Cancer 2014, 14, 978. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Zhu, L.; Lovaas, J.D.; Chmilewski, L.K.; Wang, J.; Faller, D.V.; Dai, Y. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 2012, 31, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Kokura, K.; Izumi, V.; Koomen, J.M.; Seto, E.; Chen, J.; Fang, J. MPP8 and SIRT1 crosstalk in E-cadherin gene silencing and epithelial-mesenchymal transition. EMBO Rep. 2015, 16, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Anoopkumar-Dukie, S.; Arora, D.; Davey, A.K. Review of the anti-inflammatory effect of SIRT1 and SIRT2 modulators on neurodegenerative diseases. Eur. J. Pharmacol. 2020, 867, 172847. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G. The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Qi, B.; Shi, C.; Meng, J.; Xu, S.; Liu, J. Resveratrol alleviates ethanol-induced neuroinflammation in vivo and in vitro: Involvement of TLR2-MyD88-NF-kappaB pathway. Int. J. Biochem. Cell Biol. 2018, 103, 56–64. [Google Scholar] [CrossRef]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Kalampokini, S.; Becker, A.; Fassbender, K.; Lyros, E.; Unger, M.M. Nonpharmacological Modulation of Chronic Inflammation in Parkinson’s Disease: Role of Diet Interventions. Parkinsons Dis. 2019, 2019, 7535472. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.F.; Li, N.; Wang, Q.; Cheng, X.J.; Li, X.M.; Liu, T.T. Resveratrol decreases the insoluble Abeta1-42 level in hippocampus and protects the integrity of the blood-brain barrier in AD rats. Neuroscience 2015, 310, 641–649. [Google Scholar] [CrossRef]
- Qi, Y.; Shang, L.; Liao, Z.; Su, H.; Jing, H.; Wu, B.; Bi, K.; Jia, Y. Intracerebroventricular injection of resveratrol ameliorated Abeta-induced learning and cognitive decline in mice. Metab. Brain Dis. 2019, 34, 257–266. [Google Scholar] [CrossRef]
- Feng, X.; Liang, N.; Zhu, D.; Gao, Q.; Peng, L.; Dong, H.; Yue, Q.; Liu, H.; Bao, L.; Zhang, J.; et al. Resveratrol inhibits beta-amyloid-induced neuronal apoptosis through regulation of SIRT1-ROCK1 signaling pathway. PLoS ONE 2013, 8, e59888. [Google Scholar]
- Ai, Z.; Li, C.; Li, L.; He, G. Resveratrol inhibits beta-amyloid-induced neuronal apoptosis via regulation of p53 acetylation in PC12 cells. Mol. Med. Rep. 2015, 11, 2429–2434. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Feng, X.; Wu, J.; Xu, H.; Li, G.; Zhu, D.; Yue, Q.; Liu, H.; Zhang, Y.; Sun, D.; et al. Neuroprotective effects of resveratrol on damages of mouse cortical neurons induced by beta-amyloid through activation of SIRT1/Akt1 pathway. Biofactors 2014, 40, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Zheng, L.J.; Shen, J.; Li, X.Y.; Zhang, Q.; Bai, X.; Wang, Q.S.; Ji, J.G. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural. Regen. Res. 2018, 13, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Gimenez-Llort, L.; Sarkis, C.; Pallas, M.; Sanfeliu, C. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement Through Proteostatic and Neurotrophic Mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, T.; Dong, S.Y.; Guo, Y.J.; Jankovic, J.; Xu, H.; Wu, Y.C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef]
- Singh, P.; Hanson, P.S.; Morris, C.M. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 2017, 18, 46. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Dong, S.Y.; Cui, X.X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.H.; Tan, E.K.; Zhao, W.J.; Wu, Y.C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of alpha-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef]
- Kanneganti, T.D. The inflammasome: Firing up innate immunity. Immunol. Rev. 2015, 265, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albornoz, E.A.; Woodruff, T.M.; Gordon, R. Inflammasomes in CNS Diseases. Exp. Suppl. 2018, 108, 41–60. [Google Scholar] [PubMed]
- Fan, Z.; Pan, Y.T.; Zhang, Z.Y.; Yang, H.; Yu, S.Y.; Zheng, Y.; Ma, J.H.; Wang, X.M. Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J. Neuroinflammation 2020, 17, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates alpha-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Gustot, A.; Gallea, J.I.; Sarroukh, R.; Celej, M.S.; Ruysschaert, J.M.; Raussens, V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 2015, 471, 323–333. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated alpha-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.S.; Thome, A.D.; Yan, Z.; Schonhoff, A.M.; Williams, G.P.; Li, X.; Liu, Y.; Qin, H.; Benveniste, E.N.; Standaert, D.G. Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp. Neurol. 2018, 300, 179–187. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Sluyter, R.; Gu, B.J.; Stokes, L.; Fuller, S.J. The human P2X7 receptor and its role in innate immunity. Tissue Antigens 2011, 78, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef]
- Sandberg, M.; Patil, J.; D’Angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: Implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered Nrf2 signaling mediates hypoglycemia-induced blood-brain barrier endothelial dysfunction in vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes. Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Aseervatham G, S.B.; Choi, S.; Krishnan, J.; Ruckmani, K. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86. [Google Scholar]
- Vomhof-Dekrey, E.E.; Picklo, M.J., Sr. The Nrf2-antioxidant response element pathway: A target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorato, N.G.; Rojo, A.I.; Garcia-Yague, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Asher, G.; Shaul, Y. Ubiquitin-independent degradation: Lessons from the p53 model. Isr. Med. Assoc. J. 2006, 8, 229–232. [Google Scholar] [PubMed]
- Zhao, C.; Gillette, D.D.; Li, X.; Zhang, Z.; Wen, H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014, 289, 17020–17029. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohe, R.; Flohe, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Magilnick, N.; Ou, X.; Lu, S.C. Tumour necrosis factor alpha induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor kappaB and activator protein-1. Biochem. J. 2005, 391, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-kappaB nuclear translocation via HO-1 activation underlies alpha-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J. Neuropathol. Exp. Neurol. 2010, 69, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rabano, A.; Kirik, D.; Cuadrado, A. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Sutter, T.R.; Kensler, T.W. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol. Med. 2001, 7, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar]
- Pietsch, E.C.; Chan, J.Y.; Torti, F.M.; Torti, S.V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003, 278, 2361–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, K.; Sun, J.; Taketani, S.; Nakajima, O.; Nishitani, C.; Sassa, S.; Hayashi, N.; Yamamoto, M.; Shibahara, S.; Fujita, H.; et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001, 20, 2835–2843. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M.; et al. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; de Serrano, V.; Liu, A. Pirin is an iron-dependent redox regulator of NF-kappaB. Proc. Natl. Acad. Sci. USA 2013, 110, 9722–9727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, H.; Bartlam, M.; Zeng, Q.; Miyatake, H.; Hisano, T.; Miki, K.; Wong, L.L.; Gao, G.F.; Rao, Z. Crystal structure of human pirin: An iron-binding nuclear protein and transcription cofactor. J. Biol. Chem. 2004, 279, 1491–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzoska, K.; Stepkowski, T.M.; Kruszewski, M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol. Cell. Biochem. 2014, 389, 99–111. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Hyun, D.-H. The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants 2023, 12, 918. https://doi.org/10.3390/antiox12040918
Lee J, Hyun D-H. The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants. 2023; 12(4):918. https://doi.org/10.3390/antiox12040918
Chicago/Turabian StyleLee, Jaewang, and Dong-Hoon Hyun. 2023. "The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases" Antioxidants 12, no. 4: 918. https://doi.org/10.3390/antiox12040918
APA StyleLee, J., & Hyun, D.-H. (2023). The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants, 12(4), 918. https://doi.org/10.3390/antiox12040918