
Central Facial Nervous System Biomolecules Involved in Peripheral Facial Nerve Injury Responses and Potential Therapeutic Strategies

Abstract
:1. Introduction
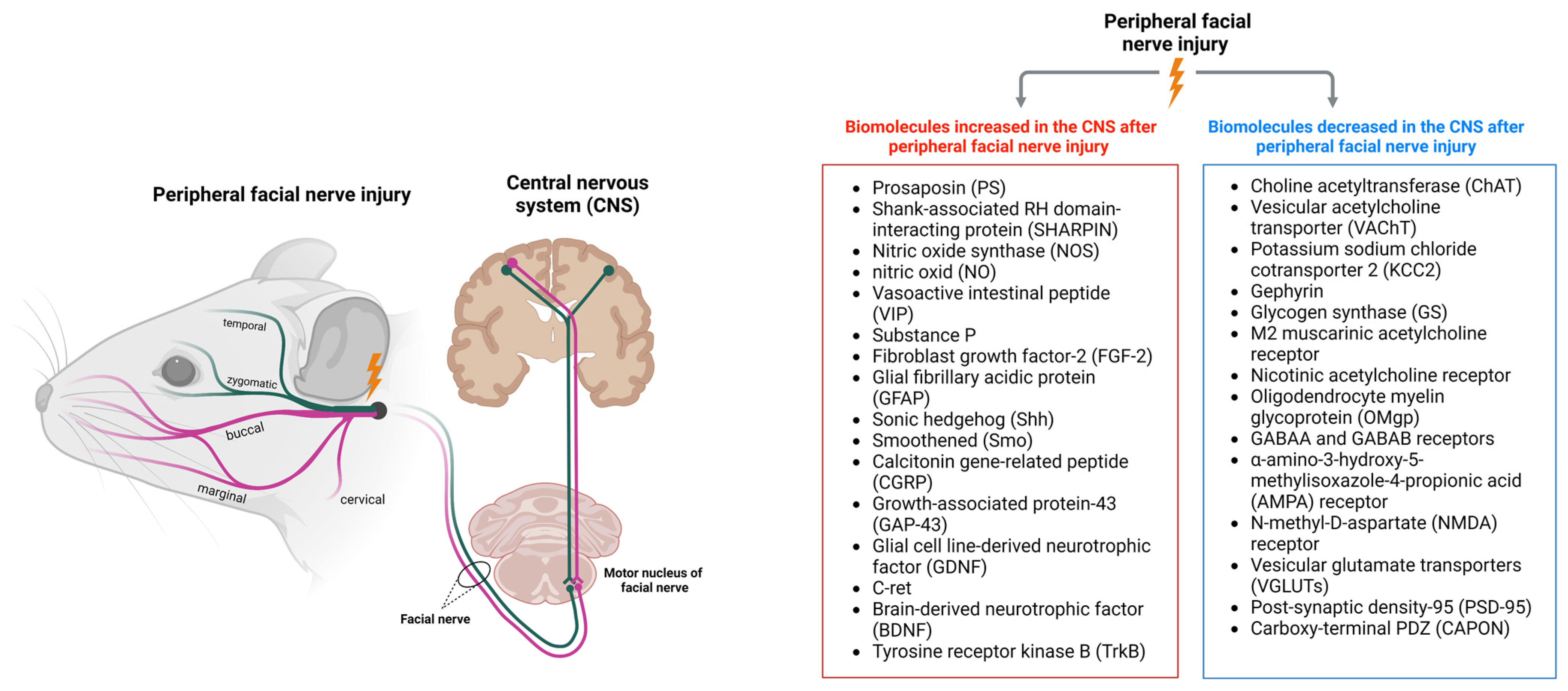
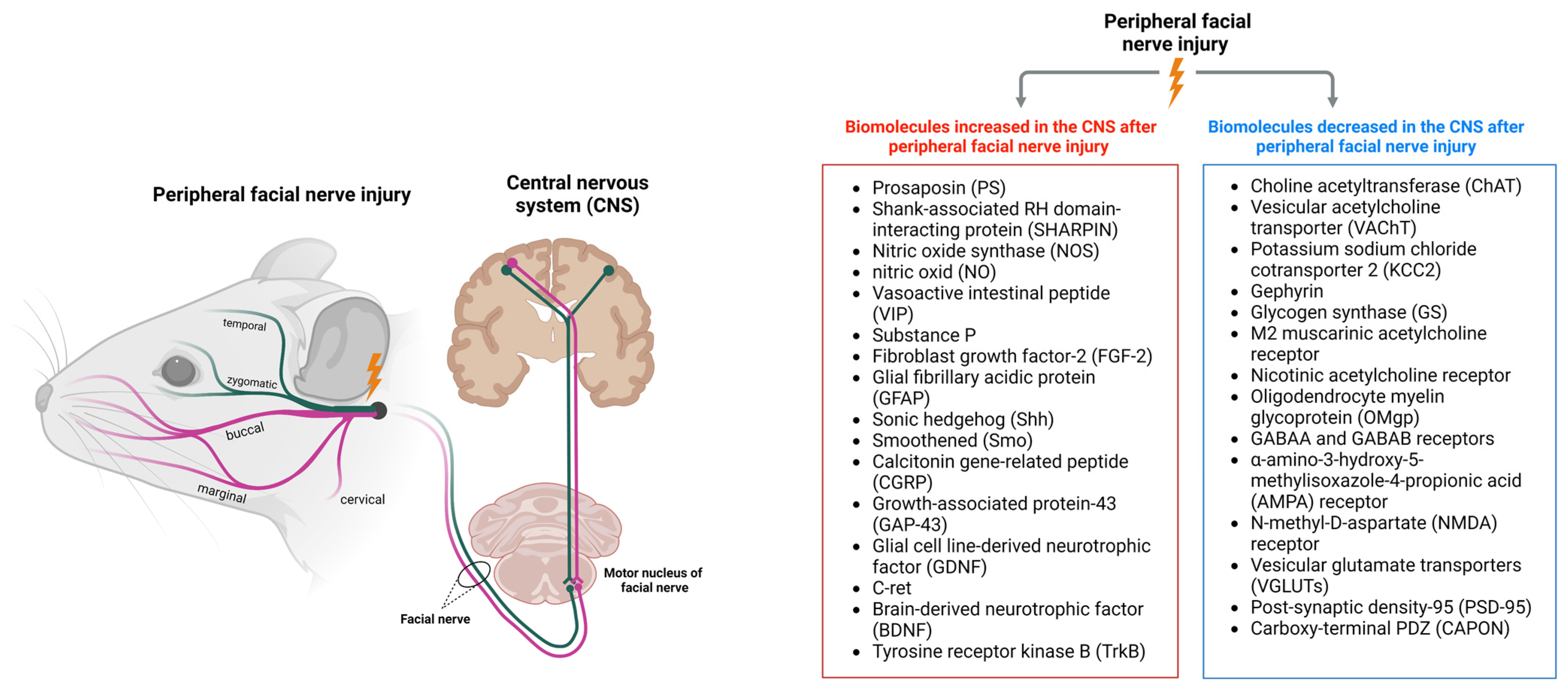
2. Changes in the CNS after Peripheral Facial Nerve Injury
2.1. Changes in Facial Motor Nerves
2.2. Cortical Reorganization
3. Biomolecules Increased in the CNS after Peripheral Facial Nerve Injury
3.1. Prosaposin
3.2. SHARPIN
3.3. Nitric Oxide Synthase and Nitric Oxide
3.4. Vasoactive Intestinal Peptide and Substance P
3.5. Fibroblast Growth Factor-2 and Glial Fibrillary Acidic Protein
3.6. Sonic Hedgehog and Smoothened
3.7. Calcitonin Gene-Related Peptide and Growth-Associated Protein-43
3.8. GDNF Family Receptor Alpha-1 and C-Ret
3.9. Brain-Derived Neurotrophic Factor and Tyrosine Receptor Kinase B
4. Biomolecules Decreased in the CNS after Peripheral Facial Nerve Injury
4.1. Choline Acetyltransferase and Vesicular Acetylcholine Transporter
4.2. Potassium Sodium Chloride Cotransporter 2 and Gephyrin
4.3. Glycogen Synthase
4.4. M2 Muscarinic Acetylcholine Receptor and Nicotinic Acetylcholine Receptor
4.5. Oligodendrocyte Myelin Glycoprotein
4.6. GABAA and GABAB Receptors
4.7. α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid Receptor and N-Methyl-D-as Partate Receptor
4.8. Vesicular Glutamate Transporter
4.9. Post-Synaptic Density-95 and Carboxy-Terminal PDZ
5. Central Facial Nerve Biomolecules and Processes Involved in Peripheral Facial Nerve Damage
5.1. Autophagy
5.2. Reactive Oxygen Species
5.3. Interleukin-10
5.4. Calcium
6. Changes in the CNS Resulting from Facial Nerve Injury
7. Discussion
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Takezawa, K.; Townsend, G.; Ghabriel, M. The Facial Nerve: Anatomy and Associated Disorders for Oral Health Professionals. Odontology 2018, 106, 103–116. [Google Scholar] [CrossRef] [PubMed]
- De Seta, D.; Mancini, P.; Minni, A.; Prosperini, L.; De Seta, E.; Attanasio, G.; Covelli, E.; De Carlo, A.; Filipo, R. Bell’s Palsy: Symptoms Preceding and Accompanying the Facial Paresis. Sci. World J. 2014, 2014, 801971. [Google Scholar] [CrossRef]
- Mavrikakis, I. Facial Nerve Palsy: Anatomy, Etiology, Evaluation, and Management. Orbit 2008, 27, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.; Gardner, D.; Miles, A.; Copley, A.; Wenke, R.; Coulson, S. A Systematic Review of Physical Rehabilitation of Facial Palsy. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Si-Yi, H.; Ling, W.; Hai-Bo, Y.; Yan-Hua, G.; Wei-Zheng, Z.; Xing-Xian, H.; Shao-Yun, Z.; Yong-Feng, L.; Yi-Rong, C. The Research for the Function Evaluation of Facial Nerve and the Mechanisms of Rehabilitation Training. Medicine 2021, 100, E25430. [Google Scholar] [CrossRef]
- Menorca, R.M.G.; Fussell, T.S.; Elfar, J.C. Nerve Physiology: Mechanisms of Injury and Recovery. Hand Clin. 2013, 29, 317–330. [Google Scholar] [CrossRef]
- Finsterer, J. Management of Peripheral Facial Nerve Palsy. Eur. Arch. Otorhinolaryngol. 2008, 265, 743–752. [Google Scholar] [CrossRef]
- Holland, N.J.; Weiner, G.M. Recent Developments in Bell’s Palsy. BMJ 2004, 329, 553–557. [Google Scholar] [CrossRef]
- Mustafa, A.H.K.; Sulaiman, A.M. The Epidemiology and Management of Bell’s Palsy in the Sudan. Open. Dent. J. 2018, 12, 827–836. [Google Scholar] [CrossRef]
- Shi, S.; Xu, L.; Li, J.; Han, Y.; Wang, H. Different Discharge Properties of Facial Nucleus Motoneurons Following Neurotmesis in a Rat Model. Neurosci. Lett. 2016, 629, 180–185. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X.; Yi, S. Pathophysiological Changes of Physical Barriers of Peripheral Nerves After Injury. Front. Neurosci. 2018, 12, 597. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.B.; Graeber, M.B. The Facial Nerve Axotomy Model. Brain Res. Rev. 2004, 44, 154–178. [Google Scholar] [CrossRef] [PubMed]
- Rotshenker, S. Wallerian Degeneration: The Innate-Immune Response to Traumatic Nerve Injury. J. Neuroinflammat. 2011, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Dailey, A.T.; Avellino, A.M.; Benthem, L.; Silver, J.; Kliot, M. Complement Depletion Reduces Macrophage Infiltration and Activation during Wallerian Degeneration and Axonal Regeneration. J. Neurosci. 1998, 18, 6713–6722. [Google Scholar] [CrossRef] [PubMed]
- Min, Q.; Parkinson, D.B.; Dun, X.-P. Migrating Schwann Cells Direct Axon Regeneration within the Peripheral Nerve Bridge. Glia 2021, 69, 235–254. [Google Scholar] [CrossRef]
- Guertin, A.D.; Zhang, D.P.; Mak, K.S.; Alberta, J.A.; Kim, H.A. Microanatomy of Axon/Glial Signaling during Wallerian Degeneration. J. Neurosci. 2005, 25, 3478–3487. [Google Scholar] [CrossRef]
- Tallon, C.; Farah, M.H. Beta Secretase Activity in Peripheral Nerve Regeneration. Neural Regen. Res. 2017, 12, 1565–1574. [Google Scholar] [CrossRef]
- Navarro, X.; Vivó, M.; Valero-Cabré, A. Neural Plasticity after Peripheral Nerve Injury and Regeneration. Prog. Neurobiol. 2007, 82, 163–201. [Google Scholar] [CrossRef]
- Jia, X.; Romero-Ortega, M.I.; Teng, Y.D. Peripheral Nerve Regeneration: Mechanism, Cell Biology, and Therapies. Biomed. Res. Int. 2014, 2014, 145304. [Google Scholar] [CrossRef]
- Sulaiman, W.; Gordon, T. Neurobiology of Peripheral Nerve Injury, Regeneration, and Functional Recovery: From Bench Top Research to Bedside Application. Ochsner J. 2013, 13, 100–108. [Google Scholar]
- Nocera, G.; Jacob, C. Mechanisms of Schwann Cell Plasticity Involved in Peripheral Nerve Repair after Injury. Cell. Mol. Life Sci. 2020, 77, 3977–3989. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, H. Peripheral Nerve Injury Induced Changes in the Spinal Cord and Strategies to Counteract/Enhance the Changes to Promote Nerve Regeneration. Neural Regen. Res. 2020, 15, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.B. Molecular Mechanisms, Biological Actions, and Neuropharmacology of the Growth-Associated Protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Mohri, D.; Satomi, F.; Kondo, E.; Fukuoka, T.; Sakagami, M.; Noguchi, K. Change in Gene Expression in Facial Nerve Nuclei and the Effect of Superoxide Dismutase in a Rat Model of Ischemic Facial Paralysis. Brain Res. 2001, 893, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ichimiya, T.; Yamamoto, S.; Honda, Y.; Kikuchi, R.; Kohsaka, S.; Nakajima, K. Functional Down-Regulation of Axotomized Rat Facial Motoneurons. Brain Res. 2013, 1507, 35–44. [Google Scholar] [CrossRef]
- Villacampa, N.; Almolda, B.; Vilella, A.; Campbell, I.L.; González, B.; Castellano, B. Astrocyte-Targeted Production of IL-10 Induces Changes in Microglial Reactivity and Reduces Motor Neuron Death after Facial Nerve Axotomy. Glia 2015, 63, 1166–1184. [Google Scholar] [CrossRef]
- Graeber, M.B.; López-Redondo, F.; Ikoma, E.; Ishikawa, M.; Imai, Y.; Nakajima, K.; Kreutzberg, G.W.; Kohsaka, S. The Microglia/Macrophage Response in the Neonatal Rat Facial Nucleus Following Axotomy. Brain Res. 1998, 813, 241–253. [Google Scholar] [CrossRef]
- Cattaneo, L.; Pavesi, G. The Facial Motor System. Neurosci. Biobehav. Rev. 2014, 38, 135–159. [Google Scholar] [CrossRef]
- Jemec, B.; Grobbelaar, A.O.; Harrison, D.H. The Abnormal Nucleus as a Cause of Congenital Facial Palsy. Arch. Dis. Child. 2000, 83, 256–258. [Google Scholar] [CrossRef]
- Ohki, M.; Takeuchi, N. Recovery of Motor Neuron Excitability after Facial Nerve Impairment in Rats. Neuroreport 2014, 25, 458–463. [Google Scholar] [CrossRef]
- Mateos-Aparicio, P.; Rodríguez-Moreno, A. The Impact of Studying Brain Plasticity. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.S.; Anastakis, D.J.; Davis, K.D. Cutting Your Nerve Changes Your Brain. Brain 2009, 132, 3122–3133. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ji, Q.; Xin, R.; Zhang, D.; Na, X.; Peng, R.; Li, K. Contralesional Cortical Structural Reorganization Contributes to Motor Recovery after Sub-Cortical Stroke: A Longitudinal Voxel-Based Morphometry Study. Front. Hum. Neurosci. 2016, 10. [Google Scholar] [CrossRef]
- Li, C.; Liu, S.-Y.; Pi, W.; Zhang, P.-X. Cortical Plasticity and Nerve Regeneration after Peripheral Nerve Injury. Neural Regen. Res. 2021, 16, 1518. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Cohen, L.G.; Hallett, M. Nervous System Reorganization Following Injury. Neuroscience 2002, 111, 761–773. [Google Scholar] [CrossRef]
- Galván, A. Neural Plasticity of Development and Learning. Hum. Brain Mapp. 2010, 31, 879–890. [Google Scholar] [CrossRef]
- Hosp, J.A.; Luft, A.R. Cortical Plasticity during Motor Learning and Recovery after Ischemic Stroke. Neural Plast. 2011, 2011, 871296. [Google Scholar] [CrossRef]
- Lee, J.; Yang, J.; Li, C.; Yuan, A.; Wu, H.; Wang, A.; Xue, Q.; Wang, T.; Wang, L.; Gao, T. Cortical Reorganization in Patients Recovered from Bell’s Palsy: An Orofacial and Finger Movements Task-State FMRI Study. Neural Plast. 2016, 2016, 8231726. [Google Scholar] [CrossRef]
- Hu, S.; Wu, Y.; Li, C.; Park, K.; Lu, G.; Mohamed, A.Z.; Wu, H.; Xu, C.; Zhang, W.; Wang, L.; et al. Increasing Functional Connectivity of the Anterior Cingulate Cortex during the Course of Recovery from Bell’s Palsy. Neuroreport 2015, 26, 6–12. [Google Scholar] [CrossRef]
- Medford, N.; Critchley, H.D. Conjoint Activity of Anterior Insular and Anterior Cingulate Cortex: Awareness and Response. Brain Struct. Funct. 2010, 214, 535–549. [Google Scholar] [CrossRef]
- Klingner, C.M.; Volk, G.F.; Maertin, A.; Brodoehl, S.; Burmeister, H.P.; Guntinas-Lichius, O.; Witte, O.W. Cortical Reorganization in Bell’s Palsy. Restor. Neurol. Neurosci. 2011, 29, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.; van der Geest, J.; Metselaar, M.; van der Lugt, A.; VanderWerf, F.; De Zeeuw, C. Long-Term Changes in Cerebellar Activation during Functional Recovery from Transient Peripheral Motor Paralysis. Exp. Neurol. 2010, 226, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hiraiwa, M.; Campana, W.M.; Mizisin, A.P.; Mohiuddin, L.; O’Brien, J.S. Prosaposin: A Myelinotrophic Protein That Promotes Expression of Myelin Constituents and Is Secreted after Nerve Injury. Glia 1999, 26, 353–360. [Google Scholar] [CrossRef]
- Kretz, K.A.; Carson, G.S.; Morimoto, S.; Kishimoto, Y.; Fluharty, A.L.; O’Brien, J.S. Characterization of a Mutation in a Family with Saposin B Deficiency: A Glycosylation Site Defect. Proc. Natl. Acad. Sci. USA 1990, 87, 2541–2544. [Google Scholar] [CrossRef]
- Sano, A.; Hineno, T.; Mizuno, T.; Kondoh, K.; Ueno, S.; Kakimoto, Y.; Inui, K. Sphingolipid Hydrolase Activator Proteins and Their Precursors. Biochem. Biophys. Res. Commun. 1989, 165, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Hiraiwa, M.; O’Brien, J. Saposins: Structure, Function, Distribution, and Molecular Genetics. J. Lipid Res. 1992, 33, 1255–1267. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Martin, B.M.; Kishimoto, Y.; Conner, G.E.; Tsuji, S.; O’Brien, J.S. Lysosomal Proteolysis of Prosaposin, the Precursor of Saposins (Sphingolipid Activator Proteins): Its Mechanism and Inhibition by Ganglioside. Arch. Biochem. Biophys. 1997, 341, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Unuma, K.; Chen, J.; Saito, S.; Kobayashi, N.; Sato, K.; Saito, K.; Wakisaka, H.; Mominoki, K.; Sano, A.; Matsuda, S. Changes in Expression of Prosaposin in the Rat Facial Nerve Nucleus after Facial Nerve Transection. Neurosci. Res. 2005, 52, 220–227. [Google Scholar] [CrossRef]
- Kotani, Y.; Matsuda, S.; Sakanaka, M.; Kondoh, K.; Ueno, S.; Sano, A. Prosaposin Facilitates Sciatic Nerve Regeneration In Vivo. J. Neurochem. 2002, 66, 2019–2025. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Liu, J.; Lu, A.-G.; Wang, C.-Y.; Misasi, R.; Yamauchi, T.; Hozumi, I.; Inuzuka, T.; O’Brien, J.S. Regulation of Gene Expression in Response to Brain Injury: Enhanced Expression and Alternative Splicing of Rat Prosaposin (SGP-1) MRNA in Injured Brain. J. Neurotrauma 2003, 20, 755–765. [Google Scholar] [CrossRef]
- Meyer, R.C.; Giddens, M.M.; Coleman, B.M.; Hall, R.A. The Protective Role of Prosaposin and Its Receptors in the Nervous System. Brain Res. 2014, 1585, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.C.; Giddens, M.M.; Schaefer, S.A.; Hall, R.A. GPR37 and GPR37L1 Are Receptors for the Neuroprotective and Glioprotective Factors Prosaptide and Prosaposin. Proc. Natl. Acad. Sci. 2013, 110, 9529–9534. [Google Scholar] [CrossRef] [PubMed]
- Kunihiro, J.; Nabeka, H.; Wakisaka, H.; Unuma, K.; Khan, M.S.I.; Shimokawa, T.; Islam, F.; Doihara, T.; Yamamiya, K.; Saito, S.; et al. Prosaposin and Its Receptors GRP37 and GPR37L1 Show Increased Immunoreactivity in the Facial Nucleus Following Facial Nerve Transection. PLoS ONE 2020, 15, e0241315. [Google Scholar] [CrossRef]
- Nabeka, H.; Uematsu, K.; Takechi, H.; Shimokawa, T.; Yamamiya, K.; Li, C.; Doihara, T.; Saito, S.; Kobayashi, N.; Matsuda, S. Prosaposin Overexpression Following Kainic Acid-Induced Neurotoxicity. PLoS ONE 2014, 9, e110534. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Mao, Y.; Li, X.; Liu, W.; Xu, L.; Han, Y.; Wang, H. The Alteration of SHARPIN Expression in the Mouse Brainstem during Herpes Simplex Virus 1-Induced Facial Palsy. Neurosci. Lett. 2015, 586, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.-H.; Ruan, R.-S.; Leong, S.-K.; Yeoh, K.-H. Compression of the Facial Nerve Caused Increased Nitric Oxide Synthase Activity in the Facial Motor Nucleus. Neuroscience 1995, 67, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Fan, Z.; Han, Y.; Liu, W.; Xu, L.; Jiang, Z.; Li, J.; Wang, H. The Alterations of Inducible Nitric Oxide Synthase in the Mouse Brainstem during Herpes Simplex Virus Type 1-Induced Facial Palsy. Neurol. Res. 2012, 34, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Mignini, F.; Giovannetti, F.; Cocchioni, M.; Ingrid, R.; Iannetti, G. Neuropeptide Expression and T-Lymphocyte Recruitment in Facial Nucleus after Facial Nerve Axotomy. J. Craniofacial Surg. 2012, 23, 1479–1483. [Google Scholar] [CrossRef]
- Coracini, K.; Fernandes, C.; Barbarini, A.; Silva, C.; Scabello, R.; Oliveira, G.; Chadi, G. Differential Cellular FGF-2 Upregulation in the Rat Facial Nucleus Following Axotomy, Functional Electrical Stimulation and Corticosterone: A Possible Therapeutic Target to Bell’s Palsy. J. Brachial Plex. Peripher. Nerve Inj. 2014, 05, e82–e96. [Google Scholar] [CrossRef]
- Akazawa, C.; Tsuzuki, H.; Nakamura, Y.; Sasaki, Y.; Ohsaki, K.; Nakamura, S.; Arakawa, Y.; Kohsaka, S. The Upregulated Expression of Sonic Hedgehog in Motor Neurons after Rat Facial Nerve Axotomy. J. Neurosci. 2004, 24, 7923–7930. [Google Scholar] [CrossRef]
- Ni, Y.; Chen, D.; Jiang, Y.; Qiu, D.; Li, W.; Li, H. The Regenerative Potential of Facial Nerve Motoneurons Following Chronic Axotomy in Rats. Neural Plast. 2020, 2020, 8884511. [Google Scholar] [CrossRef] [PubMed]
- Burazin, T.C.D.; Gundlach, A.L. Up-Regulation of GDNFR-α and c-Ret MRNA in Facial Motor Neurons Following Facial Nerve Injury in the Rat. Mol. Brain Res. 1998, 55, 331–336. [Google Scholar] [CrossRef]
- Kobayashi, N.R.; Bedard, A.M.; Hincke, M.T.; Tetzlaff, W. Increased Expression of BDNF and TrkB MRNA in Rat Facial Motoneurons after Axotomy. Eur. J. Neurosci. 1996, 8, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J. Herpes Simplex Virus Infections of the Central Nervous System. CONTINUUM Lifelong Learn. Neurol. 2015, 21, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, B.; Liu, F.; Wang, J.; Yang, Y.; Zheng, Y.; Tan, S. Shank-associated RH Domain-interacting Protein Expression Is Upregulated in Entodermal and Mesodermal Cancer or Downregulated in Ectodermal Malignancy. Oncol. Lett. 2018, 16, 7180–7188. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Potter, C.S.; Sundberg, J.P.; Hogenesch, H. SHARPIN Is a Key Regulator of Immune and Inflammatory Responses. J. Cell. Mol. Med. 2012, 16, 2271–2279. [Google Scholar] [CrossRef]
- Sieber, S.; Lange, N.; Kollmorgen, G.; Erhardt, A.; Quaas, A.; Gontarewicz, A.; Sass, G.; Tiegs, G.; Kreienkamp, H.-J. Sharpin Contributes to TNFα Dependent NFκB Activation and Anti-Apoptotic Signalling in Hepatocytes. PLoS ONE 2012, 7, e29993. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal. Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Liang, Y.; Seymour, R.E.; Sundberg, J.P. Inhibition of NF-ΚB Signaling Retards Eosinophilic Dermatitis in SHARPIN-Deficient Mice. J. Investig. Dermatol. 2011, 131, 141–149. [Google Scholar] [CrossRef]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.L.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN Forms a Linear Ubiquitin Ligase Complex Regulating NF-ΚB Activity and Apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef]
- Esplugues, J. V NO as a Signalling Molecule in the Nervous System. Br. J. Pharmacol. 2002, 135, 1079–1095. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Zamora, R.; Vodovotz, Y.; Billiar, T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Mol. Med. 2000, 6, 347–373. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Nitric Oxide and Cell Death. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1411, 401–414. [Google Scholar] [CrossRef]
- Cymerys, J.; Kowalczyk, A.; Mikołajewicz, K.; Słońska, A.; Krzyżowska, M. Nitric Oxide Influences HSV-1-Induced Neuroinflammation. Oxid. Med. Cell. Longev. 2019, 2019, 2302835. [Google Scholar] [CrossRef]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The Role of Nitric Oxide in Inflammatory Reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric Oxide and Redox Mechanisms in the Immune Response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- Hato, N.; Kohno, H.; Yamada, H.; Takahashi, H.; Gyo, K. Role of Nitric Oxide in the Onset of Facial Nerve Palsy by HSV-1 Infection. JAMA Otolaryngol. -Head Neck Surg. 2013, 139, 1339. [Google Scholar] [CrossRef]
- Ruan, R.S.; Leong, S.K.; Yeoh, K.H. Expression of NADPH-Diaphorase Activity in the Facial Motoneurons after Compression of the Facial Nerve in the Albino Rat. Brain Res. 1994, 652, 350–352. [Google Scholar] [CrossRef]
- Faraci, F.M.; Breese, K.R. Nitric Oxide Mediates Vasodilatation in Response to Activation of N-Methyl-D-Aspartate Receptors in Brain. Circ. Res. 1993, 72, 476–480. [Google Scholar] [CrossRef]
- Iwasaki, M.; Akiba, Y.; Kaunitz, J.D. Recent Advances in Vasoactive Intestinal Peptide Physiology and Pathophysiology: Focus on the Gastrointestinal System. F1000Research 2019, 8, 1629. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J. VIP and PACAP: Neuropeptide Modulators of CNS Inflammation, Injury, and Repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Abad, C.; Martinez, C.; Leceta, J.; Gomariz, R.P. Vasoactive Intestinal Peptide Prevents Experimental Arthritis by Downregulating Both Autoimmune and Inflammatory Components of the Disease. Nat. Med. 2001, 7, 563–568. [Google Scholar] [CrossRef]
- Bains, M.; Laney, C.; Wolfe, A.E.; Orr, M.; Waschek, J.A.; Ericsson, A.C.; Dorsam, G.P. Vasoactive Intestinal Peptide Deficiency Is Associated With Altered Gut Microbiota Communities in Male and Female C57BL/6 Mice. Front. Microbiol. 2019, 10, 2689. [Google Scholar] [CrossRef]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a Major Cytokine in the Central Nervous System. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide Substance P and the Immune Response. Cell. Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef]
- Galiano, M.; Liu, Z.Q.; Kalla, R.; Bohatschek, M.; Koppius, A.; Gschwendtner, A.; Xu, S.; Werner, A.; Kloss, C.U.A.; Jones, L.L.; et al. Interleukin-6 (IL6) and Cellular Response to Facial Nerve Injury: Effects on Lymphocyte Recruitment, Early Microglial Activation and Axonal Outgrowth in IL6-Deficient Mice. Eur. J. Neurosci. 2001, 14, 327–341. [Google Scholar] [CrossRef]
- Yun, Y.-R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.-H.; Shin, U.S.; Kim, H.-W. Fibroblast Growth Factors: Biology, Function, and Application for Tissue Regeneration. J. Tissue Eng. 2010, 1, 218142. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast Growth Factors. Genome Biol. 2001, 2, reviews3005.1. [Google Scholar] [CrossRef]
- Bikfalvi, A.; Klein, S.; Pintucci, G.; Rifkin, D.B. Biological Roles of Fibroblast Growth Factor-2*. Endocr. Rev. 1997, 18, 26–45. [Google Scholar] [CrossRef]
- Qian, X.; Davis, A.A.; Goderie, S.K.; Temple, S. FGF2 Concentration Regulates the Generation of Neurons and Glia from Multipotent Cortical Stem Cells. Neuron 1997, 18, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Leadbeater, W.E.; Gonzalez, A.-M.; Logaras, N.; Berry, M.; Turnbull, J.E.; Logan, A. Intracellular Trafficking in Neurones and Glia of Fibroblast Growth Factor-2, Fibroblast Growth Factor Receptor 1 and Heparan Sulphate Proteoglycans in the Injured Adult Rat Cerebral Cortex. J. Neurochem. 2006, 96, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Haastert, K.; Grosheva, M.; Angelova, S.K.; Guntinas-Lichius, O.; Skouras, E.; Michael, J.; Grothe, C.; Dunlop, S.A.; Angelov, D.N. Schwann Cells Overexpressing FGF-2 Alone or Combined with Manual Stimulation Do Not Promote Functional Recovery after Facial Nerve Injury. J. Biomed. Biotechnol. 2009, 2009, 408794. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Grothe, C. Differential Regulation of Fibroblast Growth Factor (FGF)-2 and FGF Receptor 1 MRNAs and FGF-2 Isoforms in Spinal Ganglia and Sciatic Nerve After Peripheral Nerve Lesion. J. Neurochem. 2002, 68, 1150–1158. [Google Scholar] [CrossRef]
- Fahmy, G.H.; Moftah, M.Z. Fgf-2 in Astroglial Cells during Vertebrate Spinal Cord Recovery. Front. Cell. Neurosci. 2010, 4, 129. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.P.; Duobles, T.; Castelucci, P.; Chadi, G. Differential Regulation of FGF-2 in Neurons and Reactive Astrocytes of Axotomized Rat Hypoglossal Nucleus. A Possible Therapeutic Target for Neuroprotection in Peripheral Nerve Pathology. Acta Histochem. 2010, 112, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Tan, D.; Wang, X.; Li, L.; Wen, J.; Pan, M.; Li, Y.; Wu, W.; Guo, J. Peripheral Nerve Injury-Induced Astrocyte Activation in Spinal Ventral Horn Contributes to Nerve Regeneration. Neural Plast. 2018, 2018, 8561704. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef]
- Cuevas, P.; Carceller, F.; Giménez-Gallego, G. Acidic Fibroblast Growth Factor Prevents Post-Axotomy Neuronal Death of the Newborn Rat Facial Nerve. Neurosci. Lett. 1995, 197, 183–186. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and Mechanisms. Genes. Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, Y.; Li, S.; Li, H.; Yang, C.; Lin, J. The Role of Shh Signalling Pathway in Central Nervous System Development and Related Diseases. Cell. Biochem. Funct. 2021, 39, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, Z.; Rikani, A.A.; Choudhry, A.M.; Tariq, S.; Zakaria, F.; Asghar, M.W.; Sarfraz, M.K.; Haider, K.; Shafiq, A.A.; Mobassarah, N.J. Sonic Hedgehog Signalling Pathway: A Complex Network. Ann. Neurosci. 2014, 21. [Google Scholar] [CrossRef] [PubMed]
- Dave, R.K.; Ellis, T.; Toumpas, M.C.; Robson, J.P.; Julian, E.; Adolphe, C.; Bartlett, P.F.; Cooper, H.M.; Reynolds, B.A.; Wainwright, B.J. Sonic Hedgehog and Notch Signaling Can Cooperate to Regulate Neurogenic Divisions of Neocortical Progenitors. PLoS ONE 2011, 6, e14680. [Google Scholar] [CrossRef]
- Lai, K.; Kaspar, B.K.; Gage, F.H.; Schaffer, D.V. Sonic Hedgehog Regulates Adult Neural Progenitor Proliferation in Vitro and in Vivo. Nat. Neurosci. 2003, 6, 21–27. [Google Scholar] [CrossRef]
- Gao, F.J.; Klinedinst, D.; Fernandez, F.-X.; Cheng, B.; Savonenko, A.; Devenney, B.; Li, Y.; Wu, D.; Pomper, M.G.; Reeves, R.H. Forebrain Shh Overexpression Improves Cognitive Function and Locomotor Hyperactivity in an Aneuploid Mouse Model of Down Syndrome and Its Euploid Littermates. Acta Neuropathol. Commun. 2021, 9, 137. [Google Scholar] [CrossRef]
- Kim, I.; Kim, Y.; Kang, D.; Jung, J.; Kim, S.; Rim, H.; Kim, S.; Yeo, S.-G. Neuropeptides Involved in Facial Nerve Regeneration. Biomedicines 2021, 9, 1575. [Google Scholar] [CrossRef]
- Amara, S.G.; Arriza, J.L.; Leff, S.E.; Swanson, L.W.; Evans, R.M.; Rosenfeld, M.G. Expression in Brain of a Messenger RNA Encoding a Novel Neuropeptide Homologous to Calcitonin Gene-Related Peptide. Science (1979) 1985, 229, 1094–1097. [Google Scholar] [CrossRef]
- Arvidsson, U.; Johnson, H.; Piehl, F.; Cullheim, S.; H kfelt, T.; Risling, M.; Terenius, L.; Ulfhake, B. Peripheral Nerve Section Induces Increased Levels of Calcitonin Gene-Related Peptide (CGRP)-like Immunoreactivity in Axotomized Motoneurons. Exp. Brain Res. 1990, 79, 212–216. [Google Scholar] [CrossRef]
- Ma, W.; Bisby, M.A. Differential Expression of Galanin Immunoreactivities in the Primary Sensory Neurons Following Partial and Complete Sciatic Nerve Injuries. Neuroscience 1997, 79, 1183–1195. [Google Scholar] [CrossRef]
- Kim, J.; Kobayashi, S.; Shimizu-Okabe, C.; Okabe, A.; Moon, C.; Shin, T.; Takayama, C. Changes in the Expression and Localization of Signaling Molecules in Mouse Facial Motor Neurons during Regeneration of Facial Nerves. J. Chem. Neuroanat. 2018, 88, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-J.; Zhang, F.-G.; Li, J.; Song, H.-X.; Zhou, L.-B.; Yao, B.-C.; Li, F.; Li, W.-C. Expression of Calcitonin Gene-Related Peptide in Anterior and Posterior Horns of the Spinal Cord after Brachial Plexus Injury. J. Clin. Neurosci. 2010, 17, 87–91. [Google Scholar] [CrossRef]
- Bendotti, C.; Pende, M.; Samanin, R. Expression of GAP-43 in the Granule Cells of Rat Hippocampus After Seizure-Induced Sprouting of Mossy Fibres: In Situ Hybridization and Immunocytochemical Studies. Eur. J. Neurosci. 1994, 6, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.; Stewart, H.J.; Hall, S.M.; Wilkin, G.P.; Mirsky, R.; Jessen, K.R. GAP-43 Is Expressed by Nonmyelin-Forming Schwann Cells of the Peripheral Nervous System. J. Cell. Biol. 1992, 116, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, L.C.; Verhaagen, J.; Edwards, P.M.; Hol, E.M.; Bär, P.R.; Gispen, W.H. The Expression of B-50/GAP-43 in Schwann Cells Is Upregulated in Degenerating Peripheral Nerve Stumps Following Nerve Injury. Brain Res. 1993, 602, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.; Shum, A.; Caraveo, G. GAP-43 and BASP1 in Axon Regeneration: Implications for the Treatment of Neurodegenerative Diseases. Front. Cell. Dev. Biol. 2020, 8, 567537. [Google Scholar] [CrossRef]
- Treanor, J.J.S.; Goodman, L.; de Sauvage, F.; Stone, D.M.; Poulsen, K.T.; Beck, C.D.; Gray, C.; Armanini, M.P.; Pollock, R.A.; Hefti, F.; et al. Characterization of a Multicomponent Receptor for GDNF. Nature 1996, 382, 80–83. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Houenou, L.J.; Johnson, J.E.; Lin, L.-F.H.; Li, L.; Lo, A.C.; Newsome, A.L.; Prevette, D.M.; Wang, S. Developing Motor Neurons Rescued from Programmed and Axotomy-Induced Cell Death by GDNF. Nature 1995, 373, 344–346. [Google Scholar] [CrossRef]
- Glazner, G.W.; Mu, X.; Springer, J.E. Localization of Glial Cell Line-Derived Neurotrophic Factor Receptor Alpha and c-Ret MRNA in Rat Central Nervous System. J. Comp. Neurol. 1998, 391, 42–49. [Google Scholar] [CrossRef]
- Zhao, Z.; Alam, S.; Oppenheim, R.W.; Prevette, D.M.; Evenson, A.; Parsadanian, A. Overexpression of Glial Cell Line-Derived Neurotrophic Factor in the CNS Rescues Motoneurons from Programmed Cell Death and Promotes Their Long-Term Survival Following Axotomy. Exp. Neurol. 2004, 190, 356–372. [Google Scholar] [CrossRef]
- Koeberle, P.D.; Ball, A.K. Neurturin Enhances the Survival of Axotomized Retinal Ganglion Cells in Vivo: Combined Effects with Glial Cell Line-Derived Neurotrophic Factor and Brain-Derived Neurotrophic Factor. Neuroscience 2002, 110, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Brain-Derived Neurotrophic Factor: Regulation, Effects, and Potential Clinical Relevance. Neurology 2015, 84, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Frostick, S.P.; Yin, Q.; Kemp, G.J. Schwann Cells, Neurotrophic Factors, and Peripheral Nerve Regeneration. Microsurgery 1998, 18, 397–405. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, I.N. Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed]
- Alcántara, S.; Frisén, J.; del Río, J.A.; Soriano, E.; Barbacid, M.; Silos-Santiago, I. TrkB Signaling Is Required for Postnatal Survival of CNS Neurons and Protects Hippocampal and Motor Neurons from Axotomy-Induced Cell Death. J. Neurosci. 1997, 17, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Gilmor, M.; Nash, N.; Roghani, A.; Edwards, R.; Yi, H.; Hersch, S.; Levey, A. Expression of the Putative Vesicular Acetylcholine Transporter in Rat Brain and Localization in Cholinergic Synaptic Vesicles. J. Neurosci. 1996, 16, 2179–2190. [Google Scholar] [CrossRef]
- Arvidsson, U.; Riedl, M.; Elde, R.; Meister, B. Vesicular Acetylcholine Transporter (VAChT) Protein: A Novel and Unique Marker for Cholinergic Neurons in the Central and Peripheral Nervous Systems. J. Comp. Neurol. 1997, 378, 454–467. [Google Scholar] [CrossRef]
- Ichikawa, T.; Ajiki, K.; Matsuura, J.; Misawa, H. Localization of Two Cholinergic Markers, Choline Acetyltransferase and Vesicular Acetylcholine Transporter in the Central Nervous System of the Rat: In Situ Hybridization Histochemistry and Immunohistochemistry. J. Chem. Neuroanat. 1997, 13, 23–39. [Google Scholar] [CrossRef]
- Takezawa, Y.; Kohsaka, S.; Nakajima, K. Transient Down-Regulation and Restoration of Glycogen Synthase Levels in Axotomized Rat Facial Motoneurons. Brain Res. 2014, 1586, 34–45. [Google Scholar] [CrossRef]
- Ishijima, T.; Nakajima, K. Changes of Signaling Molecules in the Axotomized Rat Facial Nucleus. J. Chem. Neuroanat. 2022, 126, 102179. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.M.; Brady, R.; Hersh, L.B.; Hayes, R.C.; Wiley, R.G. Expression of Choline Acetyltransferase and Nerve Growth Factor Receptor within Hypoglossal Motoneurons Following Nerve Injury. J. Comp. Neurol. 1991, 304, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Lazo, O.M.; Mauna, J.C.; Pissani, C.A.; Inestrosa, N.C.; Bronfman, F.C. Axotomy-Induced Neurotrophic Withdrawal Causes the Loss of Phenotypic Differentiation and Downregulation of NGF Signalling, but Not Death of Septal Cholinergic Neurons. Mol. Neurodegener. 2010, 5, 5. [Google Scholar] [CrossRef]
- Koyama, Y.; Fujiwara, T.; Kubo, T.; Tomita, K.; Yano, K.; Hosokawa, K.; Tohyama, M. Reduction of Oligodendrocyte Myelin Glycoprotein Expression Following Facial Nerve Transection. J. Chem. Neuroanat. 2008, 36, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Vassias, I.; Lecolle, S.; Vidal, P.P.; De Waele, C. Modulation of GABA Receptor Subunits in Rat Facial Motoneurons after Axotomy. Mol. Brain Res. 2005, 135, 260–275. [Google Scholar] [CrossRef] [PubMed]
- Eleore, L.; Vassias, I.; Vidal, P.P.; De Waele, C. Modulation of the Glutamatergic Receptors (AMPA and NMDA) and of Glutamate Vesicular Transporter 2 in the Rat Facial Nucleus after Axotomy. Neuroscience 2005, 136, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.H.; Tamatani, M.; Tohyama, M. Changes in MRNA for Post-Synaptic Density-95 (PSD-95) and Carboxy-Terminal PDZ Ligand of Neuronal Nitric Oxide Synthase Following Facial Nerve Transection. Mol. Brain Res. 2000, 76, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Senba, E.; Simmons, D.M.; Wada, E.; Wada, K.; Swanson, L.W. RNA Levels of Neuronal Nicotinic Acetylcholine Receptor Subunits Are Differentially Regulated in Axotomized Facial Motoneurons: An in Situ Hybridization Study. Mol. Brain Res. 1990, 8, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Boulenguez, P.; Liabeuf, S.; Bos, R.; Bras, H.; Jean-Xavier, C.; Brocard, C.; Stil, A.; Darbon, P.; Cattaert, D.; Delpire, E.; et al. Down-Regulation of the Potassium-Chloride Cotransporter KCC2 Contributes to Spasticity after Spinal Cord Injury. Nat. Med. 2010, 16, 302–307. [Google Scholar] [CrossRef]
- Nabekura, J.; Ueno, T.; Okabe, A.; Furuta, A.; Iwaki, T.; Shimizu-Okabe, C.; Fukuda, A.; Akaike, N. Reduction of KCC2 Expression and GABA A Receptor-Mediated Excitation after In Vivo Axonal Injury. J. Neurosci. 2002, 22, 4412–4417. [Google Scholar] [CrossRef]
- Choii, G.; Ko, J. Gephyrin: A Central GABAergic Synapse Organizer. Exp. Mol. Med. 2015, 47, e158. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Jiang, M.; Miralles, C.P.; Li, R.; Chen, G.; de Blas, A.L. Gephyrin Clustering Is Required for the Stability of GABAergic Synapses. Mol. Cell. Neurosci. 2007, 36, 484–500. [Google Scholar] [CrossRef]
- Toyoda, H.; Ohno, K.; Yamada, J.; Ikeda, M.; Okabe, A.; Sato, K.; Hashimoto, K.; Fukuda, A. Induction of NMDA and GABA A Receptor-Mediated Ca 2+ Oscillations With KCC2 MRNA Downregulation in Injured Facial Motoneurons. J. Neurophysiol. 2003, 89, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F. Contributions of Glycogen to Astrocytic Energetics during Brain Activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic Glycogen Metabolism in the Healthy and Diseased Brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [PubMed]
- Wayllace, N.Z.; Valdez, H.A.; Merás, A.; Ugalde, R.A.; Busi, M.V.; Gomez-Casati, D.F. An Enzyme-Coupled Continuous Spectrophotometric Assay for Glycogen Synthases. Mol. Biol. Rep. 2012, 39, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J.; Cheng, C.; Huang, D.; Lin, A.; Mu, J.; Skurat, A.V.; Wilson, W.; Zhai, L. Novel Aspects of the Regulation of Glycogen Storage. J. Basic. Clin. Physiol. Pharmacol. 1998, 9. [Google Scholar] [CrossRef] [PubMed]
- Saez, I.; Duran, J.; Sinadinos, C.; Beltran, A.; Yanes, O.; Tevy, M.F.; Martínez-Pons, C.; Milán, M.; Guinovart, J.J. Neurons Have an Active Glycogen Metabolism That Contributes to Tolerance to Hypoxia. J. Cereb. Blood Flow. Metab. 2014, 34, 945–955. [Google Scholar] [CrossRef]
- Woolf, C.J.; Chong, M.S.; Ainsworth, A. Axotomy Increases Glycogen Phosphorylase Activity in Motoneurones. Neuroscience 1984, 12, 1261–1269. [Google Scholar] [CrossRef]
- Obel, L.F.; Müller, M.S.; Walls, A.B.; Sickmann, H.M.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A. Brain Glycogen-New Perspectives on Its Metabolic Function and Regulation at the Subcellular Level. Front. Neuroenergetics 2012, 4, 3. [Google Scholar] [CrossRef]
- Resende, R.R.; Adhikari, A. Cholinergic Receptor Pathways Involved in Apoptosis, Cell Proliferation and Neuronal Differentiation. Cell. Commun. Signal. 2009, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, M.P.; Birdsall, N.J. International Union of Pharmacology. XVII. Classification of Muscarinic Acetylcholine Receptors. Pharmacol. Rev. 1998, 50, 279–290. [Google Scholar] [PubMed]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the Human M2 Muscarinic Acetylcholine Receptor Bound to an Antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, Y. G Protein Regulation of Cardiac Muscarinic Potassium Channel. Am. J. Physiol. -Cell. Physiol. 1995, 269, C821–C830. [Google Scholar] [CrossRef]
- Eglen, R.M.; Reddy, H.; Watson, N.; Challiss, R.A.J. Muscarinic Acetylcholine Receptor Subtypes in Smooth Muscle. Trends Pharmacol. Sci. 1994, 15, 114–119. [Google Scholar] [CrossRef]
- Wingler, L.M.; Lefkowitz, R.J. Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell. Biol. 2020, 30, 736–747. [Google Scholar] [CrossRef]
- Ishii, M.; Kurachi, Y. Muscarinic Acetylcholine Receptors. Curr. Pharm. Des. 2006, 12, 3573–3581. [Google Scholar] [CrossRef]
- Swanson, L.; Simmons, D.; Whiting, P.; Lindstrom, J. Immunohistochemical Localization of Neuronal Nicotinic Receptors in the Rodent Central Nervous System. J. Neurosci. 1987, 7, 3334–3342. [Google Scholar] [CrossRef]
- Yeh, J.J.; Ferreira, M.; Ebert, S.; Yasuda, R.P.; Kellar, K.J.; Wolfe, B.B. Axotomy and Nerve Growth Factor Regulate Levels of Neuronal Nicotinic Acetylcholine Receptor A3 Subunit Protein in the Rat Superior Cervical Ganglion. J. Neurochem. 2008, 79, 258–265. [Google Scholar] [CrossRef]
- Wada, E.; Wada, K.; Boulter, J.; Deneris, E.; Heinemann, S.; Patrick, J.; Swanson, L.W. Distribution of Alpha2, Alpha3, Alpha4, and Beta2 Neuronal Nicotinic Receptor Subunit MRNAs in the Central Nervous System: A Hybridization Histochemical Study in the Rat. J. Comp. Neurol. 1989, 284, 314–335. [Google Scholar] [CrossRef]
- Morioka, N.; Hisaoka-Nakashima, K.; Nakata, Y. Regulation by Nicotinic Acetylcholine Receptors of Microglial Glutamate Transporters: Role of Microglia in Neuroprotection. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Springer: Singapore, 2018; pp. 73–88. [Google Scholar]
- Vourc’h, P.; Andres, C. Oligodendrocyte Myelin Glycoprotein (OMgp): Evolution, Structure and Function. Brain Res. Brain Res. Rev. 2004, 45, 115–124. [Google Scholar] [CrossRef]
- Krämer, E.M.; Koch, T.; Niehaus, A.; Trotter, J. Oligodendrocytes Direct Glycosyl Phosphatidylinositol-Anchored Proteins to the Myelin Sheath in Glycosphingolipid-Rich Complexes. J. Biol. Chem. 1997, 272, 8937–8945. [Google Scholar] [CrossRef]
- Ghit, A.; Assal, D.; Al-Shami, A.S.; Hussein, D.E.E. GABAA Receptors: Structure, Function, Pharmacology, and Related Disorders. J. Genet. Eng. Biotechnol. 2021, 19, 123. [Google Scholar] [CrossRef] [PubMed]
- Kasaragod, V.B.; Mortensen, M.; Hardwick, S.W.; Wahid, A.A.; Dorovykh, V.; Chirgadze, D.Y.; Smart, T.G.; Miller, P.S. Mechanisms of Inhibition and Activation of Extrasynaptic Aβ GABAA Receptors. Nature 2022, 602, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.R.; Della-Flora Nunes, G.; Weaver, M.R.; Frick, L.R.; Feltri, M.L. Schwann Cell Interactions during the Development of the Peripheral Nervous System. Dev. Neurobiol. 2021, 81, 464–489. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Hamanoue, M.; Koshimoto, M.; Kohsaka, S.; Nakajima, K. Response of the GABAergic System to Axotomy of the Rat Facial Nerve. Neurochem. Res. 2018, 43, 324–339. [Google Scholar] [CrossRef]
- Kuhn, S.A.; van Landeghem, F.K.H.; Zacharias, R.; Färber, K.; Rappert, A.; Pavlovic, S.; Hoffmann, A.; Nolte, C.; Kettenmann, H. Microglia Express GABA B Receptors to Modulate Interleukin Release. Mol. Cell. Neurosci. 2004, 25, 312–322. [Google Scholar] [CrossRef]
- Rink-Notzon, S.; Reuscher, J.; Nohroudi, K.; Manthou, M.; Gordon, T.; Angelov, D.N. Trigeminal Sensory Supply Is Essential for Motor Recovery after Facial Nerve Injury. Int. J. Mol. Sci. 2022, 23, 15101. [Google Scholar] [CrossRef]
- Balasuriya, D.; Goetze, T.A.; Barrera, N.P.; Stewart, A.P.; Suzuki, Y.; Edwardson, J.M. α-Amino-3-Hydroxy-5-Methyl-4-Isoxazole Propionic Acid (AMPA) and N-Methyl-D-Aspartate (NMDA) Receptors Adopt Different Subunit Arrangements. J. Biol. Chem. 2013, 288, 21987–21998. [Google Scholar] [CrossRef]
- Scheefhals, N.; MacGillavry, H.D. Functional Organization of Postsynaptic Glutamate Receptors. Mol. Cell. Neurosci. 2018, 91, 82–94. [Google Scholar] [CrossRef]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Aizawa, S.; Inoue, Y.; Sugiyama, H.; et al. Reduced Hippocampal LTP and Spatial Learning in Mice Lacking NMDA Receptor Ε1 Subunit. Nature 1995, 373, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Xing, G.-G.; Wang, R.; Yang, B.; Zhang, D. Postnatal Switching of NMDA Receptor Subunits from NR2B to NR2A in Rat Facial Motor Neurons. Eur. J. Neurosci. 2006, 24, 2987–2992. [Google Scholar] [CrossRef] [PubMed]
- Gras, C.; Herzog, E.; Bellenchi, G.C.; Bernard, V.; Ravassard, P.; Pohl, M.; Gasnier, B.; Giros, B.; El Mestikawy, S. A Third Vesicular Glutamate Transporter Expressed by Cholinergic and Serotoninergic Neurons. J. Neurosci. 2002, 22, 5442–5451. [Google Scholar] [CrossRef] [PubMed]
- Vigneault, É.; Poirel, O.; Riad, M.; Prud’homme, J.; Dumas, S.; Turecki, G.; Fasano, C.; Mechawar, N.; El Mestikawy, S. Distribution of Vesicular Glutamate Transporters in the Human Brain. Front. Neuroanat. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Brumovsky, P.; Watanabe, M.; Hökfelt, T. Expression of the Vesicular Glutamate Transporters-1 and -2 in Adult Mouse Dorsal Root Ganglia and Spinal Cord and Their Regulation by Nerve Injury. Neuroscience 2007, 147, 469–490. [Google Scholar] [CrossRef]
- Hughes, D.I.; Polgár, E.; Shehab, S.A.S.; Todd, A.J. Peripheral Axotomy Induces Depletion of the Vesicular Glutamate Transporter VGLUT1 in Central Terminals of Myelinated Afferent Fibres in the Rat Spinal Cord. Brain Res. 2004, 1017, 69–76. [Google Scholar] [CrossRef]
- López-Redondo, F.; Nakajima, K.; Honda, S.; Kohsaka, S. Glutamate Transporter GLT-1 Is Highly Expressed in Activated Microglia Following Facial Nerve Axotomy. Mol. Brain Res. 2000, 76, 429–435. [Google Scholar] [CrossRef]
- Lin, Y.; Skeberdis, V.A.; Francesconi, A.; Bennett, M.V.L.; Zukin, R.S. Postsynaptic Density Protein-95 Regulates NMDA Channel Gating and Surface Expression. J. Neurosci. 2004, 24, 10138–10148. [Google Scholar] [CrossRef]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms That Modulate Metabolism. Cell. Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Repair Schwann Cell and Its Function in Regenerating Nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Li, R.; Li, D.; Wu, C.; Ye, L.; Wu, Y.; Yuan, Y.; Yang, S.; Xie, L.; Mao, Y.; Jiang, T.; et al. Nerve Growth Factor Activates Autophagy in Schwann Cells to Enhance Myelin Debris Clearance and to Expedite Nerve Regeneration. Theranostics 2020, 10, 1649–1677. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, L.; Zhang, H.; Li, S.; Liu, P.; Zhao, T.; Li, C. Autophagy Promotes Peripheral Nerve Regeneration and Motor Recovery Following Sciatic Nerve Crush Injury in Rats. J. Mol. Neurosci. 2016, 58, 416–423. [Google Scholar] [CrossRef]
- Gao, D.; Tang, T.; Zhu, J.; Tang, Y.; Sun, H.; Li, S. CXCL12 Has Therapeutic Value in Facial Nerve Injury and Promotes Schwann Cells Autophagy and Migration via PI3K-AKT-MTOR Signal Pathway. Int. J. Biol. Macromol. 2019, 124, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhang, H.; Xu, M.; Li, L.; Wu, M.; Zhang, S.; Liu, X.; Xia, W.; Xu, K.; Xiao, J.; et al. Delivery of Basic Fibroblast Growth Factor Through an In Situ Forming Smart Hydrogel Activates Autophagy in Schwann Cells and Improves Facial Nerves Generation via the PAK-1 Signaling Pathway. Front. Pharmacol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Guan, H.; Wang, X.; Chen, Z.; Zhu, W.; Wei, X.; Li, S. Cox4i2 Triggers an Increase in Reactive Oxygen Species, Leading to Ferroptosis and Apoptosis in HHV7 Infected Schwann Cells. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Mattsson, P.; Aldskogius, H.; Svensson, M. Nimodipine-Induced Survival Rate of Facial Motor Neurons Following Intracranial Transection of the Facial Nerve in the Adult Rat. J. Neurosurg. 1999, 90, 760–765. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Sun, M.-S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.-L.; Guo, Z.-N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxid. Med. Cell. Longev. 2018, 2018, 3804979. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive Oxygen Species and the Central Nervous System. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Burgaya, F.; Acarin, L.; Peluffo, H.; Castellano, B.; Gonzalez, B. Interleukin-10 and Interleukin Refeceptor-I Are Upregulated in Glial Cells After an Excitotoxic Injury to the Postnatal Rat Brain. J. Neuropathol. Exp. Neurol. 2009, 68, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Silva, D.; Carriche, G.M.; Castro, A.G.; Roque, S.; Saraiva, M. Balancing the Immune Response in the Brain: IL-10 and Its Regulation. J. Neuroinflammat. 2016, 13, 297. [Google Scholar] [CrossRef]
- Siqueira Mietto, B.; Kroner, A.; Girolami, E.I.; Santos-Nogueira, E.; Zhang, J.; David, S. Role of IL-10 in Resolution of Inflammation and Functional Recovery after Peripheral Nerve Injury. J. Neurosci. 2015, 35, 16431–16442. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.; Wainwright, D.A.; Mesnard, N.A.; Serpe, C.J.; Sanders, V.M.; Jones, K.J. IL-10 within the CNS Is Necessary for CD4+ T Cells to Mediate Neuroprotection. Brain Behav. Immun. 2011, 25, 820–829. [Google Scholar] [CrossRef]
- Runge, E.M.; Setter, D.O.; Iyer, A.K.; Regele, E.J.; Kennedy, F.M.; Sanders, V.M.; Jones, K.J. Cellular Sources and Neuroprotective Roles of Interleukin-10 in the Facial Motor Nucleus after Axotomy. Cells 2022, 11, 3167. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell. 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Khaitin, A. Calcium in Neuronal and Glial Response to Axotomy. Int. J. Mol. Sci. 2021, 22, 13344. [Google Scholar] [CrossRef]
- Limbrick, D.D.; Sombati, S.; DeLorenzo, R.J. Calcium Influx Constitutes the Ionic Basis for the Maintenance of Glutamate-Induced Extended Neuronal Depolarization Associated with Hippocampal Neuronal Death. Cell. Calcium 2003, 33, 69–81. [Google Scholar] [CrossRef]
- Rudkovskii, M.V.; Fedorenko, A.G.; Khaitin, A.M.; Pitinova, M.A.; Uzdensky, A.B. The Effect of Axotomy on Firing and Ultrastructure of the Crayfish Mechanoreceptor Neurons and Satellite Glial Cells. Mol. Cell. Neurosci. 2020, 107, 103534. [Google Scholar] [CrossRef]
- Herzfeld, E.; Strauss, C.; Simmermacher, S.; Bork, K.; Horstkorte, R.; Dehghani, F.; Scheller, C. Investigation of the Neuroprotective Impact of Nimodipine on Neuro2a Cells by Means of a Surgery-like Stress Model. Int. J. Mol. Sci. 2014, 15, 18453–18465. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J.; Rotterman, T.M.; Akhter, E.T.; Lane, A.R.; English, A.W.; Cope, T.C. Synaptic Plasticity on Motoneurons After Axotomy: A Necessary Change in Paradigm. Front. Mol. Neurosci. 2020, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Angelov, D.N.; Gunkel, A.; Stennert, E.; Neiss, W.F. Recovery of Original Nerve Supply after Hypoglossal-Facial Anastomosis Causes Permanent Motor Hyperinnervation of the Whisker-Pad Muscles in the Rat. J. Comp. Neurol. 1993, 338, 214–224. [Google Scholar] [CrossRef]
- Nakao, Y.; Matsumoto, K.; Kumagami, H. Altered Distribution of Motor Neurons in Experimental Facial Nerve Paralysis. Acta Otolaryngol. 1992, 112, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- TOTOKI, T.; HARANO, K.; AIKA, Y.; KANASEKI, T. CHANGES OF THE CELL BODIES IN THE FACIAL NUCLEUS AFTER FACIAL NERVE BLOCK. Br. J. Anaesth. 1980, 52, 563–566. [Google Scholar] [CrossRef]
- Liu, P.; Peng, J.; Han, G.-H.; Ding, X.; Wei, S.; Gao, G.; Huang, K.; Chang, F.; Wang, Y. Role of Macrophages in Peripheral Nerve Injury and Repair. Neural Regen. Res. 2019, 14, 1335–1342. [Google Scholar] [CrossRef]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their Role, Activation and Polarization in Pulmonary Diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef]

{kind=link}
| Biomolecules | Reference | Animal Model | Surgical Procedures | Experimental Design | Evaluations | Results | Conclusions |
|---|---|---|---|---|---|---|---|
| Prosaposin | Kunihiro et al., 2020 [53] | Male Wistar rats (n = 16) | Left facial nerve transection | -Group 1: Postoperative day 1 (n = 4) -Group 2: Postoperative day 3 (n = 4) -Group 3: Postoperative day 7 (n = 8) | -Immunohistochemistry -Immunofluorescence | -GPR37-IR was more intense in the cytoplasm of motoneurons on the operated side than on the untreated side and was markedly higher in microglia and astrocytes on the operated side. -Microglia with a strong GPR37L1-IR signal covered the damaged neurons. | -At the same time, secreted PS stimulates microglia or astrocytes via GPR37L1 to produce neuroprotective factors that protect the damaged neurons. |
| SHARPIN | Li et al., 2015 [55] | Balb/c mice (n = 142) | Inoculation on the right side with 25 µL of HSV-1 solution (2 × 107 TCID50/mL) | -Group 1: HSV-1 (n = 108) Group 1A: HSV-1 (sacrificed at 6 h and 1, 2, 3 and 7 days) Group 1B: HSV-1 +MPSS (30 mg/kg; for 2 days) Group 1C: HSV-1 + MPSS + RU486 (20 mg/kg) -Group 2: Normal saline (n = 25) -Group 3: Normal control (n = 9) | -Real-time PCR -Western blot -Immunofluorescence | -SHARPIN mRNA and protein expression were distinctly increased and peaked at 2 days after facial paralysis, then declined to normal levels during the following 5 days. -On the second day post-HSV-1 inoculation, the detection of SHARPIN protein by immunofluorescence coincided with peak SHARPIN expression. -SHARPIN expression was detected in the brainstem of HSV-1-infected mice and was localized to the cytoplasm of the facial nerve nucleus in the brainstem. | -The enhanced activity of SHARPIN in the early phase represents an important mechanism in HSV-1-induced facial paralysis. -SHARPIN might be a new target for the treatment of HSV-1-induced facial paralysis. |
| NOS | Wong et al., 1995 [56] | Male Wistar rats (n = NA) | Left facial nerve compression | -Group 1: Left facial nerve compression -Group 2: Sham-operated | -NOS assay -Histochemistry | -NOS activity in the FMN and surrounding tissues increased markedly (by ~60%) on the same side within 5 days after compression of the facial nerve, and remained significantly increased up to day 20. NOS activity decreased to control levels by day 30 after compression. -A significant decrease in NOS activity was observed on day 40. -NADPH-diaphorase reactivity in the FMN was markedly increased between days 21 and 42, peaking on day 35. | -Endothelial NOS activity increased in the initial period after nerve compression, coinciding with the period of facial paralysis and possible neuronal damage and perikaryal reactions in the FMN. |
| iNOS | Mao et al., 2012 [57] | Balb/c male mice (n = 143) | Inoculation on the left side with 25 µL of HSV-1 solution (2 × 107 TCID50/mL) | -Group 1: HSV-1 (n = 110) Group 1A: HSV-1 at different time points (6 h and 1, 2, 3, and 7 days) Group 1B: HSV-1 +MPSS (30 mg/kg; for 2 days) Group 1C: HSV-1 + MPSS + RU486 (20 mg/kg) -Group 2: Sham-normal saline (n = 24) -Group 3: Normal control (n = 9) | -Hematoxylin and eosin (H&E) staining -Trichrome staining -RT-PCR -Western blot -Immunohistochemistry | -HSV-1 increased mRNA expression of iNOS in the brainstem of facially paralyzed mice. -The expression of iNOS was increased and peaked at 2 days post-induction of facial paralysis, and then declined to normal levels during the following 5 days. -iNOS expression in motor neurons and glial cells was upregulated dramatically after HSV-1 infection. | -The nerve cell damage induced by HSV-1 infection was related to the overproduction of NO by iNOS. -Enhanced activity of the gene encoding iNOS in the early phase represents an important mechanism in HSV-1–induced facial paralysis. |
| VIP SP | Mignini, Fiorenzo et al., 2012 [58] | Male Wistar rats (n = 30) | Right facial nerve resection | -Group 1: Nerve axotomy (n = 30); 7, 14, or 21 days -Group 2: Control left side (n = 30) | -Immunohistochemistry -RT-PCR | -VIP+ and SP+ cells increased for 7 to 14 days after axotomy, whereas CD3+ cells increased from 48 h after axotomy and peaked at day 14 after injury. -VIP and SP mRNAs were as upregulated in the axotomized side 7 and 14 days, respectively, after surgery. -IL-6 levels 48 h after axotomy were significantly higher than those at 24 h. | -Actions of the neuropeptides, VIP and SP, are beneficial in inflammation -VIP and SP expression in the facial nerve could explain the role of T cells in preventing initial neuronal death or slowing the rate of neurodegeneration and neuronal loss. |
| FGF-2 GFAP | Coracini, Karen F. et al., 2010 [59] | Male Wistar rats (n = 18) | Right facial nerve crush for 30 s or a 3 mm transection | -Group 1: Facial nerve crush (n = 6) -Group 2: Facial nerve transection (n = 6) -Group 3: Sham-operated (n = 6) | -Immunohistochemistry | -The vast majority of nuclear FGF-2 immunoreactivity was associated with GFAP-positive astrocytes in rat facial nuclei. -A higher amount of FGF-2 was found in nuclei of reactive astrocytes of axotomized facial nuclei. -The degree of astroglial activation and the magnitude of changes in astroglial FGF-2 immunoreactivity were greater after facial nerve transection (without fiber regeneration) than after crush injury. | -The presence of FGF-2 immunoreactivity in neurons and astrocytes of the facial nucleus indicates that FGF-2 may be an important growth factor for peripheral motoneurons. -Expression of astroglial/neuronal FGF-2 in the facial nucleus may be correlated with local paracrine/autocrine trophic effects on axotomized facial motoneurons. |
| Shh Smo | Akazawa et al., 2004 [60] | Wistar rats (n = NA) | Facial nerve transection | -Group 1: Control -Group 2: Axotomy -Group 3: Axotomy + AdV-Shh -Group 4: Axotomy + AdV-lacZ | -Immunofluorescence -Immunohistochemistry -Northern and Western blot | -Shh expression was upregulated beginning 24 h after axotomy and declined at 4 weeks. -Smo mRNA expression was upregulated at 24 h after axotomy. -Shh transcripts and polypeptides were not upregulated after axotomy of neonatal rats. | -Shh is identified as a key molecule in nerve regeneration and shown to play a regulatory role after nerve injury. -Shh has potential therapeutic applications for regeneration of neuronal tissues after injuries in vivo. |
| CGRP GAP-43 | Mohri et al., 2001 [24] | Male Sprague-Dawley rats (n = 48) | -Transient paralysis (ischemia) -Right facial nerve transection | -Group 1: Right side facial nerve injury (n = 36) -Group 2: Untreated left side, serving as a Control (n = 36) -Group 3: Axotomy + saline (n = 3) -Group 4: Axotomy + SOD (n = 3) -Group 5: Ischemia + saline (n = 3) -Group 6: Ischemia + SOD (n = 3) | -Confocal laser-scanning microscopy -Immunohistochemistry | -CGRP mRNA levels in Group 1 showed a first peak at postoperative day 3 and a second peak on postoperative day 14. -The first increases in CGRP mRNA expression in Group 5 were less than those in Group 3. -The time course of c-Jun mRNA expression following ischemic nerve injury was similar to that after axotomy, although axotomy produced a greater upregulation of c-Jun mRNA than ischemia. -GAP-43 mRNA levels returned to control values before postoperative day 14. -CGRP mRNA expression in Group 6 on postoperative day 3 was inhibited compared with that in Group 5 | -CGRP and c-Jun mRNA expression may be dependent upon the extent and severity of nerve damage. -A minor injury to the peripheral nerve may elicit a small regenerative change in the cell body. -Free radicals generated by ischemia may be partially responsible for ischemic nerve damage and changes in gene expression in motoneurons. |
| Shh GAP-43 | Ni et al., 2020 [61] | Male Wistar rats (n = 50) | Right facial nerve transection | -Group 1: Axotomy (n = 10) -Group 2: Reinjury involving chronic axotomy (n = 40); at 12, 20, 28, and 36 weeks after the initial facial nerve axotomy | -Immunohistochemistry -Toluidine blue staining -Transmission electron microscopy -RT-PCR -Western blot | -Following reinjury, GAP-43 mRNA and protein in facial motoneurons were initially upregulated, but then gradually decreased. -Strong Shh immunoreactivity was observed in the cell bodies of facial motoneurons (GAP43-positive cells), but was not detected in the cell bodies of astrocytes (GFAP-positive cells). -Shh protein expression decreased over time following facial nerve reinjury. | -The regeneration potential of the facial nerve peaks within 5 months after chronic facial nerve axotomy in rats and may be dependent on activation of the Shh signaling pathway. |
| GDNFR-α c-ret | Burazin et al., 1998 [62] | Male Sprague-Dawley rats (n = 24) | Right facial nerve resection or crush | -Group 1: Nerve axotomy; at 1, 3, 7, 14 or 21 days (n = 3–4) following surgery -Group 2: Sham-operated; at 1 or 21 days (n = 2) following surgery | -In situ hybridization | -c-ret mRNA increased 1.4-fold in the ipsilateral facial nucleus 1 and 3 days following unilateral facial nerve crush or resection, respectively, but returned to levels equivalent to those on the contralateral side by postoperative days 7–21. -GDNFR-α mRNA was increased 2- to 3-fold in the ipsilateral facial nucleus at 1 and 3 days after facial nerve crush, reaching levels similar to those 3–21 days after resection | -The GDNF signaling system exerts powerful and long-lasting trophic effects in damaged neurons, further suggesting the broad potential for biological and therapeutic actions of GDNF and related factors in the CNS, particularly on motor neurons. |
| BDNF TrkB | Kobayash et al., 1996 [63] | Male Sprague-Dawley rats (n = 90) | Left facial nerve transection | -Group 1: Nerve axotomy (n = 90); at 3, 8, 16 and 24 h, and 2, 3, 4, 7, 14 and 21 days (n = 10) -Group 2: Control-right facial nerve (n = 90) | -In situ hybridization -RT-PCR -Western blot | -Axotomy increased BDNF mRNA expression in axotomized facial motoneurons as early as 8 h after injury and sustained it at levels 2- to 4-fold higher than those on the contralateral side for several days. -Increased expression of BDNF mRNA and protein was followed by increased expression of TrkB mRNA encoding the BDNF receptor, starting 2 days after axotomy and persisting for 2–3 weeks. -Axotomy increased both BDNF mRNA and protein several folds in facial motoneurons, as demonstrated by their cellular localization. | -Upregulation of BDNF mRNA within axotomized facial motoneurons and the production of BDNF protein within facial motor nuclei argue for autocrine trophic support of injured motoneurons. -BDNF increases might contribute to the survival of motoneurons after target disconnection by axotomy. |
| Biomolecules | Reference | Animal Model | Surgical Procedures | Experimental Design | Evaluations | Results | Conclusions |
|---|---|---|---|---|---|---|---|
| ChAT Gephyrin KCC2 | Kim et al., 2018 [111] | Male C57BL/6J mice (n = 42) | Transection of the main trunk of the right facial nerve except for the supraorbital nerve | Observed at days 3, 7, 14, 21, 28, and 60 after operation (7 mice per time point) | -Immunohistochemistry | -Only galanin expression had returned to normal levels 1 month after surgery. In contrast, expression of the other four molecules returned to normal levels by postoperative day 60. -Galanin appeared within cell bodies after surgery and had disappeared by day 28. -The number of ChAT-positive neurons in facial nuclei was lowest at day 7 and gradually increased after day 14, and the time course of changes in the ratio of the number of ChAT-positive neurons paralleled facial motor function. -Expression of gephyrin and KCC2 decreased from day 3 to day 28 and both recovered to normal levels by day 60; the time course of their restorations paralleled the recovery of FMN function. -Micro-separations comprising irregular spaces or astroglial processes were observed between motor neurons and pre-synapses. | -ChAT and KCC2 expression change during regeneration and may be objective indicators of regenerating axons, whereas galanin may be a marker for injured axons. -Decreases in KCC2 may play a role in re-extension of injured axons, and decreases in ChAT-positive neurons may be related to functional recovery. |
| ChAT VAChT Glycogen synthase | Takezawa et al., 2014 [130] | Male Wistar rats (n = 130) | Right facial nerve transection | Divided at various time points (1, 3, 5, 7, 14, 21, 28, and 35 days) | -Immunoblotting -Immunohistochemistry -Immunofluorescence -Nissl staining | -ChAT and VAChT in the operated nucleus were downregulated between days 3 and 14 post-injury. -GS levels in the injured nucleus were reduced beginning on day 7 post-injury, ultimately decreasing to ~50% of initial levels by postoperative day 14. -GS protein expression in motoneurons was significantly decreased in the injured nucleus. -The decrease in GS levels in injured motoneurons observed at 2 weeks post-insult recovered to original levels by 5 weeks in association with restoration of motoneuron functions. | -GS levels are altered in injured motoneurons in a rat facial nerve axotomy model. -The molecular mechanisms underlying energy metabolism in motoneurons are regulated during injury and regeneration. |
| ChAT VAChT m2MAchR | Ichimiya et al., 2013 [25] | Male rat littermates (n = NA) | Right facial nerve transection | Decapitated at early time points (1, 3, 5, 7, and 14 days) or later time points (3, 4, and 5 weeks) | -Immunoblotting -Immunohistochemistry -Immunofluorescence | -ChAT protein levels in the ipsilateral nucleus dropped significantly at an early stage after facial nerve transection in association with a parallel drop in VAchT protein levels. -ChAT and VAChT levels in the axotomized facial nucleus were downregulated starting 1 day after transection and remained depressed for 14 days. -m2MAchR levels in the transected nucleus were largely sustained (82%) up to 3 days after insult, but dropped markedly at 5 days and remained low thereafter. -ChAT and VAChT levels in the transected facial nucleus returned to control levels 4–5 weeks after injury. However, m2MAchR levels in the ipsilateral nucleus had not returned to control levels even at 5 weeks after insult. | -ChAT and VAchT were found to be downregulated in transected motoneurons almost immediately (days 1–3) after injury, whereas m2MAchR levels decreased starting on day 5 after insult. -Although m2MAchR was sustained at low levels for 5 weeks after injury, ChAT and VAChT recovered in the later stage (weeks 4–5). |
| OMgp | Koyama et al., 2008 [134] | Male Sprague-Dawley rats (n = 18) | Left facial nerve axotomy | -Group 1: Left facial nerve axotomy (n = 18); 1, 3, 5, 7, 14 and 28 days after nerve transection -Group 2: Sham operated-right facial nerve (n = 18) | -RT-PCR -Western blot -Immunohistochemistry | -OMgp mRNA levels significantly decreased from 3 to 14 days following axotomy, then returned to control levels at 28 days after transection. -OMgp immunoreactivity on the surface membrane of both neuronal soma and dendrites decreased significantly after axotomy 3–14 days after transection and then returned to control levels at 28 days post-axotomy. -OMgp expression did not change in oligodendrocytes and was not detectable in astrocytes. | -Neural OMgp might be involved in reconnecting neural circuits between axotomized and upper neurons. -Downregulation of neuronal OMgp expression after peripheral nerve axotomy provides important insights into the mechanism of OMgp in reconnecting the disconnected neural circuit after axotomy. |
| GABAA and GABAB receptors | Vassias et al., 2005 [135] | Male pigmented Long-Evans rats (n = 72) | -Left facial nerve cut | -Group 1: Left nerve axotomy (n = 42); divided into subgroups after lesion: 1 day (n = 12), 3 days (n = 12), 8 days (n = 12), 30 days (n = 3) and 60 days (n = 3) -Group 2: Control non-operated (n = 12) -Group 3: Colchicine infusion (n = 3) -Group 4: TTX infusion (n = 3) -Group 5: Cardiotoxin injection (n = 3) -Group 6: Botulinum toxin injection (n = 3) -Group 7: Control-PBS injection (n = 6) | -Immunohistochemistry | -mRNAs encoding α1, β2, and γ2 subunits of GABAA receptors were strongly downregulated in axotomized facial motoneurons as early as 3 days post-lesion and remained at low levels on post-lesion day 60. -mRNAs for GABA(B1B) and GABA(B2) receptor subunits were also downregulated by axotomy whereas those of GABA(B1A) remained unchanged. These changes in mRNA were accompanied by a decrease in GABA(B2) protein but not by a decrease in GABAB(1B) protein. -Colchicine reduced GABAA α1 immunoreactivity and mRNA levels in the facial nucleus ipsilateral to the injected side. -TTX treatment decreased α1 GABAA subunit expression in the lateral facial nucleus on day post-lesion 8. -Botulinum toxin had no effect at 1 week. | -Synaptic transmission of inhibitory inputs to facial motoneurons through GABAA and GABAB receptors is severely reduced by axotomy. -The loss of the GABAA receptor α1 subunit was most likely the consequence of three phenomena: the loss of trophic factor transported from the periphery, a positive injury signal, and disruption in activity. |
| AMPAR NMDAR | Eleore et al., 2005 [136] | Male pigmented Long-Evans rats (n = 73) | -Left facial nerve section -TTX application for 8 days | -Group 1: Nerve axotomy (n = 55); divided into 1, 3, 8, 30 or 60 days after nerve injury -Group 2: TTX injection (n = 3) -Group 3: Control-sham operated (n = 11) -Group 4: Control-PBS injection (n = 4) | -In situ hybridization -Immunohistochemistry | -GLuR2-3 mRNAs were substantially reduced after facial nerve lesion; GLuR4 mRNA was downregulated less strongly. -mRNAs for NR1 and NR2A B and D subunits were lost from motoneurons following axotomy. Facial nerve axotomy resulted in a decrease in NR1 subunits in facial nuclei ipsilateral to the lesion. -VGLUT2 immunoreactivity in injured nuclei was lower 3 and 8 days after axotomy. -Catenin and pan-cadherin immunostaining was markedly decreased at the periphery of cell soma. -TTX caused facial palsy similar to that observed after facial nerve axotomy. | -Axotomy severely alters glutamatergic synaptic transmission in facial motoneurons at both post-synaptic and pre-synaptic levels. -The loss of AMPAR and NMDAR subunits is partly induced by a disruption in activity. |
| PSD-95 CAPON | Che et al., 2000 [137] | Male Sprague-Dawley rats (n = 32) | Left facial nerve transection | -Group 1: Nerve axotomy (n = 24); at postoperative days 1, 3, 5, 7, 14, 21, 28 and 35 (n = 3 for each period) -Group 2: Sham-operated (n = 8); at postoperative days 1, 3, 5, 7, 14, 21, 28 and 35 (n = 1 for each period) | -In situ hybridization -NADPH-d staining | -PSD-95 mRNA expression was decreased from postoperative day 1 to 7, gradually increased thereafter, and returned to constitutive levels at postoperative day 28. -CAPON mRNA was decreased from postoperative day 1 to 5 and increased thereafter, reaching constitutive levels at postoperative day 28. -Axotomized nerves started to reconnect to the muscles between postoperative day 7 and 14, and the number of WGA-positive neurons increased until postoperative day 35. -nNOS mRNA expression was increased from postoperative day 7 to just prior to the beginning of reinnervation of the muscle by the axotomized facial nerve. | -Recovery of PSD-95 and CAPON mRNA expression is correlated with reinnervation of muscles. -PSD-95 and CAPON are involved in synaptogenesis and recovery of synaptic function in motoneurons after axotomy. |
| nAChR α3 subunit | Senba et al., 1990 [138] | Male Sprague-Dawley rats (n = 19) | Left facial nerve transection | -Group 1: Nerve axotomy (n = 16); divided into postoperative survival times: 6 h (n = 2), 12 h (n = 3), 1 day (n = 4), 1 week (n = 3) and 2 weeks (n = 4) -Group 2: Control (n = 3) | -In situ hybridization | -α3 subunit mRNA expression on the operated side was decreased to ~1/3 of that on the control side 1 day after axotomy and completely disappeared 1 week after axotomy. -β2 subunit mRNA levels were enhanced in motoneurons on the operated side. | -The synthesis of α and β subunits of neuronal nAChRs is differentially regulated in axotomized motoneurons and the two subunits may play functionally different roles during the regeneration process. |
| Biomolecule/Process | Reference | Animal Model | Surgical Procedures | Experimental Design | Evaluations | Results | Conclusions |
|---|---|---|---|---|---|---|---|
| Autophagy | Hu et al., 2022 [39] | Male Sprague Dawley rats (n = 50) | Main trunk of the left facial nerve clamped for 60 s | -Group 1: Sham group (n = 10) -Group2: FNI group (n = 10) -Group 3: FNI + poloxamer (n = 10) -Group 4: FNI + bFGF (n = 10) -Group 5: FNI + P-bFGF (n = 10) | -Hematoxylin and eosin (H&E) or Masson’s trichrome staining -Immunofluorescence -Western blot | -P-bFGF improved functional recovery of early facial nerve injury. -P-bFGF upregulated the functional protein S100 in Schwann cells and boosted the remyelination of these cells. -P-bFGF treatment enhanced the fluorescence intensity of autophagy-related proteins (LC3B, f LC3B-II, Beclin1, and ATG5) and reduced the pro-apoptosis proteins cleaved caspase-3 and BAX. | -P-bFGF effectively promotes cell proliferation, myelination and functional recovery and also reduces apoptosis of nerve cells after FNI by activation of the PAK1 pathway in Schwann cells. |
| Autophagy | Gao et al., 2019 [185] | Male Sprague-Dawley rats (n = 80) | -Right extracranial facial nerve main trunk pressed for 50 s -CXCL12 injection at a dose of 4 μg/kg/d | Part I (n = 36); divided into 0, 1, 3, 7, 17 and 28 days -Group 1: Nerve injury -Group 2: Sham Part II (n = 44); divided into 3 and 28 days -Group1: Nerve injury -Group 2: Nerve injury + CXCL12 | -H&E staining -Immunofluorescence -Western blot -Transmission electron microscopy | -Facial nerve injury enhanced the expression of CXCL12. CXCL12 significantly increased the migration of Schwann cells. -CXCL12 time-dependently increased autophagy of Schwann cells. -The autophagy inhibitor 3-MA significantly decreased CXCL12-induced expression of LC3II and increased expression of p62. -CXCL12 promoted Schwann cell migration through the PI3K/AKT/mTOR pathway. -CXCL12 promoted the recovery of facial nerve function and facilitated remyelination after facial nerve injury. | -CXCL12 has a therapeutic effect on facial nerve injury -CXCL12 acts through the PI3K/AKT/mTOR pathway to enhance autophagy and play a pivotal role in regulation of Schwann cell migration. |
| ROS | Chang et al., 2021 [187] | Female Wistar rats (n = NA) | Inoculation with 0.1 mL HHV7 virus solution | -Group 1: Normal control -Group 2: HHV7 infection -Group3: HHV7 infection + shNC -Group4: HHV7 infection + shCoxi42 | -Luxol Fast Blue staining -Immunofluorescence -Western blot -Flow cytometry -Phen Green SK staining -TUNEL assay | -Increased expression of Cox4i2 in Schwann cells infected with HHV7 promoted the production of ROS, and knockdown of Cox4i2 expression in HHV7-infected Schwann cells induced a relative decrease in ROS levels. -Increased expression of Cox4i2 led to an increase in ROS production in HHV7-infected Schwann cells that subsequently induced ferroptosis. Conversely, ferroptosis was inhibited by knock down of Cox4i2 in HHV7-infected Schwann cells. | -This study revealed a new mechanism of ROS-induced and Cox4i2-mediated apoptosis and ferroptosis in HHV7-infected Schwann cells. |
| IL-10 | Villacampa et al., 2015 [26] | GFAP-IL-10Tg (n = 66) Wild-type (n = 61) | Right facial nerve 1 mm resection | -Group 1: GFAP-IL-10Tg -Group 2: GFAP-IL-10Tg + Rt facial nerve axotomy -Group 3: Wild type -Group 4: Wild type + right facial nerve axotomy | -RT-PCR -Toluidine blue staining -Immunohistochemistry | -Greater CD3 positive lymphocyte infiltration was observed in the axotomized facial nerve of GFAP-IL-10Tg mice. -Astrocyte-targeted IL-10 production showed a strong beneficial effect on neuronal survival. -FMN constitutively express IL-10R; after facial nerve axotomy, IL-10R expression was lower but was maintained at all time-points. -Expression of CD39, an ectonucleotidase highly expressed in M2 macrophages, was increased in activated microglia from wild-type and GFAP-IL-10Tg animals after facial nerve axotomy. -The number of microglial cells significantly increased at 3 and 7 dpi on the lesioned facial nerve. | -IL-10 production within the CNS can lead to significant modifications in the pattern of microglial activation and T-cell infiltration and may exert a beneficial effect on the outcome of peripheral nerve injury. |
| Calcium | Mattsson et al., 1999 [188] | Male Sprague-Dawley rats (n = 37) | -Right facial nerve transection -Nimodipine administration from 3 days before the operation until death | -Group 1: Nerve axotomy (n = 14) -Group 2: Nerve axotomy + nimodipine (n = 16) -Group 3: Sham-operated animal (n = 7) | -Immunocytochemistry | -Nimodipine, a well-known antagonist of calcium influx, was shown to be neuroprotective after various lesions in the CNS. -OX42 immunoreactivity ipsilateral to the nerve injury increased from 2 to 7 days post-injury and remained at this level up to 28 days post-injury, in Groups 1 and 2. -ED1 immunoreactivity was increased ipsilateral to the nerve lesion from 2 to 28 days post-injury in Groups 1 and 2. | -Nimodipine, a calcium channel blocker that enhances blood flow and reduces ischemia, significantly improved neuronal survival for at least 1 month after oral administration in rats with intracranial transection of the facial nerve. -Nimodipine may have potential as a neuroprotective agent for various types of nerve injury. |
| Experimental Focus | Reference | Animal Model | Surgical Procedures | Experimental Design | Evaluations | Results | Conclusions |
|---|---|---|---|---|---|---|---|
| Cortex | Lee et al., 2016 [38] | Human (n = 37) | - | -Group 1: Recovered palsy (n = 17; male, 8, female, 9) -Group 2: Control (n = 20; female, 7, male, 13) | -Siemens Symphony 1.5 T MRI whole-body scanning -3D anatomical imaging -Task-state fMRI | -Cortical reorganization persisted in patients recovered from Bell’s palsy, or the functional status of the brain had not returned to the normal condition before the disease. -Activation significantly increased in the posterior cingulate cortex (PCC), primary somatosensory cortex (SI), primary motor cortex (MI) and cingulate motor area (CMA), and decreased in the parahippocampal gyrus during finger movements. -Signals decreased in the SI, PCC, precuneus, and culmen during lip pursing movements. -Cerebral blood flow in the facial motor area of the brain in patients recovered from Bell’s palsy was reduced, whereas that in hand motor areas was enhanced. | -Regions showing changes in activation between the two groups included the motor association cortex and cerebellum. -All of these changes in the cortex might be relevant to differences in the functional status of the brain. |
| Discharge properties | Shi et al., 2016 [10] | Female Wistar rats (n = 8) | -Transection of the right trunk of the facial nerve -Implantation of a 4 × 4 electrode arrays into the brainstem on the right | -Group1: Right facial nerve transection (n = 8) -Group 2: Non-injured side (n = 8) | -Toluidine blue staining -Transmission electron microscopy | -Nerve degeneration, manifested as disorderly distributed smaller fibers with demyelination and swelling of organelles, was detected in the injured group compared with Group 2. -The sustained spike pattern of neuron A changed to a phasic pattern and the firing rate of neuron B decreased compared with its original firing rate. -The mean frequency, coefficient of variation median ISI and modal ISI were significantly changed in both neuron A and neuron B during execution of movements following neurotmesis. | -Neurotmesis attenuated nerve firing rates, and changed firing patterns throughout the duration of movements, all of which may provide a theoretical basis for observed facial palsy, synkinesia, and prosopospasm. |
| Nucleus | Jemec et al., 2000 [29] | Human (n = 21) | - | -Group: Patients with CFP (n = 21, male, 7, female, 14); includes those with CFP as their sole symptom (n = 15) and with syndromes that can include CFP (n = 5) | -MRI scan | -Five (24%) of the abnormal scans showed a lesion in the area of the facial nucleus in the pons; in two, the nucleus was completely absent, and in the other three, T1/T2 weighting in the area was abnormal. -Three (14%) of the abnormal scans showed other abnormalities, including a prominent circle of Willis, partial agenesis of the corpus callosum, and cerebellar hypoplasia. | -Developmental abnormalities of the facial nucleus itself constitute an important, and previously ignored, cause of monosymptomatic unilateral CFP. |
| Motor neurons | Nakao et al., 1992 [205] | Rabbits (n = 15) | Facial nerve crush | -Group 1: Control (n = 10) -Group 2: Nerve crush (n = 5) | -Immunohistochemistry | -After recovery from facial nerve paralysis, labeled neurons innervating the zygomatic muscle were located not only in the ventromedial portion but also partially in the dorsomedial portion. -Multipolar neurons of varying size were labeled bilaterally in the reticular formation from the pons to the medulla. These neurons contained HRP granules that were brown in color but paler than those in the facial nucleus. | -Labeled premotor neurons may have a role in muscle movements after recovery from facial nerve palsy. |
| Cell body | Totoki et al., 1980 [206] | Male Japanese monkeys (n = 4) | Block of the neural canal of the facial nerve by insertion of a 22-gauge needle | -Group 1: Control (n = 1) -Group 2: Nerve block (n = 3) | -Toluidine blue staining | -Nerve cells of the facial nucleus were circular, with their nuclei shifted to one side of the cell, 4 days after nerve block. -Nissl staining was dispersed and the size of Nissl granules was decreased in neurons. -Nissl granules were still small and nuclei were positioned towards one side of the cell 2 months after nerve block. -Nerve cells in the facial nucleus were nearly normal in the blocked side 7 months after nerve block, but recovery was incomplete and small Nissl granules were present in some cells. | -Nerve function recovered in 2 months, but 7 months were required for nerve cells in the facial nerve nucleus to recover completely. -A mechanism to account for the complete remission in patients ~6 months after nerve block is suggested. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-M.; Choi, Y.J.; Yoo, M.C.; Yeo, S.G. Central Facial Nervous System Biomolecules Involved in Peripheral Facial Nerve Injury Responses and Potential Therapeutic Strategies. Antioxidants 2023, 12, 1036. https://doi.org/10.3390/antiox12051036
Lee J-M, Choi YJ, Yoo MC, Yeo SG. Central Facial Nervous System Biomolecules Involved in Peripheral Facial Nerve Injury Responses and Potential Therapeutic Strategies. Antioxidants. 2023; 12(5):1036. https://doi.org/10.3390/antiox12051036
Chicago/Turabian StyleLee, Jae-Min, You Jung Choi, Myung Chul Yoo, and Seung Geun Yeo. 2023. "Central Facial Nervous System Biomolecules Involved in Peripheral Facial Nerve Injury Responses and Potential Therapeutic Strategies" Antioxidants 12, no. 5: 1036. https://doi.org/10.3390/antiox12051036
APA StyleLee, J.-M., Choi, Y. J., Yoo, M. C., & Yeo, S. G. (2023). Central Facial Nervous System Biomolecules Involved in Peripheral Facial Nerve Injury Responses and Potential Therapeutic Strategies. Antioxidants, 12(5), 1036. https://doi.org/10.3390/antiox12051036