

Metabolic Priming as a Tool in Redox and Mitochondrial Theragnostics




Abstract
:1. Introduction
2. Mitochondrial Alterations in Health and Disease
2.1. Mitochondria as Dynamic Cellular Information Hubs
2.2. Mitochondrial Stress
2.3. Mitochondrial Dysfunction
2.4. Mitochondrial Function as an Important End-Point in Drug Development
3. Mitochondrial Stress Leading to Cellular Redox and Metabolic Remodeling
3.1. Cellular Responses to Metabolic Stress
3.2. Protein Distribution and Density in Response to Metabolic Stress
3.3. Cellular Redox Alterations
3.4. Redox In Vitro Models
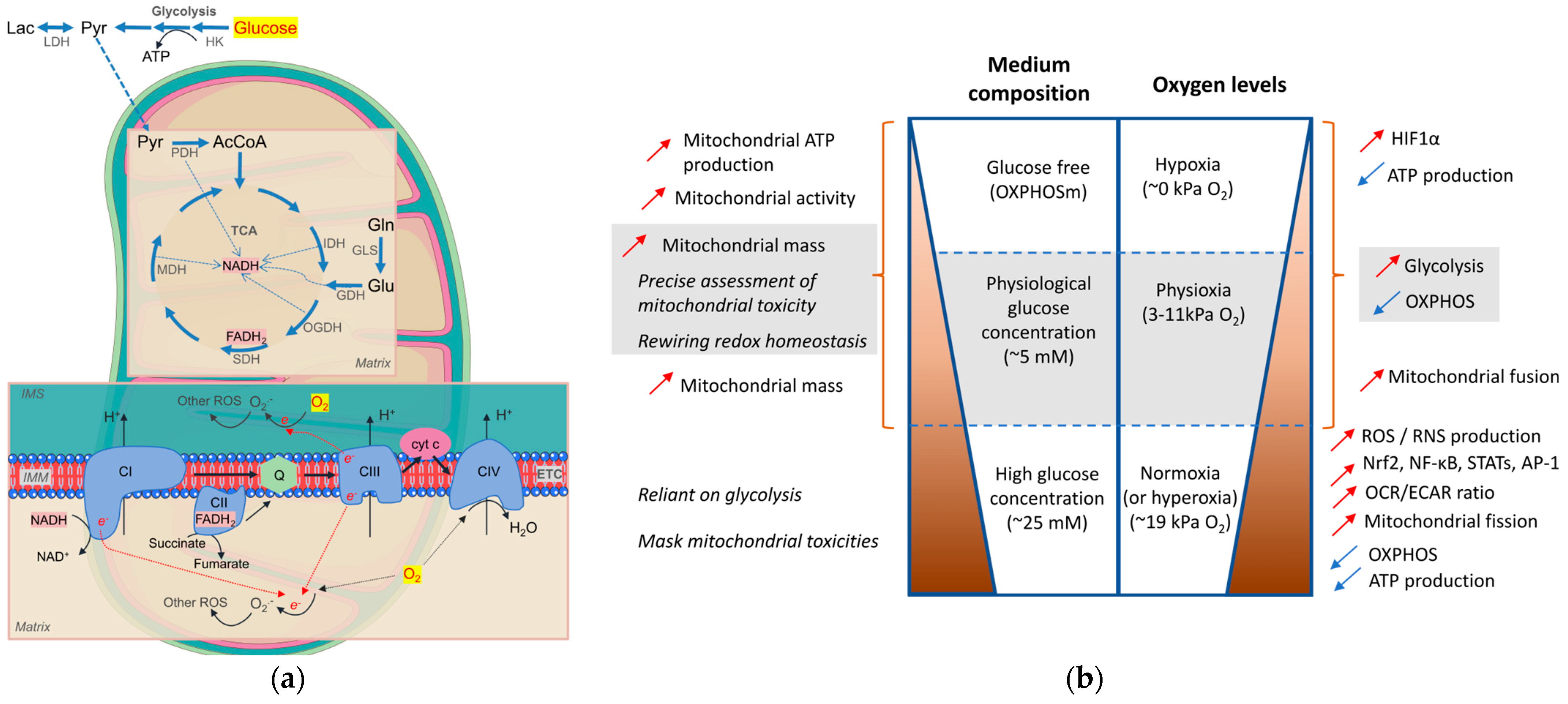
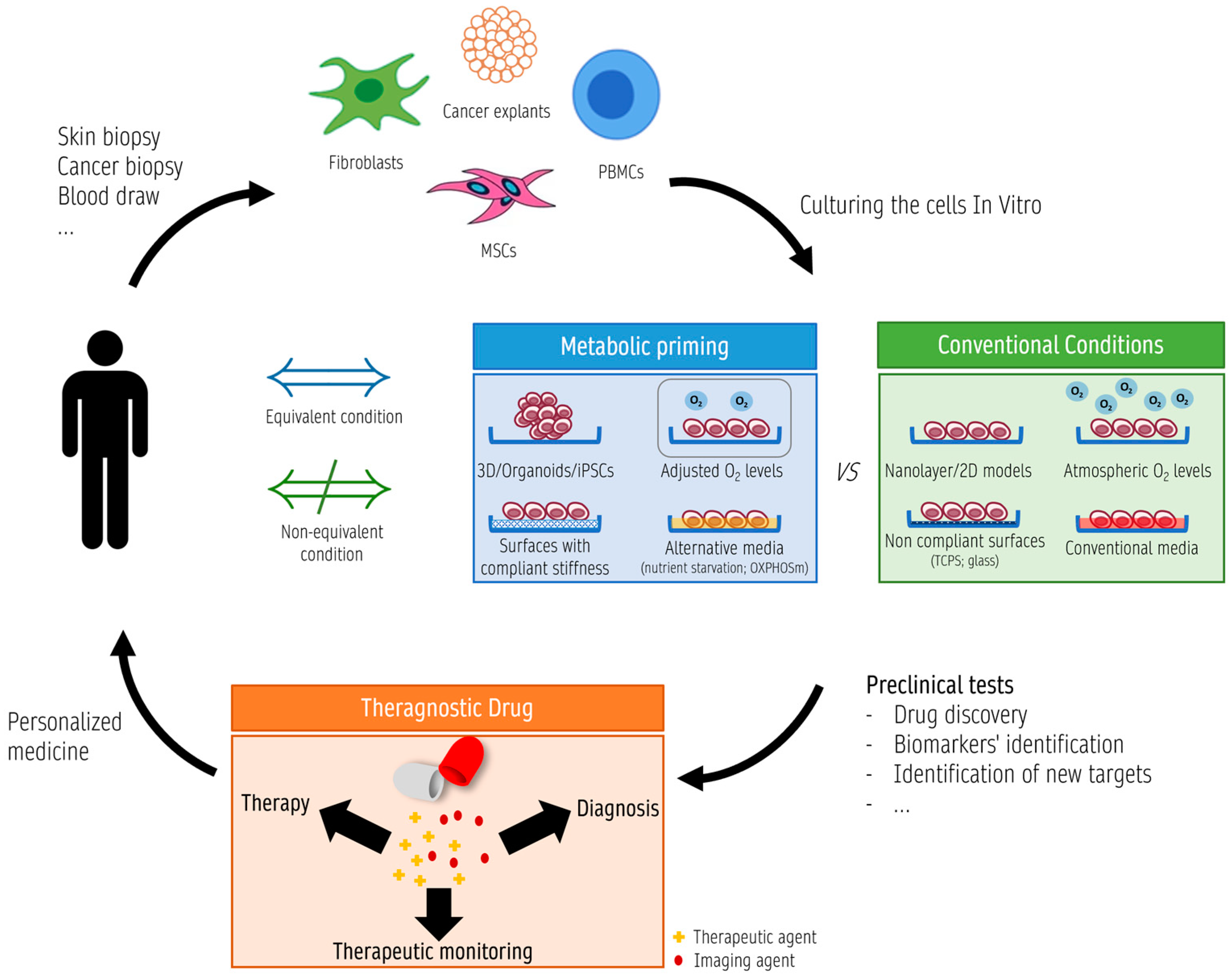
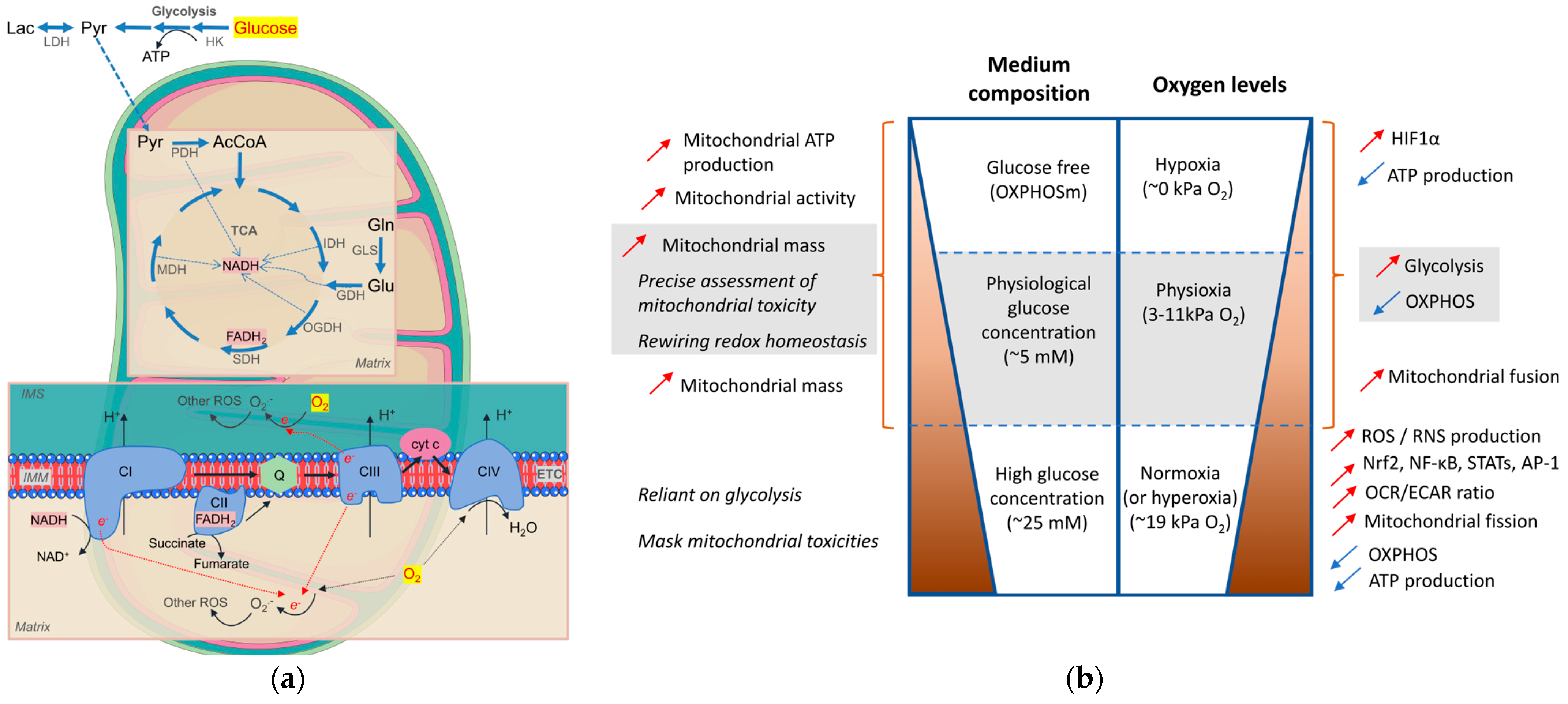
4. Metabolic Priming In Vitro/Ex Vivo for Mitochondrial Theragnostics
4.1. Media Composition
4.2. O2 Levels
4.3. Evaluation Tools for Mitochondrial Health Status
4.4. How Mitochondria Can Be Used as a Biosensor for Drug Development
4.4.1. Redox Biomarkers
4.4.2. Mitochondria-Targeted Drugs
4.4.3. Mitochondrial Theragnostics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rusu, M.E.; Fizesan, I.; Vlase, L.; Popa, D.S. Antioxidants in Age-Related Diseases and Anti-Aging Strategies. Antioxidants 2022, 11, 1868. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Rahman, H.S. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharm. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Carvajal, F.; Sanhueza, M. The Mitochondrial Unfolded Protein Response: A Hinge Between Healthy and Pathological Aging. Front. Aging Neurosci. 2020, 12, 581849. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Seyhan, A.A. Lost in translation: The valley of death across preclinical and clinical divide–identification of problems and overcoming obstacles. Transl. Med. Commun. 2019, 4, 18. [Google Scholar] [CrossRef]
- Diogo, C.V.; Yambire, K.F.; Mosquera, L.F.; Branco, F.T.; Raimundo, N. Mitochondrial adventures at the organelle society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef]
- Oliveira, P.J. Mitochondria as a drug target in health and disease. Curr. Drug Targets 2011, 12, 761. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Liu, Y.; Birsoy, K. Metabolic sensing and control in mitochondria. Mol. Cell 2023, 83, 877–889. [Google Scholar] [CrossRef]
- Raimundo, N. Mitochondrial pathology: Stress signals from the energy factory. Trends Mol. Med. 2014, 20, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Passmore, J.B.; Pinho, S.; Gomez-Lazaro, M.; Schrader, M. The respiratory chain inhibitor rotenone affects peroxisomal dynamics via its microtubule-destabilising activity. Histochem. Cell Biol. 2017, 148, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Garcia-Poyatos, C.; Martin-Garcia, E.; Jedrychowski, M.; Gygi, S.P.; Enriquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2alpha Axis. Mol. Cell 2019, 74, 877–890. [Google Scholar] [CrossRef]
- Peng, W.; Wong, Y.C.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc. Natl. Acad. Sci. USA 2020, 117, 19266–19275. [Google Scholar] [CrossRef] [PubMed]
- Savu, D.I.; Moisoi, N. Mitochondria-Nucleus communication in neurodegenerative disease. Who talks first, who talks louder? Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148588. [Google Scholar] [CrossRef]
- Soledad, R.B.; Charles, S.; Samarjit, D. The secret messages between mitochondria and nucleus in muscle cell biology. Arch. Biochem. Biophys. 2019, 666, 52–62. [Google Scholar] [CrossRef]
- Janssen, J.J.E.; Grefte, S.; Keijer, J.; de Boer, V.C.J. Mito-Nuclear Communication by Mitochondrial Metabolites and Its Regulation by B-Vitamins. Front. Physiol. 2019, 10, 78. [Google Scholar] [CrossRef]
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef]
- Belli, M.; Zhang, L.; Liu, X.; Donjacour, A.; Ruggeri, E.; Palmerini, M.G.; Nottola, S.A.; Macchiarelli, G.; Rinaudo, P. Oxygen concentration alters mitochondrial structure and function in in vitro fertilized preimplantation mouse embryos. Hum. Reprod. 2019, 34, 601–611. [Google Scholar] [CrossRef]
- Baker, N.; Patel, J.; Khacho, M. Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef] [PubMed]
- A census of complexes formed by mitochondrial proteins. Nature 2023. [CrossRef]
- Vartak, R.; Porras, C.A.; Bai, Y. Respiratory supercomplexes: Structure, function and assembly. Protein Cell 2013, 4, 582–590. [Google Scholar] [CrossRef]
- Dudkina, N.V.; Kouril, R.; Peters, K.; Braun, H.P.; Boekema, E.J. Structure and function of mitochondrial supercomplexes. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 664–670. [Google Scholar] [CrossRef]
- Hirst, J. Open questions: Respiratory chain supercomplexes-why are they there and what do they do? BMC Biol. 2018, 16, 111. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Lopez-Calcerrada, S.; Sierra-Magro, A.; Perez-Perez, R.; Formosa, L.E.; Hock, D.H.; Illescas, M.; Penas, A.; Brischigliaro, M.; Ding, S.; et al. Two independent respiratory chains adapt OXPHOS performance to glycolytic switch. Cell Metab. 2022, 34, 1792–1808. [Google Scholar] [CrossRef]
- Kadenbach, B.; Huttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef]
- Reguera, D.P.; Cunatova, K.; Vrbacky, M.; Pecinova, A.; Houstek, J.; Mracek, T.; Pecina, P. Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9, 443. [Google Scholar] [CrossRef]
- Maycotte, P.; Marin-Hernandez, A.; Goyri-Aguirre, M.; Anaya-Ruiz, M.; Reyes-Leyva, J.; Cortes-Hernandez, P. Mitochondrial dynamics and cancer. Tumour Biol. 2017, 39, 1010428317698391. [Google Scholar] [CrossRef]
- Diaz-Vegas, A.; Sanchez-Aguilera, P.; Krycer, J.R.; Morales, P.E.; Monsalves-Alvarez, M.; Cifuentes, M.; Rothermel, B.A.; Lavandero, S. Is Mitochondrial Dysfunction a Common Root of Noncommunicable Chronic Diseases? Endocr. Rev. 2020, 41, bnaa005. [Google Scholar] [CrossRef]
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell. Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ziv, R.; Bolas, T.; Dillin, A. Systemic effects of mitochondrial stress. EMBO Rep. 2020, 21, e50094. [Google Scholar] [CrossRef] [PubMed]
- Topf, U.; Wrobel, L.; Chacinska, A. Chatty Mitochondria: Keeping Balance in Cellular Protein Homeostasis. Trends Cell Biol. 2016, 26, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, B.P.; Haynes, C.M. Mitochondrial distress call moves to the cytosol to trigger a response to stress. Nature 2020, 579, 348–349. [Google Scholar] [CrossRef] [PubMed]
- Bordon, Y. Protect the species with mitohormesis? Nat. Rev. Immunol. 2021, 21, 407. [Google Scholar] [CrossRef] [PubMed]
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Ferreira, A.; Serafim, T.L.; Sardao, V.A.; Cunha-Oliveira, T. Role of mtDNA-related mitoepigenetic phenomena in cancer. Eur. J. Clin. Investig. 2015, 45, 44–49. [Google Scholar] [CrossRef]
- Dong, Z.; Pu, L.; Cui, H. Mitoepigenetics and Its Emerging Roles in Cancer. Front. Cell Dev. Biol. 2020, 8, 4. [Google Scholar] [CrossRef]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar] [CrossRef]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharm. 2020, 182, 114282. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Pinho, S.A.; Afonso, G.J.M.; Oliveira, P.J.; Cunha-Oliveira, T.; Saso, L. NRF2 and Mitochondrial Function in Cancer and Cancer Stem Cells. Cells 2022, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166016. [Google Scholar] [CrossRef]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Neufer, P.D. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 2012, 23, 142–153. [Google Scholar] [CrossRef]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- de Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Amorim, R.; Simoes, I.C.M.; Teixeira, J.; Cagide, F.; Potes, Y.; Soares, P.; Carvalho, A.; Tavares, L.C.; Benfeito, S.; Pereira, S.P.; et al. Mitochondria-targeted anti-oxidant AntiOxCIN(4) improved liver steatosis in Western diet-fed mice by preventing lipid accumulation due to upregulation of fatty acid oxidation, quality control mechanism and antioxidant defense systems. Redox Biol. 2022, 55, 102400. [Google Scholar] [CrossRef] [PubMed]
- Dandare, A.; Khan, M.J.; Naeem, A.; Liaquat, A. Clinical relevance of circulating non-coding RNAs in metabolic diseases: Emphasis on obesity, diabetes, cardiovascular diseases and metabolic syndrome. Genes Dis. 2022. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Mercado-Ayon, E.; Mercado-Ayon, Y.; Dong, Y.N.; Halawani, S.; Ngaba, L.; Lynch, D.R. Mitochondrial dysfunction in the development and progression of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 702, 108698. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Carvalho, M.; Sardao, V.; Ferreiro, E.; Mena, D.; Pereira, F.B.; Borges, F.; Oliveira, P.J.; Silva, F.S.G. Integrative Profiling of Amyotrophic Lateral Sclerosis Lymphoblasts Identifies Unique Metabolic and Mitochondrial Disease Fingerprints. Mol. Neurobiol. 2022, 59, 6373–6396. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxidative Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Simoes, R.F.; Pino, R.; Moreira-Soares, M.; Kovarova, J.; Neuzil, J.; Travasso, R.; Oliveira, P.J.; Cunha-Oliveira, T.; Pereira, F.B. Quantitative analysis of neuronal mitochondrial movement reveals patterns resulting from neurotoxicity of rotenone and 6-hydroxydopamine. FASEB J. 2021, 35, e22024. [Google Scholar] [CrossRef]
- Farkhondeh, T.; Mehrpour, O.; Forouzanfar, F.; Roshanravan, B.; Samarghandian, S. Oxidative stress and mitochondrial dysfunction in organophosphate pesticide-induced neurotoxicity and its amelioration: A review. Env. Sci. Pollut. Res. Int. 2020, 27, 24799–24814. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.M.; Frost, B.; Scherzer, C.; Feany, M.B. Biotin rescues mitochondrial dysfunction and neurotoxicity in a tauopathy model. Proc. Natl. Acad. Sci. USA 2020, 117, 33608–33618. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Park, G.S.; Shin, S.M.; Park, C.E.; Kim, S.; Han, S.J.; Pham, H.Q.; Shin, J.H.; Lee, D.W. Mitochondrial Mutations in Cholestatic Liver Disease with Biliary Atresia. Sci. Rep. 2018, 8, 905. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Xu, J.; Du, W.; Zhao, Y.; Lim, K.; Lu, L.; Zhang, C.; Li, L. Mitochondria targeting drugs for neurodegenerative diseases-Design, mechanism and application. Acta Pharm. Sin. B 2022, 12, 2778–2789. [Google Scholar] [CrossRef]
- Groh, C.; Haberkant, P.; Stein, F.; Filbeck, S.; Pfeffer, S.; Savitski, M.M.; Boos, F.; Herrmann, J.M. Mitochondrial dysfunction rapidly modulates the abundance and thermal stability of cellular proteins. Life Sci. Alliance 2023, 6, e202201805. [Google Scholar] [CrossRef]
- Vuda, M.; Kamath, A. Drug induced mitochondrial dysfunction: Mechanisms and adverse clinical consequences. Mitochondrion 2016, 31, 63–74. [Google Scholar] [CrossRef]
- Ramachandran, A.; Visschers, R.G.J.; Duan, L.; Akakpo, J.Y.; Jaeschke, H. Mitochondrial dysfunction as a mechanism of drug-induced hepatotoxicity: Current understanding and future perspectives. J. Clin. Transl. Res. 2018, 4, 75–100. [Google Scholar] [CrossRef]
- Will, Y.; Shields, J.E.; Wallace, K.B. Drug-Induced Mitochondrial Toxicity in the Geriatric Population: Challenges and Future Directions. Biology 2019, 8, 32. [Google Scholar] [CrossRef]
- Betiu, A.M.; Noveanu, L.; Hancu, I.M.; Lascu, A.; Petrescu, L.; Maack, C.; Elmer, E.; Muntean, D.M. Mitochondrial Effects of Common Cardiovascular Medications: The Good, the Bad and the Mixed. Int. J. Mol. Sci. 2022, 23, 13653. [Google Scholar] [CrossRef] [PubMed]
- Afonso, G.J.M.; Simões, R.F.; Pinho, S.L.C.; Oliveira, P.J.; Cunha-Oliveira, T. 5-Challenges in mitochondrial profiling during pre-clinical studies. In Mitochondrial Intoxication; de Oliveira, M.R., Ed.; Academic Press Elsevier Science: Amsterdam, The Netherlands, 2023; pp. 101–131. [Google Scholar]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, P.; Zou, M.H. AMP-activated protein kinase, stress responses and cardiovascular diseases. Clin. Sci. 2012, 122, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Storey, K.B. mTOR Signaling in Metabolic Stress Adaptation. Biomolecules 2021, 11, 681. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Thompson, C.B. Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 2010, 40, 323–332. [Google Scholar] [CrossRef]
- Gross, A.S.; Graef, M. Mechanisms of Autophagy in Metabolic Stress Response. J. Mol. Biol. 2020, 432, 28–52. [Google Scholar] [CrossRef]
- Domblides, C.; Lartigue, L.; Faustin, B. Metabolic Stress in the Immune Function of T Cells, Macrophages and Dendritic Cells. Cells 2018, 7, 68. [Google Scholar] [CrossRef]
- Kalucka, J.; Missiaen, R.; Georgiadou, M.; Schoors, S.; Lange, C.; De Bock, K.; Dewerchin, M.; Carmeliet, P. Metabolic control of the cell cycle. Cell Cycle 2015, 14, 3379–3388. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Y.; Bozi, L.H.M.; Fischer, P.; Jedrychowski, M.P.; Xiao, H.; Wu, T.; Darabedian, N.; He, X.; Mills, E.L.; et al. Lactate regulates cell cycle by remodeling the anaphase promoting complex. Nature 2023, 616, 790–797. [Google Scholar] [CrossRef]
- Baker, S.A.; Rutter, J. Metabolites as signalling molecules. Nat. Rev. Mol. Cell Biol. 2023, 24, 355–374. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Beninca, C.; Del Rio, L.F.; Shu, C.; Baghdasarian, S.; Zanette, V.; Gerle, C.; Jiko, C.; Khairallah, R.; Khan, S.; et al. Inhibition of ATP synthase reverse activity restores energy homeostasis in mitochondrial pathologies. EMBO J. 2023, e111699. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta 2016, 1857, 1167–1182. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals-role of cytochrome c oxidase. J. Mol. Med. 2020, 98, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Huttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta 2014, 1842, 1579–1586. [Google Scholar] [CrossRef]
- Hart, M.L.; Quon, E.; Vigil, A.B.G.; Engstrom, I.A.; Newsom, O.J.; Davidsen, K.; Hoellerbauer, P.; Carlisle, S.M.; Sullivan, L.B. Mitochondrial redox adaptations enable alternative aspartate synthesis in SDH-deficient cells. Elife 2023, 12, e78654. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S. The power of life--cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 2012, 12, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple Mechanisms Regulate Eukaryotic Cytochrome C Oxidase. Cells 2021, 10, 514. [Google Scholar] [CrossRef]
- Hicks, K.G.; Cluntun, A.A.; Schubert, H.L.; Hackett, S.R.; Berg, J.A.; Leonard, P.G.; Aleixo, M.A.A.; Zhou, Y.; Bott, A.J.; Salvatore, S.R.; et al. Protein-metabolite interactomics of carbohydrate metabolism reveal regulation of lactate dehydrogenase. Science 2023, 379, 996–1003. [Google Scholar] [CrossRef]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef]
- Fewell, S.W.; Brodsky, J.L. Entry into the Endoplasmic Reticulum: Protein Translocation, Folding and Quality Control. In Madame Curie Bioscience Database; Landes Bioscience: London, UK, 2013. [Google Scholar]
- Wagner, K.; Mick, D.U.; Rehling, P. Protein transport machineries for precursor translocation across the inner mitochondrial membrane. Biochim. Biophys. Acta 2009, 1793, 52–59. [Google Scholar] [CrossRef]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPR(mt). Cell Rep. 2019, 28, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free. Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Dhara, A.; Aier, I.; Paladhi, A.; Varadwaj, P.K.; Hira, S.K.; Sen, N. PGC1 alpha coactivates ERG fusion to drive antioxidant target genes under metabolic stress. Commun. Biol. 2022, 5, 416. [Google Scholar] [CrossRef]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886. [Google Scholar] [CrossRef]
- Li, K.K.; Pang, J.C.; Lau, K.M.; Zhou, L.; Mao, Y.; Wang, Y.; Poon, W.S.; Ng, H.K. MiR-383 is downregulated in medulloblastoma and targets peroxiredoxin 3 (PRDX3). Brain Pathol. 2013, 23, 413–425. [Google Scholar] [CrossRef]
- Gohel, D.; Singh, R. Mitohormesis; Potential implications in neurodegenerative diseases. Mitochondrion 2021, 56, 40–46. [Google Scholar] [CrossRef]
- Macgregor-Das, A.M.; Das, S. A microRNA’s journey to the center of the mitochondria. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H206–H215. [Google Scholar] [CrossRef]
- Jeandard, D.; Smirnova, A.; Tarassov, I.; Barrey, E.; Smirnov, A.; Entelis, N. Import of Non-Coding RNAs into Human Mitochondria: A Critical Review and Emerging Approaches. Cells 2019, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, S.; Wang, P.; Wang, G. Non-coding RNA Regulated Cross-Talk Between Mitochondria and Other Cellular Compartments. Front. Cell Dev. Biol. 2021, 9, 688523. [Google Scholar] [CrossRef] [PubMed]
- Xin, N.; Durieux, J.; Yang, C.; Wolff, S.; Kim, H.E.; Dillin, A. The UPRmt preserves mitochondrial import to extend lifespan. J. Cell Biol. 2022, 221, e202201071. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Jezek, P.; Jaburek, M.; Holendova, B.; Engstova, H.; Dlaskova, A. Mitochondrial cristae morphology reflecting metabolism, superoxide formation, redox homeostasis, and pathology. Antioxid Redox Signal. 2023. [Google Scholar] [CrossRef]
- Ramirez-Camacho, I.; Garcia-Nino, W.R.; Flores-Garcia, M.; Pedraza-Chaverri, J.; Zazueta, C. Alteration of mitochondrial supercomplexes assembly in metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165935. [Google Scholar] [CrossRef]
- Ikeda, K.; Horie-Inoue, K.; Suzuki, T.; Hobo, R.; Nakasato, N.; Takeda, S.; Inoue, S. Mitochondrial supercomplex assembly promotes breast and endometrial tumorigenesis by metabolic alterations and enhanced hypoxia tolerance. Nat. Commun. 2019, 10, 4108. [Google Scholar] [CrossRef]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes As Networking Platforms and Regulators of Cancer Cell Fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, C.; Ott, M.; Buttner, S. Mitochondria orchestrate proteostatic and metabolic stress responses. EMBO Rep. 2019, 20, e47865. [Google Scholar] [CrossRef] [PubMed]
- Richman, T.R.; Ermer, J.A.; Siira, S.J.; Kuznetsova, I.; Brosnan, C.A.; Rossetti, G.; Baker, J.; Perks, K.L.; Szappanos, H.C.; Viola, H.M.; et al. Mitochondrial mistranslation modulated by metabolic stress causes cardiovascular disease and reduced lifespan. Aging Cell 2021, 20, e13408. [Google Scholar] [CrossRef] [PubMed]
- Callegari, S.; Dennerlein, S. Sensing the Stress: A Role for the UPR(mt) and UPR(am) in the Quality Control of Mitochondria. Front. Cell Dev. Biol. 2018, 6, 31. [Google Scholar] [CrossRef]
- De Jesus, A.; Keyhani-Nejad, F.; Pusec, C.M.; Goodman, L.; Geier, J.A.; Stoolman, J.S.; Stanczyk, P.J.; Nguyen, T.; Xu, K.; Suresh, K.V.; et al. Hexokinase 1 cellular localization regulates the metabolic fate of glucose. Mol. Cell 2022, 82, 1261–1277.e9. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Thorne, R.F.; Shi, R.; Zhang, Q.; Wu, M.; Liu, L. Mitochondrial SENP2 regulates the assembly of SDH complex under metabolic stress. Cell Rep. 2023, 42, 112041. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic reticulum and the unfolded protein response: Dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 11. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal 2009, 11, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Bassot, A.; Chen, J.; Takahashi-Yamashiro, K.; Yap, M.C.; Gibhardt, C.S.; Le, G.N.T.; Hario, S.; Nasu, Y.; Moore, J.; Gutierrez, T.; et al. The endoplasmic reticulum kinase PERK interacts with the oxidoreductase ERO1 to metabolically adapt mitochondria. Cell Rep. 2023, 42, 111899. [Google Scholar] [CrossRef]
- Cogliati, S.; Cabrera-Alarcon, J.L.; Enriquez, J.A. Regulation and functional role of the electron transport chain supercomplexes. Biochem. Soc. Trans. 2021, 49, 2655–2668. [Google Scholar] [CrossRef] [PubMed]
- Yong, L.; Tang, S.; Yu, H.; Zhang, H.; Zhang, Y.; Wan, Y.; Cai, F. The role of hypoxia-inducible factor-1 alpha in multidrug-resistant breast cancer. Front. Oncol. 2022, 12, 964934. [Google Scholar] [CrossRef]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2021, 599, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Lionaki, E.; Gkikas, I.; Tavernarakis, N. Differential Protein Distribution between the Nucleus and Mitochondria: Implications in Aging. Front. Genet. 2016, 7, 162. [Google Scholar] [CrossRef]
- Chen, J.; Guccini, I.; Di Mitri, D.; Brina, D.; Revandkar, A.; Sarti, M.; Pasquini, E.; Alajati, A.; Pinton, S.; Losa, M.; et al. Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat. Genet. 2018, 50, 219–228. [Google Scholar] [CrossRef]
- Nunes-Xavier, C.E.; Mingo, J.; Emaldi, M.; Flem-Karlsen, K.; Maelandsmo, G.M.; Fodstad, O.; Llarena, R.; Lopez, J.I.; Pulido, R. Heterogeneous Expression and Subcellular Localization of Pyruvate Dehydrogenase Complex in Prostate Cancer. Front. Oncol. 2022, 12, 873516. [Google Scholar] [CrossRef]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Env. Health Perspect 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, P.A.; Struhl, K. Nutrient Deprivation Elicits a Transcriptional and Translational Inflammatory Response Coupled to Decreased Protein Synthesis. Cell Rep. 2018, 24, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Bredesen, D.E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 2004, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef]
- Walker, B.R.; Moraes, C.T. Nuclear-Mitochondrial Interactions. Biomolecules 2022, 12, 427. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genom. Hum. Genet. 2010, 11, 25–44. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Lightfoot, A.P.; Earl, K.E.; Stofanko, M.; McDonagh, B. Redox homeostasis and age-related deficits in neuromuscular integrity and function. J. Cachexia Sarcopenia Muscle 2017, 8, 881–906. [Google Scholar] [CrossRef] [PubMed]
- Shields, H.J.; Traa, A.; Van Raamsdonk, J.M. Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental Studies. Front. Cell Dev. Biol. 2021, 9, 628157. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Schmidt, E.E.; Sayin, V.I. Cellular Redox Homeostasis. Antioxidants 2021, 10, 1377. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M.; Forman, H.J. Redox homeostasis: The Golden Mean of healthy living. Redox Biol. 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Meng, J.; Lv, Z.; Zhang, Y.; Wang, Y.; Qiao, X.; Sun, C.; Chen, Y.; Guo, M.; Han, W.; Ye, A.; et al. Precision Redox: The Key for Antioxidant Pharmacology. Antioxid Redox Signal. 2021, 34, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2021, 23, 106. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Deus, C.M.; Teixeira, J.; Raimundo, N.; Tucci, P.; Borges, F.; Saso, L.; Oliveira, P.J. Modulation of cellular redox environment as a novel therapeutic strategy for Parkinson’s disease. Eur. J. Clin. Investig. 2022, 52, e13820. [Google Scholar] [CrossRef]
- Ransy, C.; Vaz, C.; Lombes, A.; Bouillaud, F. Use of H(2)O(2) to Cause Oxidative Stress, the Catalase Issue. Int. J. Mol. Sci. 2020, 21, 9149. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev. Mol. Cell Biol. 2007, 8, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Masaki, H.; Okano, Y.; Sakurai, H. Differential role of catalase and glutathione peroxidase in cultured human fibroblasts under exposure of H2O2 or ultraviolet B light. Arch. Dermatol. Res. 1998, 290, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Molavian, H.; Tonekaboni, A.M.; Kohandel, M.; Sivaloganathan, S. The synergetic coupling among the cellular antioxidants glutathione peroxidase/peroxiredoxin and other antioxidants and its effect on the concentration of H2O2. Sci. Rep. 2015, 5, srep13620. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef]
- Urbanski, N.K.; Beresewicz, A. Generation of *OH initiated by interaction of Fe2+ and Cu+ with dioxygen; comparison with the Fenton chemistry. Acta Biochim. Pol. 2000, 47, 951–962. [Google Scholar] [CrossRef]
- Bianco, C.L.; Toscano, J.P.; Fukuto, J.M. Chapter 2-An Integrated View of the Chemical Biology of NO, CO, H2S, and O2. In Nitric Oxide (Third Edition); Ignarro, L.J., Freeman, B.A., Eds.; Academic Press Elsevier Science: Amsterdam, The Netherlands, 2017; pp. 9–21. [Google Scholar]
- Kucera, O.; Endlicher, R.; Rousar, T.; Lotkova, H.; Garnol, T.; Drahota, Z.; Cervinkova, Z. The effect of tert-butyl hydroperoxide-induced oxidative stress on lean and steatotic rat hepatocytes in vitro. Oxidative Med. Cell. Longev. 2014, 2014, 752506. [Google Scholar] [CrossRef]
- Yang, H.C.; Yu, H.; Ma, T.H.; Tjong, W.Y.; Stern, A.; Chiu, D.T. tert-Butyl Hydroperoxide (tBHP)-Induced Lipid Peroxidation and Embryonic Defects Resemble Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency in C. elegans. Int. J. Mol. Sci. 2020, 21, 8688. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Iv, C.D.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 2020, 37, 101696. [Google Scholar] [CrossRef]
- Jonsson, M.K.; van Veen, T.A.; Synnergren, J.; Becker, B. Towards Creating the Perfect In Vitro Cell Model. Stem Cells Int. 2016, 2016, 3459730. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.A.; Costa, C.F.; Deus, C.M.; Pinho, S.L.C.; Miranda-Santos, I.; Afonso, G.; Bagshaw, O.; Stuart, J.A.; Oliveira, P.J.; Cunha-Oliveira, T. Mitochondrial and metabolic remodelling in human skin fibroblasts in response to glucose availability. FEBS J. 2022, 289, 5198–5217. [Google Scholar] [CrossRef]
- Peck, S.H.; Bendigo, J.R.; Tobias, J.W.; Dodge, G.R.; Malhotra, N.R.; Mauck, R.L.; Smith, L.J. Hypoxic Preconditioning Enhances Bone Marrow-Derived Mesenchymal Stem Cell Survival in a Low Oxygen and Nutrient-Limited 3D Microenvironment. Cartilage 2021, 12, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Leite, S.B.; Cipriano, M.; Carpi, D.; Coecke, S.; Holloway, M.; Corvi, R.; Worth, A.; Barroso, J.; Whelan, M. Establishing the scientific validity of complex in vitro models: Results of a EURL ECVAM survey. Publ. Off. Eur. Union 2021, 2021, 1–28. [Google Scholar]
- Idrees, A.; Chiono, V.; Ciardelli, G.; Shah, S.; Viebahn, R.; Zhang, X.; Salber, J. Validation of in vitro assays in three-dimensional human dermal constructs. Int. J. Artif. Organs 2018, 41, 779–788. [Google Scholar] [CrossRef]
- Hassanzadeh, A.; Shamlou, S.; Yousefi, N.; Nikoo, M.; Verdi, J. Genetically-modified Stem Cell in Regenerative Medicine and Cancer Therapy; A New Era. Curr. Gene Ther. 2022, 22, 23–39. [Google Scholar] [CrossRef]
- Katti, A.; Diaz, B.J.; Caragine, C.M.; Sanjana, N.E.; Dow, L.E. CRISPR in cancer biology and therapy. Nat. Rev. Cancer 2022, 22, 259–279. [Google Scholar] [CrossRef]
- Shin, S.M.; Baek, E.J.; Oh, D.Y.; Kim, K.H.; Kim, K.J.; Park, E.J. Functional validation of co-culture model of human keratinocytes and neuronal cell line for sensitive skin by using transient receptor potential channel vanilloid subfamily member 1 antagonist. Skin. Res. Technol. 2023, 29, e13275. [Google Scholar] [CrossRef]
- Li, Y.; Nowak, C.M.; Pham, U.; Nguyen, K.; Bleris, L. Cell morphology-based machine learning models for human cell state classification. NPJ Syst. Biol. Appl. 2021, 7, 23. [Google Scholar] [CrossRef]
- Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener. 2022, 11, 36. [Google Scholar] [CrossRef]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Pereira, F.B.; Raimundo, N.; Oliveira, P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic male Parkinson’s disease patients uncovers metabolic and mitochondrial bioenergetic defects. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165615. [Google Scholar] [CrossRef] [PubMed]
- Vis, M.A.M.; Ito, K.; Hofmann, S. Impact of Culture Medium on Cellular Interactions in in vitro Co-culture Systems. Front. Bioeng. Biotechnol. 2020, 8, 911. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, H.G.; Bengtson, C.P.; Weiss, U.; Hellwig, A.; Garcia-Vilela, C.; Zaremba, B.; Kaessmann, H.; Pruunsild, P.; Bading, H. N-methyl-d-aspartate Receptor-mediated Preconditioning Mitigates Excitotoxicity in Human Induced Pluripotent Stem Cell-derived Brain Organoids. Neuroscience 2022, 484, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, J.A.; McDevitt, T.C. Pre-conditioning mesenchymal stromal cell spheroids for immunomodulatory paracrine factor secretion. Cytotherapy 2014, 16, 331–345. [Google Scholar] [CrossRef]
- Nikravesh, N.; Cox, S.C.; Birdi, G.; Williams, R.L.; Grover, L.M. Calcium pre-conditioning substitution enhances viability and glucose sensitivity of pancreatic beta-cells encapsulated using polyelectrolyte multilayer coating method. Sci. Rep. 2017, 7, 43171. [Google Scholar] [CrossRef]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Widgerow, A.D.; Fabi, S.G.; Palestine, R.F.; Rivkin, A.; Ortiz, A.; Bucay, V.W.; Chiu, A.; Naga, L.; Emer, J.; Chasan, P.E. Extracellular Matrix Modulation: Optimizing Skin Care and Rejuvenation Procedures. J. Drugs Dermatol. 2016, 15, s63–s71. [Google Scholar]
- Batenburg, K.L.; Sestito, C.; Cornelissen-Steijger, P.; van Weering, J.R.T.; Price, L.S.; Heine, V.M.; Scheper, W. A 3D human co-culture to model neuron-astrocyte interactions in tauopathies. Biol. Proced. Online 2023, 25, 4. [Google Scholar] [CrossRef]
- Bauman, E.; Feijao, T.; Carvalho, D.T.O.; Granja, P.L.; Barrias, C.C. Xeno-free pre-vascularized spheroids for therapeutic applications. Sci. Rep. 2018, 8, 230. [Google Scholar] [CrossRef]
- Bialkowska, K.; Komorowski, P.; Bryszewska, M.; Milowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures-Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Velletri, T.; Villa, C.E.; Cilli, D.; Barzaghi, B.; Lo Riso, P.; Lupia, M.; Luongo, R.; Lopez-Tobon, A.; De Simone, M.; Bonnal, R.J.P.; et al. Single cell-derived spheroids capture the self-renewing subpopulations of metastatic ovarian cancer. Cell Death Differ. 2022, 29, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Nuschke, A.; Rodrigues, M.; Wells, A.W.; Sylakowski, K.; Wells, A. Mesenchymal stem cells/multipotent stromal cells (MSCs) are glycolytic and thus glucose is a limiting factor of in vitro models of MSC starvation. Stem Cell Res. Ther. 2016, 7, 179. [Google Scholar] [CrossRef]
- Tanaka, K.; Ogino, R.; Yamakawa, S.; Suda, S.; Hayashida, K. Role and Function of Mesenchymal Stem Cells on Fibroblast in Cutaneous Wound Healing. Biomedicines 2022, 10, 1391. [Google Scholar] [CrossRef]
- Yang, Y.; Lee, E.H.; Yang, Z. Hypoxia-Conditioned Mesenchymal Stem Cells in Tissue Regeneration Application. Tissue Eng. Part B Rev. 2022, 28, 966–977. [Google Scholar] [CrossRef]
- Kusuma, G.D.; Li, A.; Zhu, D.; McDonald, H.; Inocencio, I.M.; Chambers, D.C.; Sinclair, K.; Fang, H.; Greening, D.W.; Frith, J.E.; et al. Effect of 2D and 3D Culture Microenvironments on Mesenchymal Stem Cell-Derived Extracellular Vesicles Potencies. Front. Cell Dev. Biol. 2022, 10, 819726. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M. 3D Sponge-Matrix Histoculture: An Overview. Methods Mol. Biol. 2018, 1760, 11–17. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef]
- Cantor, J.R.; Abu-Remaileh, M.; Kanarek, N.; Freinkman, E.; Gao, X.; Louissaint, A., Jr.; Lewis, C.A.; Sabatini, D.M. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 2017, 169, 258–272 e217. [Google Scholar] [CrossRef]
- Skolik, R.A.; Solocinski, J.; Konkle, M.E.; Chakraborty, N.; Menze, M.A. Global changes to HepG2 cell metabolism in response to galactose treatment. Am. J. Physiol. Cell Physiol. 2021, 320, C778–C793. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.F. Experimental Enhancement of Cellular Oxphos Reliance for Mitochondrial Health Assessments Development and Characterization of a Rapid and Efficient Method to Induce OXPHOS in Skin Fibroblasts, for the Assessment of Mitochondiral Toxicity and Protection. Master’s Thesis, Universidade de Coimbra, Coimbra, Portugal, 2019. [Google Scholar]
- Pereira, S.P.; Deus, C.M.; Serafim, T.L.; Cunha-Oliveira, T.; Oliveira, P.J. Metabolic and Phenotypic Characterization of Human Skin Fibroblasts After Forcing Oxidative Capacity. Toxicol. Sci. 2018, 164, 191–204. [Google Scholar] [CrossRef]
- Mantripragada, V.P.; Kaplevatsky, R.; Bova, W.A.; Boehm, C.; Obuchowski, N.A.; Midura, R.J.; Muschler, G.F. Influence of Glucose Concentration on Colony-Forming Efficiency and Biological Performance of Primary Human Tissue-Derived Progenitor Cells. Cartilage 2021, 13, 95S–106S. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J. Mitochondrial toxicity assessment in industry-a decade of technology development and insight. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Marroquin, L.D.; Hynes, J.; Dykens, J.A.; Jamieson, J.D.; Will, Y. Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 2007, 97, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 2011, 6, e28536. [Google Scholar] [CrossRef] [PubMed]
- de Kok, M.J.C.; Schaapherder, A.F.; Wust, R.C.I.; Zuiderwijk, M.; Bakker, J.A.; Lindeman, J.H.N.; Le Devedec, S.E. Circumventing the Crabtree effect in cell culture: A systematic review. Mitochondrion 2021, 59, 83–95. [Google Scholar] [CrossRef]
- Dragovic, S.; Vermeulen, N.P.; Gerets, H.H.; Hewitt, P.G.; Ingelman-Sundberg, M.; Park, B.K.; Juhila, S.; Snoeys, J.; Weaver, R.J. Evidence-based selection of training compounds for use in the mechanism-based integrated prediction of drug-induced liver injury in man. Arch. Toxicol. 2016, 90, 2979–3003. [Google Scholar] [CrossRef]
- Kamalian, L.; Chadwick, A.E.; Bayliss, M.; French, N.S.; Monshouwer, M.; Snoeys, J.; Park, B.K. The utility of HepG2 cells to identify direct mitochondrial dysfunction in the absence of cell death. Toxicol. Vitr. 2015, 29, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Gilkerson, R.; Aggeler, R.; Yamagata, K.; Remington, S.J.; Capaldi, R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004, 64, 985–993. [Google Scholar] [CrossRef]
- Stuart, J.A.; Fonseca, J.; Moradi, F.; Cunningham, C.; Seliman, B.; Worsfold, C.R.; Dolan, S.; Abando, J.; Maddalena, L.A. How Supraphysiological Oxygen Levels in Standard Cell Culture Affect Oxygen-Consuming Reactions. Oxidative Med. Cell. Longev. 2018, 2018, 8238459. [Google Scholar] [CrossRef]
- Keeley, T.P.; Mann, G.E. Defining Physiological Normoxia for Improved Translation of Cell Physiology to Animal Models and Humans. Physiol. Rev. 2019, 99, 161–234. [Google Scholar] [CrossRef]
- Jagannathan, L.; Cuddapah, S.; Costa, M. Oxidative stress under ambient and physiological oxygen tension in tissue culture. Curr. Pharmacol. Rep. 2016, 2, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Alva, R.; Mirza, M.; Baiton, A.; Lazuran, L.; Samokysh, L.; Bobinski, A.; Cowan, C.; Jaimon, A.; Obioru, D.; Al Makhoul, T.; et al. Oxygen toxicity: Cellular mechanisms in normobaric hyperoxia. Cell Biol. Toxicol. 2023, 39, 111–143. [Google Scholar] [CrossRef]
- Sebestyen, A.; Kopper, L.; Danko, T.; Timar, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef]
- Villeneuve, L.; Tiede, L.M.; Morsey, B.; Fox, H.S. Quantitative proteomics reveals oxygen-dependent changes in neuronal mitochondria affecting function and sensitivity to rotenone. J. Proteome Res. 2013, 12, 4599–4606. [Google Scholar] [CrossRef]
- Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. [Google Scholar] [CrossRef]
- Chen, P.S.; Chiu, W.T.; Hsu, P.L.; Lin, S.C.; Peng, I.C.; Wang, C.Y.; Tsai, S.J. Pathophysiological implications of hypoxia in human diseases. J. Biomed. Sci. 2020, 27, 63. [Google Scholar] [CrossRef]
- Lages, Y.M.; Nascimento, J.M.; Lemos, G.A.; Galina, A.; Castilho, L.R.; Rehen, S.K. Low oxygen alters mitochondrial function and response to oxidative stress in human neural progenitor cells. PeerJ 2015, 3, e1486. [Google Scholar] [CrossRef]
- Tzameli, I. The evolving role of mitochondria in metabolism. Trends Endocrinol. Metab. 2012, 23, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Prather, A.A.; Puterman, E.; Cuillerier, A.; Coccia, M.; Aschbacher, K.; Burelle, Y.; Epel, E.S. A Mitochondrial Health Index Sensitive to Mood and Caregiving Stress. Biol. Psychiatry 2018, 84, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Costa, A.P.; Lima, C.N.; Fries, G.; Sanches, M.; Soares, J.; Quevedo, J. P168. Mitochondrial Health Index Correlates With Plasma Circulating Cell-Free Mitochondrial DNA in Bipolar Disorder. Biol. Psychiatry 2022, 91, S155. [Google Scholar] [CrossRef]
- Koklesova, L.; Mazurakova, A.; Samec, M.; Kudela, E.; Biringer, K.; Kubatka, P.; Golubnitschaja, O. Mitochondrial health quality control: Measurements and interpretation in the framework of predictive, preventive, and personalized medicine. EPMA J. 2022, 13, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Benavides, G.A.; Mitchell, T.; Dranka, B.P.; Ferrick, D.; Singal, A.K.; Ballinger, S.W.; Bailey, S.M.; et al. The Bioenergetic Health Index: A new concept in mitochondrial translational research. Clin. Sci. 2014, 127, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Zhi, D.; Darley-Usmar, V.M.; Mitchell, T. The Bioenergetic Health Index is a sensitive measure of oxidative stress in human monocytes. Redox Biol. 2016, 8, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Marrocco, I.; Altieri, F.; Peluso, I. Measurement and Clinical Significance of Biomarkers of Oxidative Stress in Humans. Oxidative Med. Cell. Longev. 2017, 2017, 6501046. [Google Scholar] [CrossRef]
- Thakkar, H.; Eerla, R.; Gangakhedkar, S.; Shah, R.P. Exploring unexplored biomarkers of oxidative distress and their use. Adv. Redox Res. 2021, 3, 100020. [Google Scholar] [CrossRef]
- Maciejczyk, M.; Pawlukianiec, C.; Zendzian-Piotrowska, M.; Ladny, J.R.; Zalewska, A. Salivary Redox Biomarkers in Insulin Resistance: Preclinical Studies in an Animal Model. Oxidative Med. Cell. Longev. 2021, 2021, 3734252. [Google Scholar] [CrossRef]
- Apeland, T.; Ushakova, A.; Mansoor, M.A.; Furriol, J.; Jonsson, G.; Marti, H.P. Association of redox and inflammation-related biomarkers with prognosis in IgA nephropathy: A prospective observational study. Free. Radic. Biol. Med. 2022, 188, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Kalapotharakos, G.; Murtoniemi, K.; Akerstrom, B.; Hamalainen, E.; Kajantie, E.; Raikkonen, K.; Villa, P.; Laivuori, H.; Hansson, S.R. Plasma Heme Scavengers Alpha-1-Microglobulin and Hemopexin as Biomarkers in High-Risk Pregnancies. Front. Physiol. 2019, 10, 300. [Google Scholar] [CrossRef]
- Larsson, S.; Akerstrom, B.; Gram, M.; Lohmander, L.S.; Struglics, A. Alpha(1)-Microglobulin Protects Against Bleeding-Induced Oxidative Damage in Knee Arthropathies. Front. Physiol. 2018, 9, 1596. [Google Scholar] [CrossRef]
- Szczurek, W.; Szygula-Jurkiewicz, B. Oxidative stress and inflammatory markers-the future of heart failure diagnostics? Kardiochir Torakochirurgia Pol. 2015, 12, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Balukova, A.; Pospisil, P. Triplet Excited Carbonyls and Singlet Oxygen Formation During Oxidative Radical Reaction in Skin. Front. Physiol. 2018, 9, 1109. [Google Scholar] [CrossRef]
- Fong, V.H.; Vieira, A. Transthyretin aggregates induce production of reactive nitrogen species. Neurodegener. Dis. 2013, 11, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, E.; Wygralak, Z.; Kedracka-Krok, S.; Bezara, P.; Bystranowska, D.; Dobryszycki, P.; Ozyhar, A. Deep blue autofluorescence reflects the oxidation state of human transthyretin. Redox Biol. 2022, 56, 102434. [Google Scholar] [CrossRef]
- Sharma, M.; Khan, S.; Rahman, S.; Singh, L.R. The Extracellular Protein, Transthyretin Is an Oxidative Stress Biomarker. Front. Physiol. 2019, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Sinha, S.; Vohora, D. Drug discovery and development: An overview. Pharm. Med. Transl. Clin. Res. 2018, 19–32. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Yildirim, O.; Gottwald, M.; Schuler, P.; Michel, M.C. Opportunities and Challenges for Drug Development: Public-Private Partnerships, Adaptive Designs and Big Data. Front. Pharm. 2016, 7, 461. [Google Scholar] [CrossRef]
- Nassar, S.F.; Raddassi, K.; Wu, T. Single-Cell Multiomics Analysis for Drug Discovery. Metabolites 2021, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine Learning and Integrative Analysis of Biomedical Big Data. Genes 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhan, X. Machine Learning Identifies Pan-Cancer Landscape of Nrf2 Oxidative Stress Response Pathway-Related Genes. Oxidative Med. Cell. Longev. 2022, 2022, 8450087. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Profumo, E.; Tucci, P.; Saso, L. A Perspective on Nrf2 Signaling Pathway for Neuroinflammation: A Potential Therapeutic Target in Alzheimer’s and Parkinson’s Diseases. Front. Cell Neurosci. 2021, 15, 787258. [Google Scholar] [CrossRef]
- Mazur, A.; Fangman, M.; Ashouri, R.; Arcenas, A.; Dore, S. Nrf2 as a therapeutic target in ischemic stroke. Expert Opin. Ther. Targets 2021, 25, 163–166. [Google Scholar] [CrossRef]
- Ermakov, E.A.; Dmitrieva, E.M.; Parshukova, D.A.; Kazantseva, D.V.; Vasilieva, A.R.; Smirnova, L.P. Oxidative Stress-Related Mechanisms in Schizophrenia Pathogenesis and New Treatment Perspectives. Oxidative Med. Cell. Longev. 2021, 2021, 8881770. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Li, G.; Wu, J.; Wu, X.; Wang, G.; Gu, G.; Ren, H.; Hong, Z.; Li, J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018, 9, 403. [Google Scholar] [CrossRef]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Teixeira, J.; Simoes, R.F.; Cagide, F.; Benfeito, S.; Borges, F.; Raimundo, N.; Oliveira, P.J. A mitochondria-targeted caffeic acid derivative reverts cellular and mitochondrial defects in human skin fibroblasts from male sporadic Parkinson’s disease patients. Redox Biol. 2021, 45, 102037. [Google Scholar] [CrossRef]
- Teixeira, J.; Basit, F.; Willems, P.; Wagenaars, J.A.; van de Westerlo, E.; Amorim, R.; Cagide, F.; Benfeito, S.; Oliveira, C.; Borges, F.; et al. Mitochondria-targeted phenolic antioxidants induce ROS-protective pathways in primary human skin fibroblasts. Free. Radic. Biol. Med. 2021, 163, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Cagide, F.; Simoes, J.; Pita, C.; Pereira, E.; Videira, A.J.C.; Soares, P.; Duarte, J.F.S.; Santos, A.M.S.; Oliveira, P.J.; et al. Targeting Hydroxybenzoic Acids to Mitochondria as a Strategy to Delay Skin Ageing: An In Vitro Approach. Molecules 2022, 27, 6183. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.V.; Chelombitko, M.A.; Chernyavskij, D.A.; Galkin, I.I.; Pletjushkina, O.Y.; Vasilieva, T.V.; Zinovkin, R.A.; Chernyak, B.V. Mitochondria-Targeted Antioxidant SkQ1 Prevents the Development of Experimental Colitis in Mice and Impairment of the Barrier Function of the Intestinal Epithelium. Cells 2022, 11, 3441. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef] [PubMed]
- Karkucinska-Wieckowska, A.; Simoes, I.C.M.; Kalinowski, P.; Lebiedzinska-Arciszewska, M.; Zieniewicz, K.; Milkiewicz, P.; Gorska-Ponikowska, M.; Pinton, P.; Malik, A.N.; Krawczyk, M.; et al. Mitochondria, oxidative stress and nonalcoholic fatty liver disease: A complex relationship. Eur. J. Clin. Investig. 2022, 52, e13622. [Google Scholar] [CrossRef] [PubMed]
- Modoni, S.; Frangos, S.; Iakovou, I.; Boero, M.; Mansi, L. Theragnostics before we found its name. Q. J. Nucl. Med. Mol. Imaging 2021, 65, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.; Larson, S.M.; Weber, W.A. Theranostic Concepts: More Than Just a Fashion Trend-Introduction and Overview. J. Nucl. Med. 2017, 58, 1S–2S. [Google Scholar] [CrossRef]
- Fang, H.; Cavaliere, A.; Li, Z.; Huang, Y.; Marquez-Nostra, B. Preclinical Advances in Theranostics for the Different Molecular Subtypes of Breast Cancer. Front. Pharm. 2021, 12, 627693. [Google Scholar] [CrossRef]
- Kevadiya, B.D.; Ottemann, B.M.; Thomas, M.B.; Mukadam, I.; Nigam, S.; McMillan, J.; Gorantla, S.; Bronich, T.K.; Edagwa, B.; Gendelman, H.E. Neurotheranostics as personalized medicines. Adv. Drug Deliv. Rev. 2019, 148, 252–289. [Google Scholar] [CrossRef]
- Cattaneo, M.; Froio, A.; Gallino, A. Cardiovascular Imaging and Theranostics in Cardiovascular Pharmacotherapy. Eur. Cardiol. 2019, 14, 62–64. [Google Scholar] [CrossRef]
- Assadi, M.; Jokar, N.; Yordanova, A.; Gholamrezanezhad, A.; Amini, A.; Abbasi, F.; Biersack, H.J.; Amini, A.; Nabipour, I.; Ahmadzadehfar, H. Emerging Preclinical and Clinical Applications of Theranostics for Nononcological Disorders. PET Clin. 2021, 16, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jin, C.Y. Stem cells in drug screening for neurodegenerative disease. Korean J. Physiol. Pharm. 2012, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H.; Petrova-Benedict, R.; Buncic, J.R.; Wallace, D.C. Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992, 48, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Hertig, D.; Felser, A.; Diserens, G.; Kurth, S.; Vermathen, P.; Nuoffer, J.M. Selective galactose culture condition reveals distinct metabolic signatures in pyruvate dehydrogenase and complex I deficient human skin fibroblasts. Metabolomics 2019, 15, 32. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Disease | Redox- or Metabolic-Related Alterations | References |
|---|---|---|
| Cancer | Defects in mtDNA | [39,40,41] |
| Metabolic shift from OXPHOS to glycolysis | [42] | |
| Increased mitochondrial fission | [43,44,45,46,47,48] | |
| Metabolic disorders (T2D, obesity, NAFLD) | TCA cycle and mitochondrial respiratory chain overload | [49,50,51] |
| Mitochondrial malfunction due to the accumulation of free fatty acids in adipose and peripheral tissues (lipotoxicity) | [49,50,51] | |
| Increased oxidative stress | [49,50,51] | |
| Insulin resistance | [49,50,51] | |
| Dysregulation of Ca2+ homeostasis | [50] | |
| Compromised mitochondrial bioenergetics | [52] | |
| mtDNA mutations | [53,54] | |
| Neurodegenerative diseases (AD, PD, HD, ALS) | Impaired mitochondrial biogenesis and mitochondrial dynamics | [55,56,57] |
| Excess ROS production | [58] | |
| Decreased intracellular Ca2+ buffering | [58] | |
| Decreased respiratory capacity and/or loss of mitochondrial transmembrane potential | [59] | |
| Disruption of intracellular trafficking-associated neurotoxicity | [52,60,61,62,63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinho, S.A.; Anjo, S.I.; Cunha-Oliveira, T. Metabolic Priming as a Tool in Redox and Mitochondrial Theragnostics. Antioxidants 2023, 12, 1072. https://doi.org/10.3390/antiox12051072
Pinho SA, Anjo SI, Cunha-Oliveira T. Metabolic Priming as a Tool in Redox and Mitochondrial Theragnostics. Antioxidants. 2023; 12(5):1072. https://doi.org/10.3390/antiox12051072
Chicago/Turabian StylePinho, Sónia A., Sandra I. Anjo, and Teresa Cunha-Oliveira. 2023. "Metabolic Priming as a Tool in Redox and Mitochondrial Theragnostics" Antioxidants 12, no. 5: 1072. https://doi.org/10.3390/antiox12051072
APA StylePinho, S. A., Anjo, S. I., & Cunha-Oliveira, T. (2023). Metabolic Priming as a Tool in Redox and Mitochondrial Theragnostics. Antioxidants, 12(5), 1072. https://doi.org/10.3390/antiox12051072