


PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Regulation of PGC-1α
2.1. Splice Variants of PGC-1α
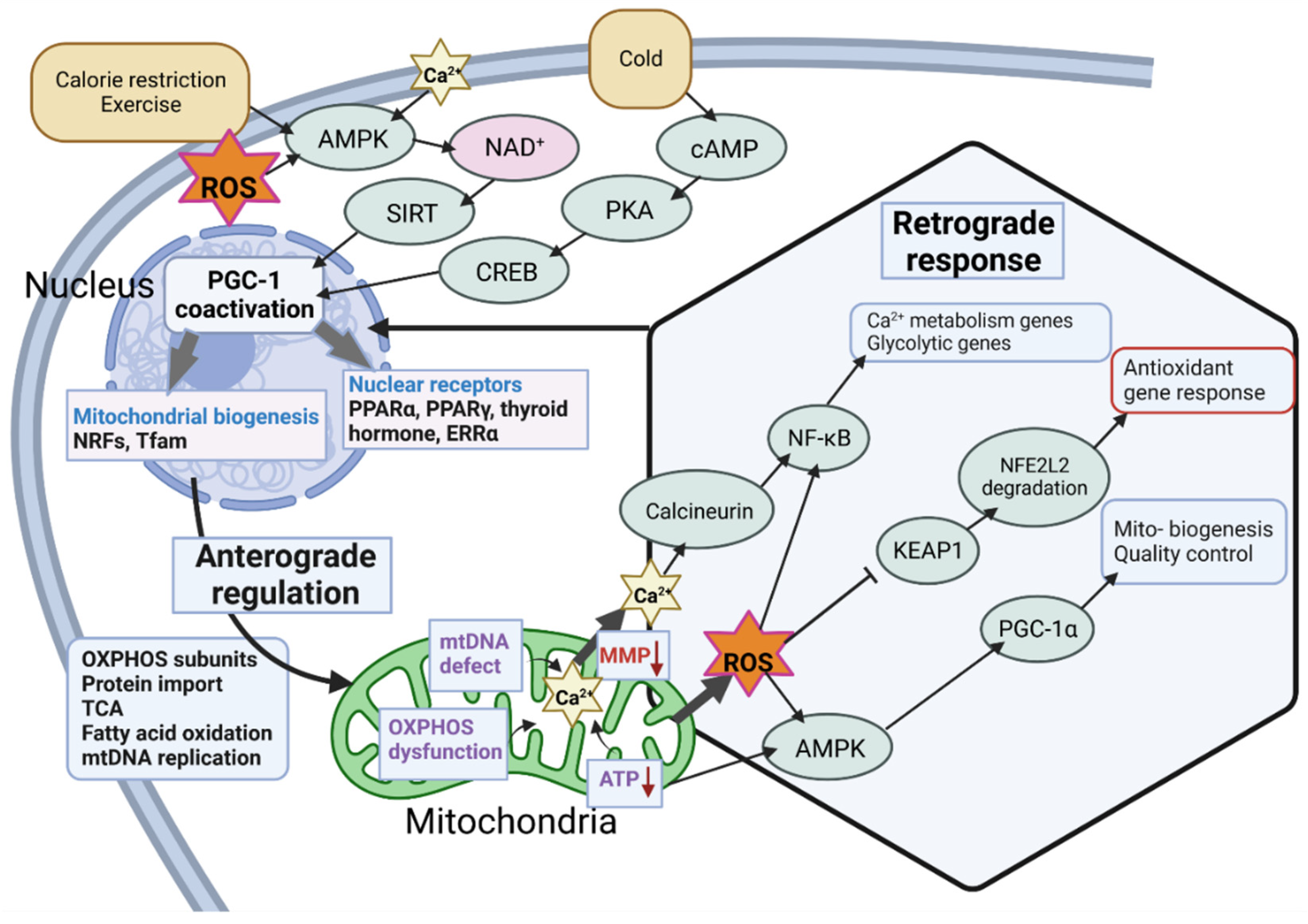
2.2. Regulation of the Master Regulator
2.3. Stress-Related Transcriptional Regulation of PGC-1α

2.4. Posttranslational Regulation of PGC-1α
3. The Link between PGC-1α and Mitochondria
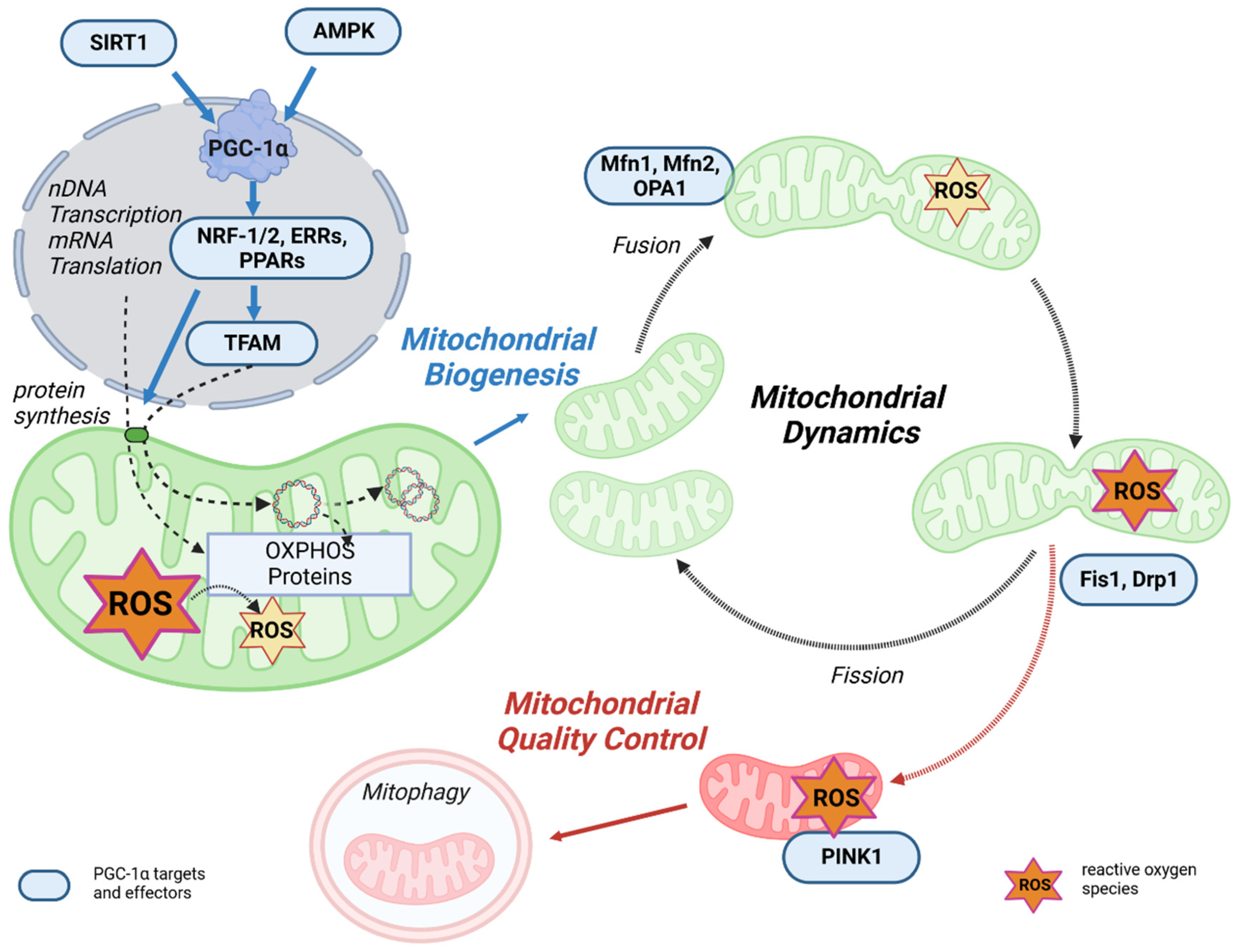
3.1. PGC-1α as the Master Regulator of Mitochondrial Biogenesis
3.2. PGC-1α Affects Mitochondrial Dynamics and Quality Control
4. PGC-1α, Mitochondria, and Oxidative Stress
4.1. Inflammation, ROS, and PGC-1α
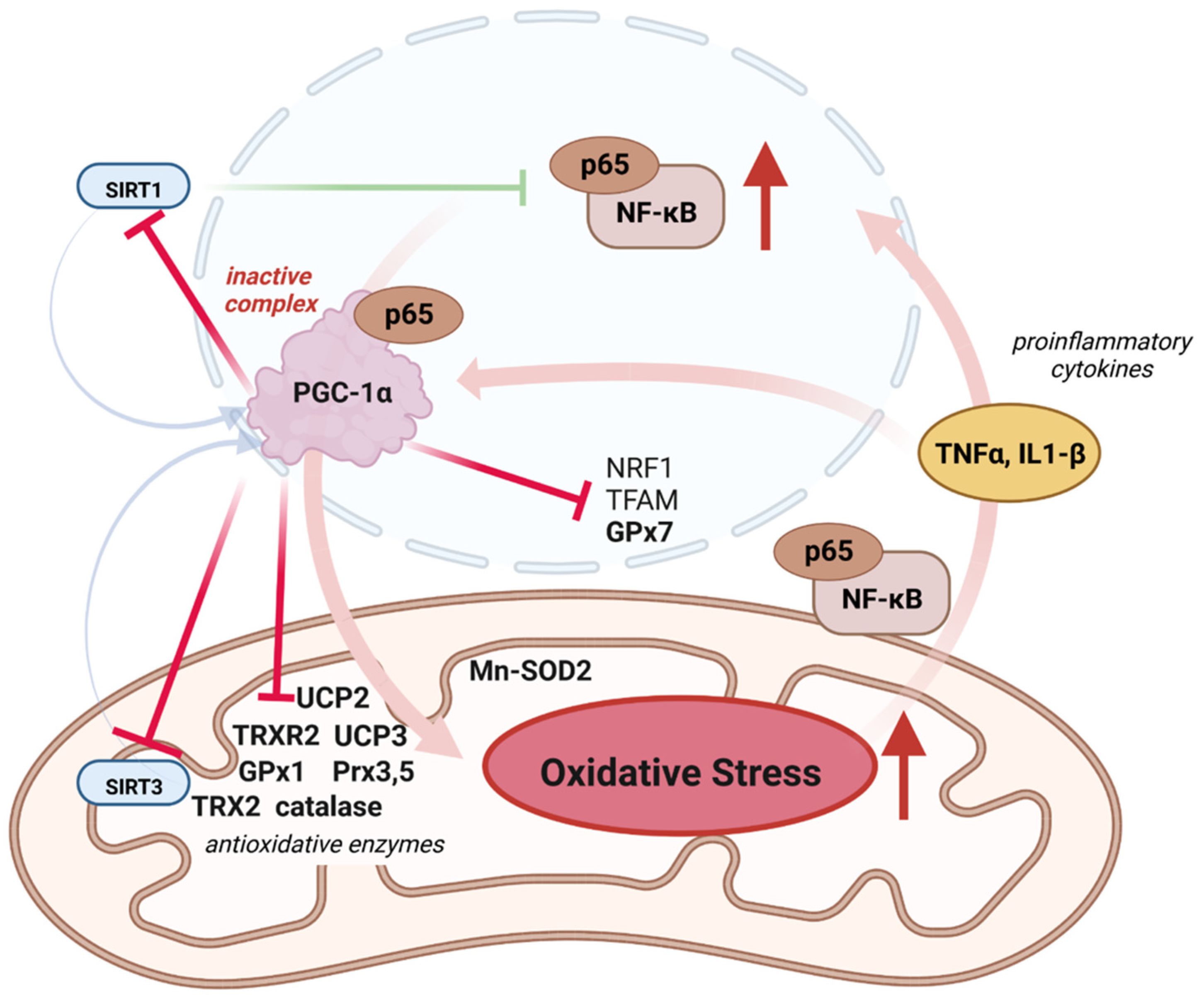
4.2. Role of PGC-1α in Mitonuclear Crosstalk for ROS Defense
4.3. PGC-1α, ROS, and Disease
4.4. PGC-1α as a Drug Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| AP | alternative promoter |
| ATF2 | activating transcription factor 2 |
| BAT | brown adipose tissue |
| CAMK2 | Ca2+/calmodulin-depend protein kinase 2 |
| Clk2 | CDC2-like kinase 2 |
| CREB | cyclic AMP response element-binding protein |
| CRTC2 | CREB-regulated transcription co-activator 2 |
| DNMT3B | DNA methyltransferase 3B |
| Drp1 | dynamin-related protein 1 |
| ER | estrogen receptor |
| ERRα | estrogen-related receptor α |
| FA | fatty acid |
| FAO | fatty acid oxidation |
| Fis1 | fission 1 protein |
| FoxO1 | forkhead box class-01 |
| GABPα | GA-binding protein-α |
| GCN5 | acetyltransferase protein acetyltransferase general control non-depressible 5 |
| GlcNAc | β-N-acetylglucosamine |
| GPx GPx1 GPx7 | glutathione peroxidase glutathione peroxidase 1 glutathione peroxidase 7 |
| GSK3β | glycogen synthase kinase 3β |
| HDACs | class II histone deacetylases |
| IGF1 | insulin-like growth factor 1 |
| IL1-β | interleukin 1β |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MEF2 | myocyte enhancer factor 2 |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| MGA | 3-Methylglutarate |
| miRNA | microRNA |
| MnSOD | Mn-dependent superoxide dismutase |
| mRNA | messenger RNA |
| mTOR | mechanistic target of rapamycin |
| NAD | nicotinamide adenine dinucleotide |
| NF-κB | nuclear factor-kappa B |
| NR | nuclear receptor |
| NRF-1 | nuclear respiratory factor 1 |
| NRF-2 | nuclear respiratory factor 2 |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| OGT | O-linked β-N-acetylglucosamine transferase |
| Opa1 | optic atrophy 1 |
| OXPHOS | oxidative phosphorylation |
| p65 | NF-κB subunit p65 |
| PGC-1α | peroxisome proliferator-activated receptor γ coactivator 1α |
| PGC-1β | peroxisome proliferator-activated receptor γ coactivator 1β |
| PKA | protein kinase A |
| PKB/Akt | protein kinase B |
| POLRMT | mtDNA promotors recruiting RNA polymerase |
| PPARs | receptor-like peroxisome proliferator-activated receptors |
| PPARα | peroxisome proliferator-activated receptor α |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PPREs | PPAR-specific response elements |
| PRC | PGC-1 related coactivator |
| PRMT1 | protein arginine methyltransferase 1 |
| Prx3 | peroxiredoxin 3 |
| Prx5 | peroxiredoxin 5 |
| ROS | reactive oxygen species |
| RRM | RNA recognition motif |
| RS | short serine/arginine-rich stretches |
| SCFCdc4 | Skp1/Cullin/F-box cell division control 4 |
| SENP1 | Sentrin/SUMO-specific protease |
| SIRT1 | sirtuin 1 |
| SIRT3 | sirtuin 3 |
| SRC-3 | steroid receptor coactivator 3 |
| SUMO | small ubiquitin-like modifier |
| TERT | telomerase reserve transcriptase |
| TFAM | mitochondrial transcription factor A |
| TFB2M | mitochondrial transcription factor B2 |
| Tfe3 | transcription factor E3 |
| TFEB | transcription factor EB |
| TNFα | tumor necrosis factor α |
| Tom70 | translocase of outer mitochondrial membrane receptor subunit 70 |
| TRX2 | thioredoxin 2 |
| TRXR2 | thioredoxin reductase 2 |
| TSS | transcription start site |
| Ub | specific ubiquitination |
| UCP-1 UCP-2 UCP-3 | uncoupling protein 1 uncoupling protein 2 uncoupling protein 3 |
| VEGFA | vascular endothelial growth factor A |
| YY1 | yin yang 1 |
| β-AR | β3-adrenergic receptor |
References
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The Role of PGC1α in Cancer Metabolism and its Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Puigserver, P.; Donovan, J.; Tarr, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 2002, 277, 1645–1648. [Google Scholar] [CrossRef]
- Jeremic, N.; Chaturvedi, P.; Tyagi, S.C. Browning of White Fat: Novel Insight Into Factors, Mechanisms, and Therapeutics. J. Cell. Physiol. 2017, 232, 61–68. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Adhihetty, P.J.; Ljubicic, V.; Hood, D.A. How is mitochondrial biogenesis affected in mitochondrial disease? Med. Sci. Sports Exerc. 2005, 37, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Andersson, U.; Scarpulla, R.C. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol. 2001, 21, 3738–3749. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Cai, B.; Nie, Q. PGC-1α affects skeletal muscle and adipose tissue development by regulating mitochondrial biogenesis. Mol. Genet. Genom. 2022, 297, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Chambers, J.M.; Wingert, R.A. PGC-1α in Disease: Recent Renal Insights into a Versatile Metabolic Regulator. Cells 2020, 9, 2234. [Google Scholar] [CrossRef]
- Mouler Rechtman, M.; Burdelova, E.O.; Bar-Yishay, I.; Ben-Yehoyada, M.; Fishman, S.; Halpern, Z.; Shlomai, A. The metabolic regulator PGC-1α links anti-cancer cytotoxic chemotherapy to reactivation of hepatitis B virus. J. Viral Hepat. 2013, 20, 34–41. [Google Scholar] [CrossRef]
- Monsalve, M.; Wu, Z.; Adelmant, G.; Puigserver, P.; Fan, M.; Spiegelman, B.M. Direct Coupling of Transcription and mRNA Processing through the Thermogenic Coactivator PGC-1. Mol. Cell 2000, 6, 307–316. [Google Scholar] [CrossRef]
- Martínez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1alpha mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Kai, Y.; Kamei, Y.; Ezaki, O. Isoform-specific increases in murine skeletal muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) mRNA in response to beta2-adrenergic receptor activation and exercise. Endocrinology 2008, 149, 4527–4533. [Google Scholar] [CrossRef]
- Yoshioka, T.; Inagaki, K.; Noguchi, T.; Sakai, M.; Ogawa, W.; Hosooka, T.; Iguchi, H.; Watanabe, E.; Matsuki, Y.; Hiramatsu, R.; et al. Identification and characterization of an alternative promoter of the human PGC-1alpha gene. Biochem. Biophys. Res. Commun. 2009, 381, 537–543. [Google Scholar] [CrossRef]
- Norrbom, J.; Sällstedt, E.K.; Fischer, H.; Sundberg, C.J.; Rundqvist, H.; Gustafsson, T. Alternative splice variant PGC-1α-b is strongly induced by exercise in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1092–E1098. [Google Scholar] [CrossRef] [PubMed]
- Ruas, J.L.; White, J.P.; Rao, R.R.; Kleiner, S.; Brannan, K.T.; Harrison, B.C.; Greene, N.P.; Wu, J.; Estall, J.L.; Irving, B.A.; et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012, 151, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Nader, G.A.; von Walden, F.; Liu, C.; Lindvall, J.; Gutmann, L.; Pistilli, E.E.; Gordon, P.M. Resistance exercise training modulates acute gene expression during human skeletal muscle hypertrophy. J. Appl. Physiol. 2014, 116, 693–702. [Google Scholar] [CrossRef]
- Tadaishi, M.; Miura, S.; Kai, Y.; Kano, Y.; Oishi, Y.; Ezaki, O. Skeletal muscle-specific expression of PGC-1α-b, an exercise-responsive isoform, increases exercise capacity and peak oxygen uptake. PLoS ONE 2011, 6, e28290. [Google Scholar] [CrossRef]
- Felder, T.K.; Soyal, S.M.; Oberkofler, H.; Hahne, P.; Auer, S.; Weiss, R.; Gadermaier, G.; Miller, K.; Krempler, F.; Esterbauer, H.; et al. Characterization of novel peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) isoform in human liver. J. Biol. Chem. 2011, 286, 42923–42936. [Google Scholar] [CrossRef]
- Soyal, S.M.; Felder, T.K.; Auer, S.; Hahne, P.; Oberkofler, H.; Witting, A.; Paulmichl, M.; Landwehrmeyer, G.B.; Weydt, P.; Patsch, W. A greatly extended PPARGC1A genomic locus encodes several new brain-specific isoforms and influences Huntington disease age of onset. Hum. Mol. Genet. 2012, 21, 3461–3473. [Google Scholar] [CrossRef]
- Zhang, Y.; Huypens, P.; Adamson, A.W.; Chang, J.S.; Henagan, T.M.; Boudreau, A.; Lenard, N.R.; Burk, D.; Klein, J.; Perwitz, N.; et al. Alternative mRNA Splicing Produces a Novel Biologically Active Short Isoform of PGC-1α*. J. Biol. Chem. 2009, 284, 32813–32826. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Suntar, I.; Sureda, A.; Belwal, T.; Sanches Silva, A.; Vacca, R.A.; Tewari, D.; Sobarzo-Sánchez, E.; Nabavi, S.F.; Shirooie, S.; Dehpour, A.R.; et al. Natural products, PGC-1α, and Duchenne muscular dystrophy. Acta Pharm. Sin. B 2020, 10, 734–745. [Google Scholar] [CrossRef]
- Cherry, A.D.; Piantadosi, C.A. Regulation of Mitochondrial Biogenesis and Its Intersection with Inflammatory Responses. Antioxid. Redox Signal. 2015, 22, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.; Blasiak, J.; Tavi, P.; Kaarniranta, K. Therapeutic potential of PGC-1α in age-related macular degeneration (AMD)—The involvement of mitochondrial quality control, autophagy, and antioxidant response. Expert Opin. Ther. Targets 2021, 25, 773–785. [Google Scholar] [CrossRef]
- Booth, F.W.; Ruegsegger, G.N.; Toedebusch, R.G.; Yan, Z. Endurance Exercise and the Regulation of Skeletal Muscle Metabolism. Prog. Mol. Biol. Transl. Sci. 2015, 135, 129–151. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca2+/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef]
- Sano, M.; Schneider, M.D. Cyclin-dependent kinase-9: An RNAPII kinase at the nexus of cardiac growth and death cascades. Circ. Res. 2004, 95, 867–876. [Google Scholar] [CrossRef]
- Salma, N.; Song, J.S.; Arany, Z.; Fisher, D.E. Transcription Factor Tfe3 Directly Regulates Pgc-1alpha in Muscle. J. Cell. Physiol. 2015, 230, 2330–2336. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Blasiak, J.; Pawlowska, E.; Sobczuk, A.; Szczepanska, J.; Kaarniranta, K. The Aging Stress Response and Its Implication for AMD Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8840. [Google Scholar] [CrossRef] [PubMed]
- Chambers, K.T.; Leone, T.C.; Sambandam, N.; Kovacs, A.; Wagg, C.S.; Lopaschuk, G.D.; Finck, B.N.; Kelly, D.P. Chronic Inhibition of Pyruvate Dehydrogenase in Heart Triggers an Adaptive Metabolic Response*. J. Biol. Chem. 2011, 286, 11155–11162. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Parsamanesh, N.; Asghari, A.; Sardari, S.; Tasbandi, A.; Jamialahmadi, T.; Xu, S.; Sahebkar, A. Resveratrol and endothelial function: A literature review. Pharmacol. Res. 2021, 170, 105725. [Google Scholar] [CrossRef]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef]
- Lustig, Y.; Ruas, J.L.; Estall, J.L.; Lo, J.C.; Devarakonda, S.; Laznik, D.; Choi, J.H.; Ono, H.; Olsen, J.V.; Spiegelman, B.M. Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes Dev. 2011, 25, 1232–1244. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Whittington, H.J.; Harding, I.; Stephenson, C.I.M.; Bell, R.; Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Cardioprotection in the aging, diabetic heart: The loss of protective Akt signalling. Cardiovasc. Res. 2013, 99, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Haas, W.; Gygi, S.P.; Puigserver, P. Cdc2-like kinase 2 is an insulin-regulated suppressor of hepatic gluconeogenesis. Cell Metab. 2010, 11, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Buck, S.W.; Gallo, C.M.; Smith, J.S. Diversity in the Sir2 family of protein deacetylases. J. Leukoc. Biol. 2004, 75, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Aquilano, K.; Vigilanza, P.; Baldelli, S.; Pagliei, B.; Rotilio, G.; Ciriolo, M.R. Peroxisome proliferator-activated receptor gamma co-activator 1alpha (PGC-1alpha) and sirtuin 1 (SIRT1) reside in mitochondria: Possible direct function in mitochondrial biogenesis. J. Biol. Chem. 2010, 285, 21590–21599. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Ciriolo, M.R. Extranuclear localization of SIRT1 and PGC-1α: An insight into possible roles in diseases associated with mitochondrial dysfunction. Curr. Mol. Med. 2013, 13, 140–154. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Cantó, C.; Jiang, L.Q.; Deshmukh, A.S.; Mataki, C.; Coste, A.; Lagouge, M.; Zierath, J.R.; Auwerx, J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010, 11, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Waldman, M.; Cohen, K.; Yadin, D.; Nudelman, V.; Gorfil, D.; Laniado-Schwartzman, M.; Kornwoski, R.; Aravot, D.; Abraham, N.G.; Arad, M.; et al. Regulation of diabetic cardiomyopathy by caloric restriction is mediated by intracellular signaling pathways involving ‘SIRT1 and PGC-1α’. Cardiovasc. Diabetol. 2018, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Trausch-Azar, J.; Leone, T.C.; Kelly, D.P.; Schwartz, A.L. Ubiquitin Proteasome-dependent Degradation of the Transcriptional Coactivator PGC-1α via the N-terminal Pathway*. J. Biol. Chem. 2010, 285, 40192–40200. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.L.; Hock, M.B.; Ekholm-Reed, S.; Wohlschlegel, J.A.; Dev, K.K.; Kralli, A.; Reed, S.I. SCFCdc4 acts antagonistically to the PGC-1alpha transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 2008, 22, 252–264. [Google Scholar] [CrossRef]
- Rytinki, M.M.; Palvimo, J.J. SUMOylation Attenuates the Function of PGC-1α*. J. Biol. Chem. 2009, 284, 26184–26193. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Yu, T.; Huang, C.; Xia, X.; Liu, X.; Gu, J.; Xue, S.; Yeh, E.T.; Cheng, J. SUMO-specific Protease 1 Regulates Mitochondrial Biogenesis through PGC-1α*. J. Biol. Chem. 2012, 287, 44464–44470. [Google Scholar] [CrossRef]
- Teyssier, C.; Ma, H.; Emter, R.; Kralli, A.; Stallcup, M.R. Activation of nuclear receptor coactivator PGC-1alpha by arginine methylation. Genes Dev. 2005, 19, 1466–1473. [Google Scholar] [CrossRef]
- Ruan, H.-B.; Han, X.; Li, M.-D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef]
- Luo, X.; Liao, C.; Quan, J.; Cheng, C.; Zhao, X.; Bode, A.M.; Cao, Y. Posttranslational regulation of PGC-1α and its implication in cancer metabolism. Int. J. Cancer 2019, 145, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1α and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Diaz-Gerevini, G.T.; Repossi, G.; Dain, A.; Tarres, M.C.; Das, U.N.; Eynard, A.R. Beneficial action of resveratrol: How and why? Nutrition 2016, 32, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Ray Hamidie, R.D.; Yamada, T.; Ishizawa, R.; Saito, Y.; Masuda, K. Curcumin treatment enhances the effect of exercise on mitochondrial biogenesis in skeletal muscle by increasing cAMP levels. Metabolism 2015, 64, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef]
- Vásquez-Reyes, S.; Velázquez-Villegas, L.A.; Vargas-Castillo, A.; Noriega, L.G.; Torres, N.; Tovar, A.R. Dietary bioactive compounds as modulators of mitochondrial function. J. Nutr. Biochem. 2021, 96, 108768. [Google Scholar] [CrossRef]
- Maissan, P.; Mooij, E.J.; Barberis, M. Sirtuins-Mediated System-Level Regulation of Mammalian Tissues at the Interface between Metabolism and Cell Cycle: A Systematic Review. Biology 2021, 10, 194. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Kirwan, A.F.; Hood, D.A. AMP-activated protein kinase-regulated activation of the PGC-1alpha promoter in skeletal muscle cells. PLoS ONE 2008, 3, e3614. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Kiilerich, K.; Pilegaard, H. PGC-1alpha-mediated adaptations in skeletal muscle. Pflugers Arch. 2010, 460, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.R.; Hernández-Yago, J. Distinct functional contributions of 2 GABP-NRF-2 recognition sites within the context of the human TOMM70 promoter. Biochem. Cell Biol. 2006, 84, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Bennett, J.P.; Stokin, G.B. Regulation of neuronal bioenergetics as a therapeutic strategy in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Pearson, A.; Levy, Y.; Cardel, B.; Handschin, C.; Ochala, J. Exploring the Role of PGC-1α in Defining Nuclear Organisation in Skeletal Muscle Fibres. J. Cell. Physiol. 2017, 232, 1270–1274. [Google Scholar] [CrossRef]
- Brüser, C.; Keller-Findeisen, J.; Jakobs, S. The TFAM-to-mtDNA ratio defines inner-cellular nucleoid populations with distinct activity levels. Cell Rep. 2021, 37, 110000. [Google Scholar] [CrossRef]
- Taherzadeh-Fard, E.; Saft, C.; Akkad, D.A.; Wieczorek, S.; Haghikia, A.; Chan, A.; Epplen, J.T.; Arning, L. PGC-1alpha downstream transcription factors NRF-1 and TFAM are genetic modifiers of Huntington disease. Mol. Neurodegener. 2011, 6, 32. [Google Scholar] [CrossRef]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Hood, D.A. Impact of Aging and Exercise on Mitochondrial Quality Control in Skeletal Muscle. Oxid. Med. Cell. Longev. 2017, 2017, 3165396. [Google Scholar] [CrossRef]
- Zhang, Q.; Lei, Y.-H.; Zhou, J.-P.; Hou, Y.-Y.; Wan, Z.; Wang, H.-L.; Meng, H. Role of PGC-1α in Mitochondrial Quality Control in Neurodegenerative Diseases. Neurochem. Res. 2019, 44, 2031–2043. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Yang, L.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Cui, Z.; Ao, L.; Liu, J.; et al. The Interaction of Mitochondrial Biogenesis and Fission/Fusion Mediated by PGC-1α Regulates Rotenone-Induced Dopaminergic Neurotoxicity. Mol. Neurobiol. 2017, 54, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1α controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging 2015, 7, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-C.; Lin, T.-K.; Yang, D.-I.; Yang, J.-L.; Liou, C.-W.; Chen, S.-D. Peroxisome proliferator-activated receptor-gamma dependent pathway reduces the phosphorylation of dynamin-related protein 1 and ameliorates hippocampal injury induced by global ischemia in rats. J. Biomed. Sci. 2016, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.; Meng, L.; Gong, M.; Li, J.; Shi, W.; Qiu, J.; Yang, Y.; Zhao, J.; Suo, Y.; et al. Pioglitazone Inhibits Diabetes-Induced Atrial Mitochondrial Oxidative Stress and Improves Mitochondrial Biogenesis, Dynamics, and Function Through the PPAR-γ/PGC-1α Signaling Pathway. Front. Pharmacol. 2021, 12, 658362. [Google Scholar] [CrossRef]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, K.Z.Q.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Pinho, R.; Gu, Y.; Radak, Z. The Role of SIRT3 in Exercise and Aging. Cells 2022, 11, 2596. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, Y.; Zhou, L.; Dorfman, R.G.; Liu, L.; Cai, R.; Jiang, C.; Tang, D.; Wang, Y.; Zou, X.; et al. SIRT3 elicited an anti-Warburg effect through HIF1α/PDK1/PDHA1 to inhibit cholangiocarcinoma tumorigenesis. Cancer Med. 2019, 8, 2380–2391. [Google Scholar] [CrossRef]
- O’Flaherty, C. Orchestrating the antioxidant defenses in the epididymis. Andrology 2019, 7, 662–668. [Google Scholar] [CrossRef]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef]
- Toda, C.; Diano, S. Mitochondrial UCP2 in the central regulation of metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 757–764. [Google Scholar] [CrossRef]
- Pérez, S.; Rius-Pérez, S.; Finamor, I.; Martí-Andrés, P.; Prieto, I.; García, R.; Monsalve, M.; Sastre, J. Obesity causes PGC-1α deficiency in the pancreas leading to marked IL-6 upregulation via NF-κB in acute pancreatitis. J. Pathol. 2019, 247, 48–59. [Google Scholar] [CrossRef]
- Qi, Y.; Yin, X.; Wang, S.; Jiang, H.; Wang, X.; Ren, M.; Su, X.-P.; Lei, S.; Feng, H. PGC-1α Silencing Compounds the Perturbation of Mitochondrial Function Caused by Mutant SOD1 in Skeletal Muscle of ALS Mouse Model. Front. Aging Neurosci. 2015, 7, 204. [Google Scholar] [CrossRef]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Laguna, J.C.; Vázquez-Carrera, M. The p65 subunit of NF-kappaB binds to PGC-1alpha, linking inflammation and metabolic disturbances in cardiac cells. Cardiovasc. Res. 2010, 87, 449–458. [Google Scholar] [CrossRef]
- Chen, S.-D.; Yang, D.-I.; Lin, T.-K.; Shaw, F.-Z.; Liou, C.-W.; Chuang, Y.-C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1α in Vascular Regulation: Implications for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.S.; Handschin, C. Functional crosstalk of PGC-1 coactivators and inflammation in skeletal muscle pathophysiology. Semin. Immunopathol. 2014, 36, 27–53. [Google Scholar] [CrossRef] [PubMed]
- Eleutherio, E.C.A.; Silva Magalhães, R.S.; de Araújo Brasil, A.; Monteiro Neto, J.R.; de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-I.; Wei, P.-C.; Hsu, J.-L.; Su, F.-Y.; Lee, W.-H. NPGPx (GPx7): A novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 2016, 8, 1626–1640. [Google Scholar] [PubMed]
- Remels, A.H.V.; Gosker, H.R.; Bakker, J.; Guttridge, D.C.; Schols, A.M.W.J.; Langen, R.C.J. Regulation of skeletal muscle oxidative phenotype by classical NF-κB signalling. Biochim. Biophys. Acta 2013, 1832, 1313–1325. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Suppression of estrogen-related receptor alpha and medium-chain acyl-coenzyme A dehydrogenase in the acute-phase response. J. Lipid Res. 2005, 46, 2282–2288. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Barger, J.L.; Boyer, B.B.; Brand, M.D.; Pan, G.; Adams, S.H. Impact of endotoxin on UCP homolog mRNA abundance, thermoregulation, and mitochondrial proton leak kinetics. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E433–E446. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Berlemann, L.A.; Bader, V.; Sehr, D.A.; Dawin, E.; Covallero, A.; Meschede, J.; Angersbach, L.; Showkat, C.; Michaelis, J.B.; et al. LUBAC assembles a ubiquitin signaling platform at mitochondria for signal amplification and transport of NF-κB to the nucleus. EMBO J. 2022, 41, e112006. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Ji, L.L. Role of PGC-1α signaling in skeletal muscle health and disease. Ann. N. Y. Acad. Sci. 2012, 1271, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Liu, X.; Zhu, X.; Qu, Z.; Gong, Z.; Li, J.; Xiao, L.; Yang, Y.; Liu, H.; Sun, L.; et al. The Role of TLR4 on PGC-1α-Mediated Oxidative Stress in Tubular Cell in Diabetic Kidney Disease. Oxid. Med. Cell. Longev. 2018, 2018, 6296802. [Google Scholar] [CrossRef] [PubMed]
- Besse-Patin, A.; Léveillé, M.; Oropeza, D.; Nguyen, B.N.; Prat, A.; Estall, J.L. Estrogen Signals Through Peroxisome Proliferator-Activated Receptor-γ Coactivator 1α to Reduce Oxidative Damage Associated With Diet-Induced Fatty Liver Disease. Gastroenterology 2017, 152, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.S.; Furrer, R.; Beer, M.; Handschin, C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem. Biophys. Res. Commun. 2015, 464, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martín-Sánchez, D.; Martinez-Moreno, J.M.; Carrasco, S.; Ruiz-Andrés, O.; Monsalve, M.; Sanchez-Ramos, C.; Gómez, M.J.; Ruiz-Ortega, M.; Sánchez-Niño, M.D.; et al. PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 2019, 249, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Keller, J.N.; Kindy, M.S.; Holtsberg, F.W.; St Clair, D.K.; Yen, H.C.; Germeyer, A.; Steiner, S.M.; Bruce-Keller, A.J.; Hutchins, J.B.; Mattson, M.P. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J. Neurosci. 1998, 18, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Noshita, N.; Sugawara, T.; Fujimura, M.; Morita-Fujimura, Y.; Chan, P.H. Manganese Superoxide Dismutase Affects Cytochrome c Release and Caspase-9 Activation After Transient Focal Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab. 2001, 21, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, D.; Mahadev Bhat, S.; Price, A.L.; Delmotte, P.; Sieck, G.C. Molecular Mechanisms Underlying TNFα-Induced Mitochondrial Biogenesis in Human Airway Smooth Muscle. Int. J. Mol. Sci. 2023, 24, 5788. [Google Scholar] [CrossRef]
- Evans, M.J.; Scarpulla, R.C. NRF-1: A trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 1990, 4, 1023–1034. [Google Scholar] [CrossRef]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; King, M.S.; Pryde, K.R. The production of reactive oxygen species by complex I. Biochem. Soc. Trans. 2008, 36, 976–980. [Google Scholar] [CrossRef]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, Y.; Matsumura, K.; Ishii, S.; Inagaki, H.; Suzuki, T.; Matsuda, Y.; Beck, K.; Kitagawa, Y. Functional domains of chicken mitochondrial transcription factor A for the maintenance of mitochondrial DNA copy number in lymphoma cell line DT40. J. Biol. Chem. 2003, 278, 31149–31158. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, I.E. Mitochondria, 2nd ed.; Wiley-Liss: Hoboken, NJ, USA, 2008; ISBN 978-0-470-04073-7. [Google Scholar]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Evans, R. PPARs and ERRs: Molecular mediators of mitochondrial metabolism. Curr. Opin. Cell Biol. 2015, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Vandenbeek, R.; Khan, N.P.; Estall, J.L. Linking Metabolic Disease With the PGC-1α Gly482Ser Polymorphism. Endocrinology 2018, 159, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Uddin, M.J.; Kim, Y.; Ko, M.; Yu, I.; Ha, H. PGC-1α, a potential therapeutic target against kidney aging. Aging Cell 2019, 18, e12994. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Kang, Y.; Zhang, H.; Zhao, Y.; Wang, Y.; Wang, W.; He, Y.; Zhang, W.; Zhang, W.; Zhu, X.; Zhou, Y.; et al. Telomere Dysfunction Disturbs Macrophage Mitochondrial Metabolism and the NLRP3 Inflammasome through the PGC-1α/TNFAIP3 Axis. Cell Rep. 2018, 22, 3493–3506. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Patrushev, N.; Forouzandeh, F.; Hilenski, L.; Alexander, R.W. PGC-1α Modulates Telomere Function and DNA Damage in Protecting against Aging-Related Chronic Diseases. Cell Rep. 2015, 12, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Sczelecki, S.; Besse-Patin, A.; Abboud, A.; Kleiner, S.; Laznik-Bogoslavski, D.; Wrann, C.D.; Ruas, J.L.; Haibe-Kains, B.; Estall, J.L. Loss of Pgc-1α expression in aging mouse muscle potentiates glucose intolerance and systemic inflammation. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E157–E167. [Google Scholar] [CrossRef]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.-J.; Liu, Z.-X.; Lee, H.-Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Leach, J.K.; van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Dirat, B.; Ader, I.; Golzio, M.; Massa, F.; Mettouchi, A.; Laurent, K.; Larbret, F.; Malavaud, B.; Cormont, M.; Lemichez, E.; et al. Inhibition of the GTPase Rac1 mediates the antimigratory effects of metformin in prostate cancer cells. Mol. Cancer Ther. 2015, 14, 586–596. [Google Scholar] [CrossRef]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, I.; Salvatore, L.; Murzilli, S.; Lo Sasso, G.; Latorre, D.; Martelli, N.; Egorova, A.V.; Polishuck, R.; Madeyski-Bengtson, K.; Lelliott, C.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC1alpha) is a metabolic regulator of intestinal epithelial cell fate. Proc. Natl. Acad. Sci. USA 2011, 108, 6603–6608. [Google Scholar] [CrossRef]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.-K.C.; Chi, J.-T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1α Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef]
- Wang, X.; Moraes, C.T. Increases in mitochondrial biogenesis impair carcinogenesis at multiple levels. Mol. Oncol. 2011, 5, 399–409. [Google Scholar] [CrossRef]
- Burchard, J.; Zhang, C.; Liu, A.M.; Poon, R.T.P.; Lee, N.P.Y.; Wong, K.-F.; Sham, P.C.; Lam, B.Y.; Ferguson, M.D.; Tokiwa, G.; et al. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol. Syst. Biol. 2010, 6, 402. [Google Scholar] [CrossRef]
- Zhang, Y.; Ba, Y.; Liu, C.; Sun, G.; Ding, L.; Gao, S.; Hao, J.; Yu, Z.; Zhang, J.; Zen, K.; et al. PGC-1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma-dependent pathway. Cell Res. 2007, 17, 363–373. [Google Scholar] [CrossRef]
- Petrocelli, J.J.; Drummond, M.J. PGC-1α-Targeted Therapeutic Approaches to Enhance Muscle Recovery in Aging. Int. J. Environ. Res. Public Health 2020, 17, 8650. [Google Scholar] [CrossRef]
- Pezzuto, J.M. Resveratrol: Twenty Years of Growth, Development and Controversy. Biomol. Ther. 2019, 27, 1–14. [Google Scholar] [CrossRef]
- Pannu, N.; Bhatnagar, A. Resveratrol: From enhanced biosynthesis and bioavailability to multitargeting chronic diseases. Biomed. Pharmacother. 2019, 109, 2237–2251. [Google Scholar] [CrossRef]
- Niu, W.; Wang, H.; Wang, B.; Mao, X.; Du, M. Resveratrol improves muscle regeneration in obese mice through enhancing mitochondrial biogenesis. J. Nutr. Biochem. 2021, 98, 108804. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res. Clin. Pract. 2016, 35, 69–77. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.-M.; Cline, G.W.; Nakahara, K.; et al. Metformin, phenformin, and galegine inhibit complex IV activity and reduce glycerol-derived gluconeogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2122287119. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef]
- Timmins, P.; Donahue, S.; Meeker, J.; Marathe, P. Steady-state pharmacokinetics of a novel extended-release metformin formulation. Clin. Pharmacokinet. 2005, 44, 721–729. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.-M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Berstein, L.M.; Popovich, I.G.; Zabezhinski, M.A.; Egormin, P.A.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Kovalenko, I.G.; et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging 2011, 3, 148–157. [Google Scholar] [CrossRef]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Antoch, M.P.; Blagosklonny, M.V. Rapamycin extends maximal lifespan in cancer-prone mice. Am. J. Pathol. 2010, 176, 2092–2097. [Google Scholar] [CrossRef]
- Fischer, K.E.; Gelfond, J.A.L.; Soto, V.Y.; Han, C.; Someya, S.; Richardson, A.; Austad, S.N. Health Effects of Long-Term Rapamycin Treatment: The Impact on Mouse Health of Enteric Rapamycin Treatment from Four Months of Age throughout Life. PLoS ONE 2015, 10, e0126644. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padrón, Á.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Fernandez, E.; Flurkey, K.; Han, M.; Javors, M.A.; Li, X.; Nadon, N.L.; Nelson, J.F.; et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 2014, 13, 468–477. [Google Scholar] [CrossRef]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.-F.; Liao, C.-Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef]
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013, 4, 913–920. [Google Scholar] [CrossRef]
- Chen, G.; Chen, H.; Wang, C.; Peng, Y.; Sun, L.; Liu, H.; Liu, F. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS ONE 2012, 7, e33626. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Wang, X.; Huang, N.; Yang, M.; Wei, D.; Tai, H.; Han, X.; Gong, H.; Zhou, J.; Qin, J.; Wei, X.; et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1α pathway-mediated mitochondria biogenesis. Cell Death Dis. 2017, 8, e2702. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Kim, H.W.; Park, C.W.; Chang, Y.S.; Choi, B.S. PPARα agonist, fenofibrate, ameliorates age-related renal injury. Exp. Gerontol. 2016, 81, 42–50. [Google Scholar] [CrossRef]
- Dillon, L.M.; Hida, A.; Garcia, S.; Prolla, T.A.; Moraes, C.T. Long-term bezafibrate treatment improves skin and spleen phenotypes of the mtDNA mutator mouse. PLoS ONE 2012, 7, e44335. [Google Scholar] [CrossRef]
- Da Rosa-Junior, N.T.; Parmeggiani, B.; Glänzel, N.M.; de Moura Alvorcem, L.; Brondani, M.; Britto, R.; Grings, M.; Ortiz, V.D.; Turck, P.; Da Rosa Araujo, A.S.; et al. Antioxidant system disturbances and mitochondrial dysfunction induced by 3-methyglutaric acid in rat heart are prevented by bezafibrate. Eur. J. Pharmacol. 2022, 924, 174950. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, J.; Zhou, F.; Wang, W.; Chen, N. PGC-1α ameliorates kidney fibrosis in mice with diabetic kidney disease through an antioxidative mechanism. Mol. Med. Rep. 2018, 17, 4490–4498. [Google Scholar] [CrossRef]
- Wang, H.; Yan, W.-J.; Zhang, J.-L.; Zhang, F.-Y.; Gao, C.; Wang, Y.-J.; Bond Law, W.; Tao, L. Adiponectin partially rescues high glucose/high fat-induced impairment of mitochondrial biogenesis and function in a PGC-1α dependent manner. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 590–599. [Google Scholar]
- Yan, W.; Zhang, H.; Liu, P.; Wang, H.; Liu, J.; Gao, C.; Liu, Y.; Lian, K.; Yang, L.; Sun, L.; et al. Impaired mitochondrial biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling contributing to increased vulnerability in diabetic heart. Basic Res. Cardiol. 2013, 108, 329. [Google Scholar] [CrossRef]
- Popov, L.-D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Chernausek, S.D. Role of microRNA-130b in placental PGC-1α/TFAM mitochondrial biogenesis pathway. Biochem. Biophys. Res. Commun. 2017, 487, 607–612. [Google Scholar] [CrossRef]
- Gorska-Ponikowska, M.; Kuban-Jankowska, A.; Eisler, S.A.; Perricone, U.; Lo Bosco, G.; Barone, G.; Nussberger, S. 2-Methoxyestradiol Affects Mitochondrial Biogenesis Pathway and Succinate Dehydrogenase Complex Flavoprotein Subunit A in Osteosarcoma Cancer Cells. Cancer Genom. Proteom. 2018, 15, 73–89. [Google Scholar] [CrossRef]
- Valerio, A.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Pisconti, A.; Palomba, L.; Cantoni, O.; Clementi, E.; Moncada, S.; et al. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J. Clin. Investig. 2006, 116, 2791–2798. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef]
- Qi, R.; Wang, D.; Xing, L.; Wu, Z. Cyclosporin A inhibits mitochondrial biogenesis in Hep G2 cells. Biochem. Biophys. Res. Commun. 2018, 496, 941–946. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants 2023, 12, 1075. https://doi.org/10.3390/antiox12051075
Abu Shelbayeh O, Arroum T, Morris S, Busch KB. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants. 2023; 12(5):1075. https://doi.org/10.3390/antiox12051075
Chicago/Turabian StyleAbu Shelbayeh, Othman, Tasnim Arroum, Silke Morris, and Karin B. Busch. 2023. "PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response" Antioxidants 12, no. 5: 1075. https://doi.org/10.3390/antiox12051075
APA StyleAbu Shelbayeh, O., Arroum, T., Morris, S., & Busch, K. B. (2023). PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants, 12(5), 1075. https://doi.org/10.3390/antiox12051075