
The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
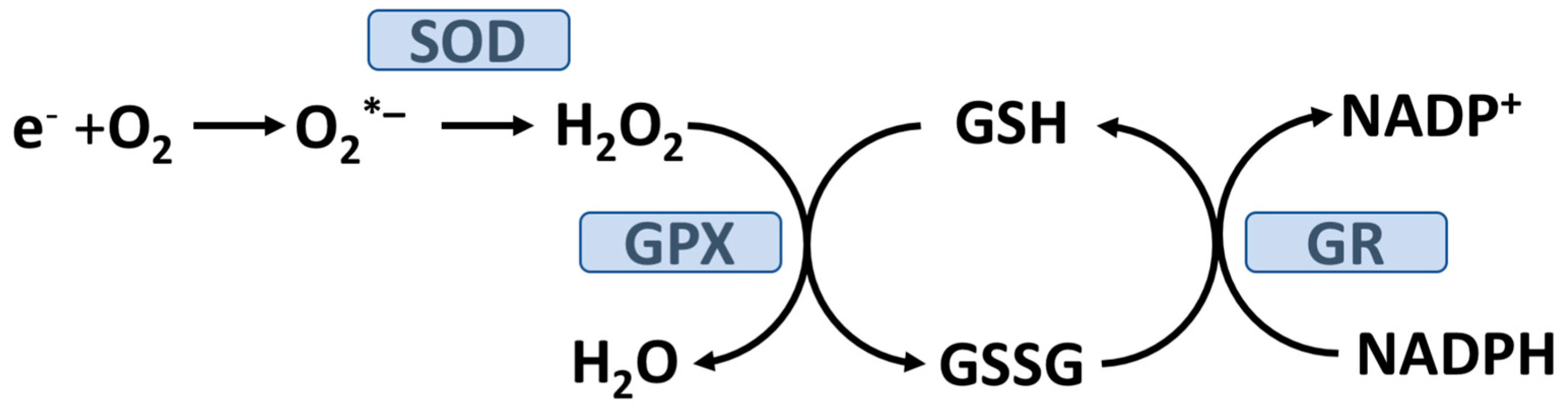
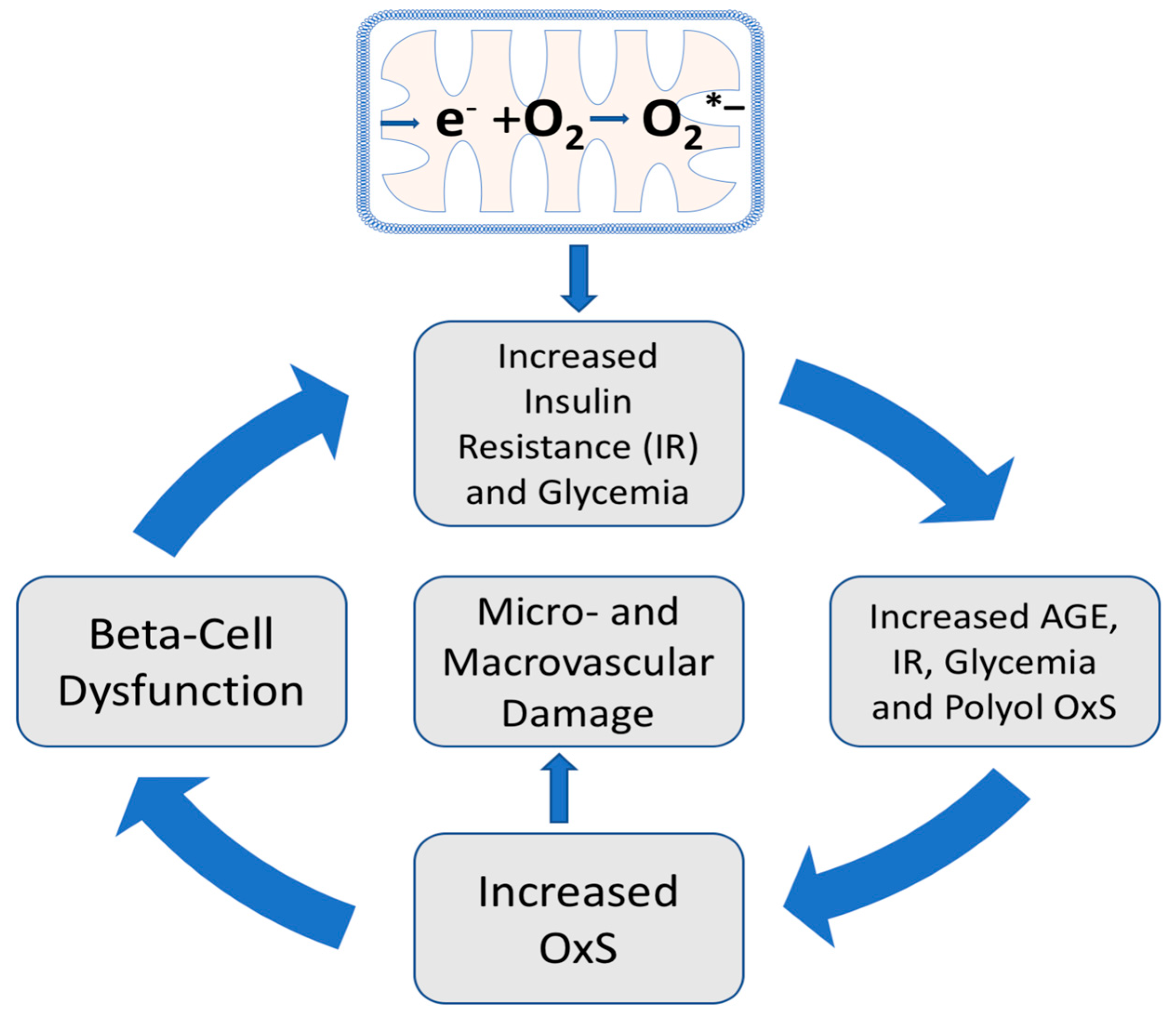
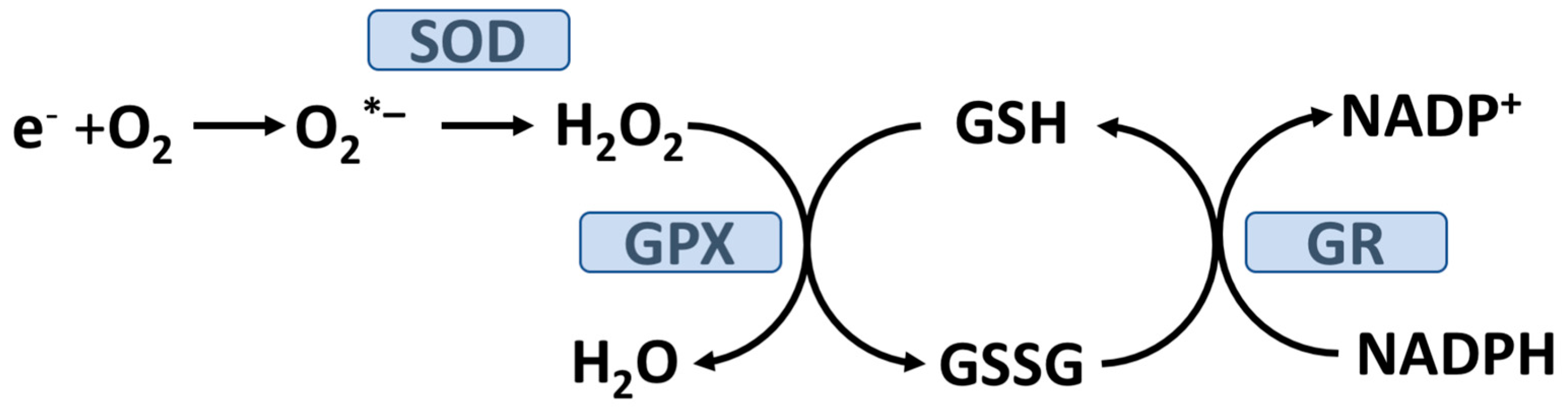
2. The Role of Oxidative Stress and T2D Pathogenesis
3. Insulin Resistance in Skeletal Muscle Is Considered the Initiating Defect Leading to T2D
4. Skeletal Muscle Mitochondrial Hydrogen Peroxide (H2O2) Emission Results in Insulin Resistance
5. Avoiding High-Fat, High-Calorie Meals Could Be an Effective OptRedox Strategy
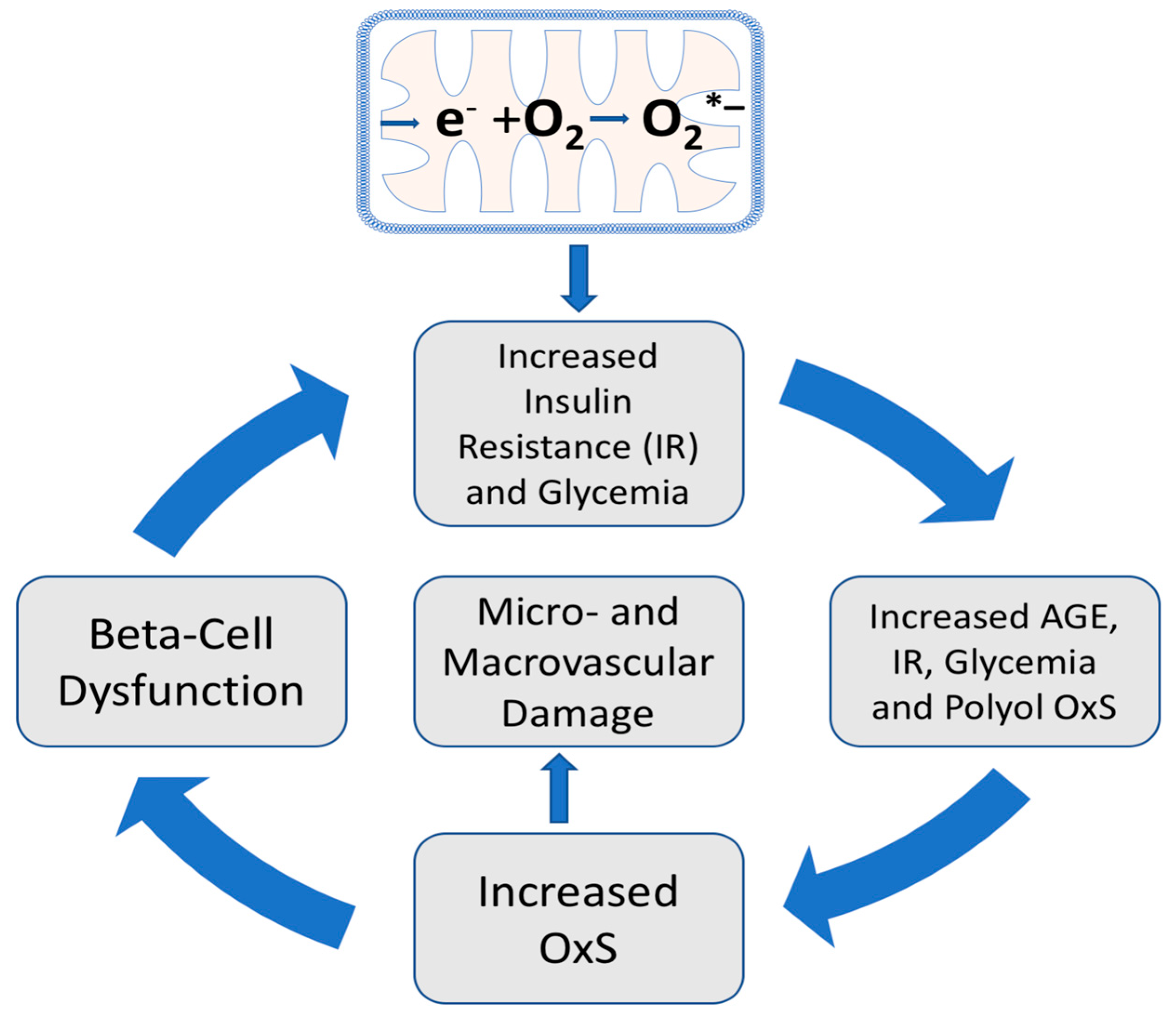
6. Hyperglycemia, Oxidative Stress, and T2D Progression
7. Hyperglycemia, AGE Formation, the Polyol Pathway, and Oxidative Stress
8. The OptRedox Strategy for Preventing or Slowing the Progression of T2D
8.1. Glycemic Control as a Natural OptRedox Process
8.2. Physical Activity/Exercise as an OptRedox Lifestyle Factor
8.3. Light-Intensity Walking as an OptRedox Lifestyle Factor That Reduces Postprandial Glycemia (PPG)
9. Exercise, Reactive Oxygen Species (ROS), and OptRedox Status
9.1. Exercise-Induced ROS Production Has a Biphasic Impact on Skeletal Muscle Force Production
9.2. Exercise-Induced Oxidative Stress and Induction of Enzymatic Antioxidants
10. Sleep and Psychological Stress as Lifestyle Factors Affecting T2D and Oxidative Stress
11. The Distinct Forms of Vitamin E and Their Effects on T2D Progression
11.1. Natural Vitamin E and the Importance of Stereochemistry
11.2. Supplementation with “Vitamin E” May Be a Valuable Strategy for Controlling Diabetes Complications
11.3. All-Racemic-Alpha-Tocopherol (All-Rac-Alpha-TOH) and Rice Bran Tocopherol Concentrate Inhibit Skeletal Muscle Generation of Hydrogen Peroxide
11.4. Vitamin E and/or Ascorbate Supplementation Improves Glycemic Control in T2D
11.5. The Tocotrienol-Rich Fraction (TRF) from Palm Oil May Be Beneficial in Both Prediabetes and T2D and in Preventing Early Diabetic Retinopathy
11.6. Delta-Tocotrienol Shows Promise in Treating Prediabetes
12. Vitamin C (L-Ascorbate or AA) and Its Role in T2D Progression and Management
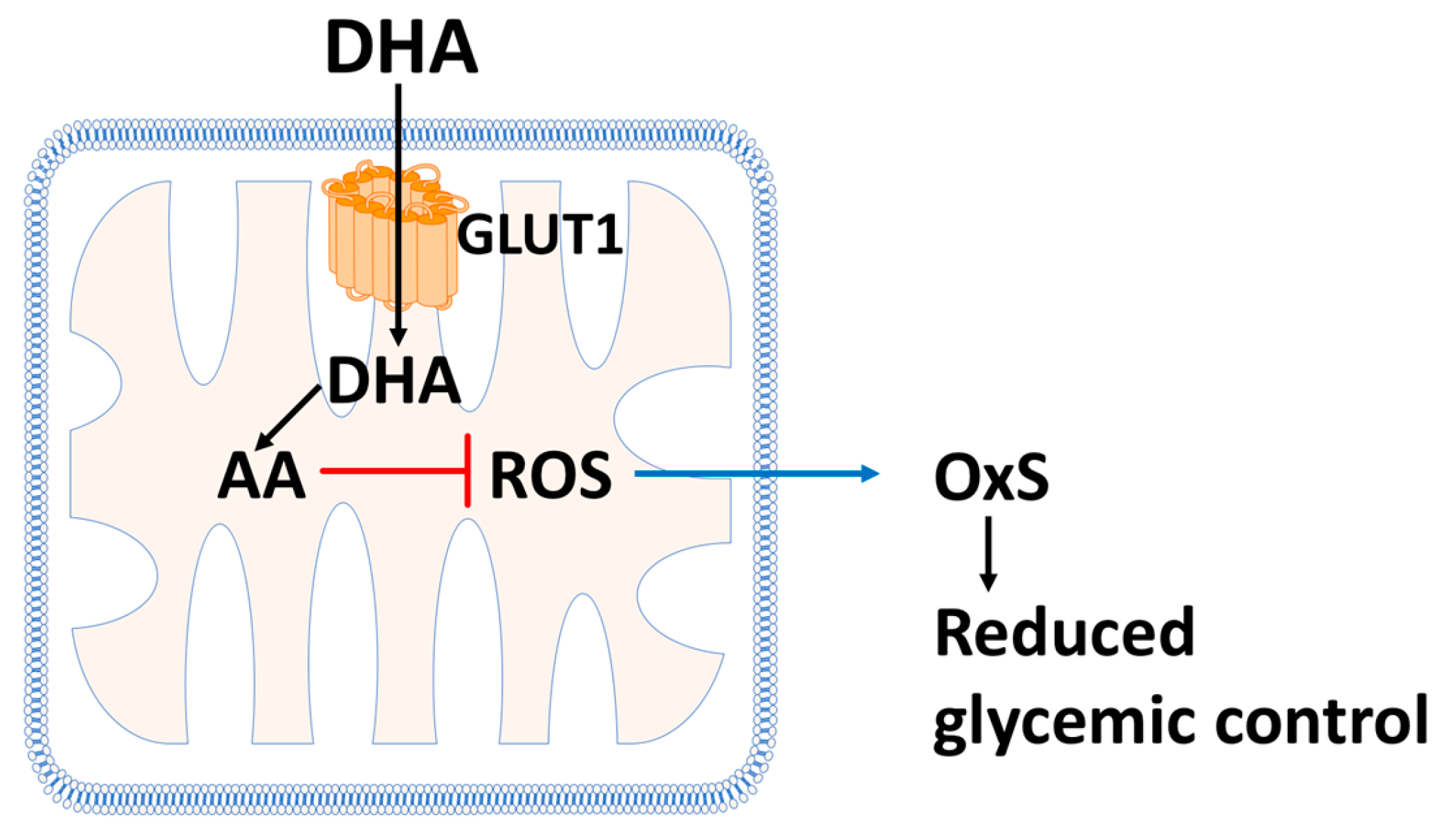
12.1. Vitamin C Protects Mitochondria from Oxidative Stress
12.2. Vitamin C and Oxidative Stress in T2D
12.3. Vitamin C Supplementation Plays a Positive Role in Adult T2D Management and Progression
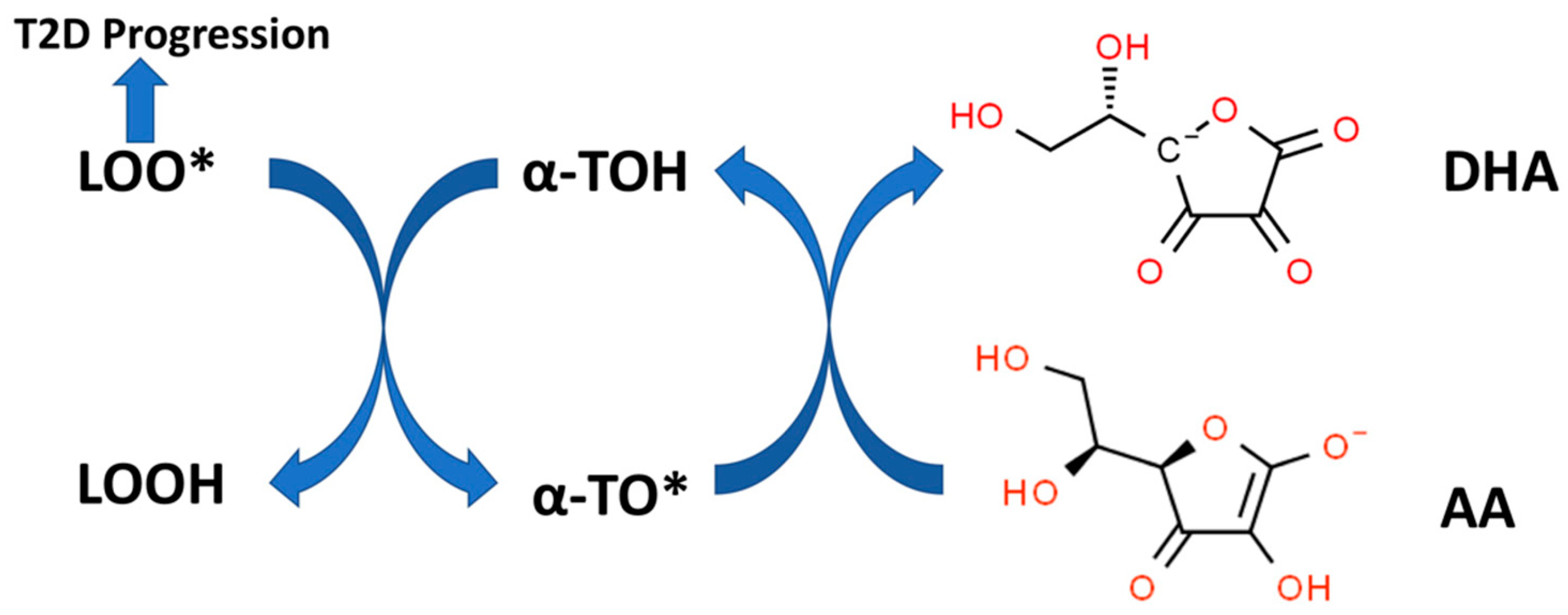
12.4. Do Synergistic Interactions between Vitamin C and Vitamin E Play a Role in Preventing T2D Pathophysiology?
13. Dietary and Supplemental Manganese and T2D
14. Selenium Supplementation and T2D
15. The Role of Beta-Carotene in T2D Prevention
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 12 July 2022).
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus—Present and future perspectives. Nat. Rev. Endocrinol. 2011, 8, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Zhang, D.; Yi, S.S. Trends in Prediabetes Among Youths in the US From 1999 Through 2018. JAMA Pediatr. 2022, 176, 608–611. [Google Scholar] [CrossRef] [PubMed]
- How Type 2 Diabetes Affects Your Workforce. Available online: https://www.cdc.gov/diabetes/prevention/how-type2-affects-workforce.htm#:~:text=Diabetes%20Is%20Costly,over%20a%205%2Dyear%20period (accessed on 13 July 2022).
- Davidson, K.W.; Barry, M.J.; Mangione, C.M.; Cabana, M.; Caughey, A.B.; Davis, E.M.; Donahue, K.E.; Doubeni, C.A.; Krist, A.H.; Kubik, M.; et al. Screening for Prediabetes and Type 2 Diabetes: US Preventive Services Task Force Recommendation Statement. JAMA 2021, 326, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Prevalence of Both Diagnosed and Undiagnosed Diabetes. Available online: https://www.cdc.gov/diabetes/data/statistics-report/diagnosed-undiagnosed-diabetes.html (accessed on 3 May 2023).
- Reinehr, T. Type 2 diabetes mellitus in children and adolescents. World J. Diabetes 2013, 4, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Pansier, B.; Schulz, P.J. School-based diabetes interventions and their outcomes: A systematic literature review. J. Public Health Res. 2015, 4, 467. [Google Scholar] [CrossRef]
- Alu, S.N.; Los, E.A.; Ford, G.A.; Stone, W.L. Oxidative Stress in Type 2 Diabetes: The Case for Future Pediatric Redoxomics Studies. Antioxidants 2022, 11, 1336. [Google Scholar] [CrossRef]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus statement by the american association of clinical endocrinologists and american college of endocrinology on the comprehensive type 2 diabetes management algorithm-2018 executive summary. Endocr. Pract. 2018, 24, 91–120. [Google Scholar] [CrossRef]
- Mechanick, J.I.; Garber, A.J.; Grunberger, G.; Handelsman, Y.; Garvey, W.T. Dysglycemia-based chronic disease: An american association of clinical endocrinologists position statement. Endocr Pract. 2018, 24, 995–1011. [Google Scholar] [CrossRef]
- Hansen, T. Type 2 diabetes mellitus—A multifactorial disease. Ann. Univ. Mariae Curie Sklodowska Med. 2002, 57, 544–549. [Google Scholar]
- Wright, E.; Scism-Bacon, J.L.; Glass, L.C. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Int J. Clin. Pract. 2006, 60, 308–314. [Google Scholar] [CrossRef]
- Chikezie, P.C.; Ojiako, O.A.; Ogbuji, A.C. Oxidative Stress in Diabetes Mellitus. Int. J. Biol. Chem. 2015, 9, 92–109. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef]
- Mueckler, M. Insulin resistance and the disruption of Glut4 trafficking in skeletal muscle. J. Clin. Investig. 2001, 107, 1211–1213. [Google Scholar] [CrossRef]
- Maier, V.H.; Gould, G.W. Long-term insulin treatment of 3T3-L1 adipocytes results in mis-targeting of GLUT4: Implications for insulin-stimulated glucose transport. Diabetologia 2000, 43, 1273–1281. [Google Scholar] [CrossRef]
- Kampmann, U.; Christensen, B.; Nielsen, T.S.; Pedersen, S.B.; Ørskov, L.; Lund, S.; Møller, N.; Jessen, N. GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS ONE 2011, 6, e27854. [Google Scholar] [CrossRef]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Fazakerley, D.J.; Minard, A.Y.; Krycer, J.R.; Thomas, K.C.; Stöckli, J.; Harney, D.J.; Burchfield, J.G.; Maghzal, G.J.; Caldwell, S.T.; Hartley, R.C.; et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 2018, 293, 7315–7328. [Google Scholar] [CrossRef]
- Lean, M.E.; Leslie, W.S.; Barnes, A.C.; Brosnahan, N.; Thom, G.; McCombie, L.; Peters, C.; Zhyzhneuskaya, S.; Al-Mrabeh, A.; Hollingsworth, K.G.; et al. Primary care-led weight management for remission of type 2 diabetes (DiRECT): An open-label, cluster-randomised trial. Lancet 2018, 391, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Juray, S.; Axen, K.V.; Trasino, S.E. Remission of Type 2 Diabetes with Very Low-Calorie Diets-A Narrative Review. Nutrients 2021, 13, 2086. [Google Scholar] [CrossRef]
- Maffettone, A.; Rinaldi, M.; Fontanella, A. Postprandial hyperglycemia: A new frontier in diabetes management? Ital. J. Med. 2018, 12, 108–115. [Google Scholar] [CrossRef]
- Sottero, B.; Gargiulo, S.; Russo, I.; Barale, C.; Poli, G.; Cavalot, F. Postprandial Dysmetabolism and Oxidative Stress in Type 2 Diabetes: Pathogenetic Mechanisms and Therapeutic Strategies. Med. Res. Rev. 2015, 35, 968–1031. [Google Scholar] [CrossRef] [PubMed]
- Cavalot, F.; Petrelli, A.; Traversa, M.; Bonomo, K.; Fiora, E.; Conti, M.; Anfossi, G.; Costa, G.; Trovati, M. Postprandial blood glucose is a stronger predictor of cardiovascular events than fasting blood glucose in type 2 diabetes mellitus, particularly in women: Lessons from the San Luigi Gonzaga Diabetes Study. J. Clin. Endocrinol. Metab. 2006, 91, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Cavalot, F.; Pagliarino, A.; Valle, M.; Di Martino, L.; Bonomo, K.; Massucco, P.; Anfossi, G.; Trovati, M. Postprandial blood glucose predicts cardiovascular events and all-cause mortality in type 2 diabetes in a 14-year follow-up: Lessons from the San Luigi Gonzaga Diabetes Study. Diabetes Care 2011, 34, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diab. Rep. 2014, 14, 453. [Google Scholar] [CrossRef] [PubMed]
- Greifenhagen, U.; Frolov, A.; Blüher, M.; Hoffmann, R. Plasma Proteins Modified by Advanced Glycation End Products (AGEs) Reveal Site-specific Susceptibilities to Glycemic Control in Patients with Type 2 Diabetes. J. Biol. Chem. 2016, 291, 9610–9616. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Beppu, M.; Kikugawa, K.; Nagai, R.; Horiuchi, S. Membrane proteins of human erythrocytes are modified by advanced glycation end products during aging in the circulation. Biochem. Biophys. Res. Commun. 1999, 258, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim. Model Exp. Med. 2018, 1, 7–13. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflug. Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef]
- Schleicher, E.; Friess, U. Oxidative stress, AGE, and atherosclerosis. Kidney Int. Suppl. 2007, 72, S17–S26. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Radak, Z.; Ji, L.L. Exercise-induced oxidative stress: Past, present and future. J. Physiol. 2016, 594, 5081–5092. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Woerle, H.J.; Neumann, C.; Zschau, S.; Tenner, S.; Irsigler, A.; Schirra, J.; Gerich, J.E.; Göke, B. Impact of fasting and postprandial glycemia on overall glycemic control in type 2 diabetes Importance of postprandial glycemia to achieve target HbA1c levels. Diabetes Res. Clin. Pract. 2007, 77, 280–285. [Google Scholar] [CrossRef]
- Gunawardena, H.P.; Silva, R.; Sivakanesan, R.; Ranasinghe, P.; Katulanda, P. Poor Glycaemic Control Is Associated with Increased Lipid Peroxidation and Glutathione Peroxidase Activity in Type 2 Diabetes Patients. Oxid. Med. Cell Longev. 2019, 2019, 9471697. [Google Scholar] [CrossRef]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical Activity/Exercise and Diabetes: A Position Statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef]
- van Dijk, J.W.; van Loon, L.J. Exercise strategies to optimize glycemic control in type 2 diabetes: A continuing glucose monitoring perspective. Diabetes Spectr. 2015, 28, 24–31. [Google Scholar] [CrossRef]
- Velicer, C. Kids and Type 2 Diabetes: How Parents and Teachers Can Help Curb the Tide. Available online: https://thrivingschools.kaiserpermanente.org/kids-and-type-2-diabetes-how-parents-and-teachers-can-help-curb-the-tide/ (accessed on 13 September 2022).
- Buffey, A.J.; Herring, M.P.; Langley, C.K.; Donnelly, A.E.; Carson, B.P. The Acute Effects of Interrupting Prolonged Sitting Time in Adults with Standing and Light-Intensity Walking on Biomarkers of Cardiometabolic Health in Adults: A Systematic Review and Meta-analysis. Sport. Med. 2022, 52, 1765–1787. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Bardoloi, A.; Parkash, A.A. Metabolic effects of obesity: A review. World J. Diabetes 2010, 1, 76–88. [Google Scholar] [CrossRef]
- Chang, T.; Li, H.; Zhang, N.; Jiang, X.; Yu, X.; Yang, Q.; Jin, Z.; Meng, H.; Chang, L. Highly integrated watch for noninvasive continual glucose monitoring. Microsyst. Nanoeng. 2022, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-León, C.; Villalonga, C.; Munoz-Torres, M.; Ruiz, J.R.; Banos, O. Mobile and Wearable Technology for the Monitoring of Diabetes-Related Parameters: Systematic Review. JMIR Mhealth Uhealth 2021, 9, e25138. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, J.W.; Venema, M.; van Mechelen, W.; Stehouwer, C.D.; Hartgens, F.; van Loon, L.J. Effect of moderate-intensity exercise versus activities of daily living on 24-hour blood glucose homeostasis in male patients with type 2 diabetes. Diabetes Care 2013, 36, 3448–3453. [Google Scholar] [CrossRef] [PubMed]
- Flores-Opazo, M.; McGee, S.L.; Hargreaves, M. Exercise and GLUT4. Exerc. Sport Sci. Rev. 2020, 48, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- Kraniou, G.N.; Cameron-Smith, D.; Hargreaves, M. Acute exercise and GLUT4 expression in human skeletal muscle: Influence of exercise intensity. J. Appl. Physiol. 2006, 101, 934–937. [Google Scholar] [CrossRef]
- Kawamura, T.; Muraoka, I. Exercise-Induced Oxidative Stress and the Effects of Antioxidant Intake from a Physiological Viewpoint. Antioxidants 2018, 7, 119. [Google Scholar] [CrossRef]
- Quindry, J.; Stone, W.; King, J.; Broeder, C. The effects of acute exercise on neutrophils and plasma oxidative stress. Med. Sci. Sport. Exerc. 2003, 35, 1139–1145. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef]
- Hancock, J.T.; Desikan, R.; Neill, S.J. Role of reactive oxygen species in cell signalling pathways. Biochem. Soc. Trans. 2001, 29, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; McHardy, H.; Morton, J.P.; Nikolaidis, M.G.; Close, G.L. Influence of vitamin C and vitamin E on redox signaling: Implications for exercise adaptations. Free Radic. Biol. Med. 2015, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B. Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Med. Sci. Sport. Exerc. 2001, 33, 371–376. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Li, J.; Liu, Z.; Chuang, C.C.; Yang, W.; Zuo, L. Redox Mechanism of Reactive Oxygen Species in Exercise. Front. Physiol. 2016, 7, 486. [Google Scholar] [CrossRef]
- Ji, L.L. Antioxidant enzyme response to exercise and aging. Med. Sci. Sport. Exerc. 1993, 25, 225–231. [Google Scholar] [CrossRef]
- Lipska, K.J. Patient Education: Exercise and Medical Care for People with Type 2 Diabetes (Beyond the Basics). Available online: www.uptodate.com/contents/exercise-and-medical-care-for-people-with-type-2-diabetes-beyond-the-basics (accessed on 3 May 2023).
- Khandelwal, D.; Dutta, D.; Chittawar, S.; Kalra, S. Sleep Disorders in Type 2 Diabetes. Indian J. Endocrinol. Metab. 2017, 21, 758–761. [Google Scholar] [CrossRef]
- Kelly, S.J.; Ismail, M. Stress and type 2 diabetes: A review of how stress contributes to the development of type 2 diabetes. Annu. Rev. Public Health 2015, 36, 441–462. [Google Scholar] [CrossRef]
- Atrooz, F.; Salim, S. Sleep deprivation, oxidative stress and inflammation. Adv. Protein Chem. Struct. Biol. 2020, 119, 309–336. [Google Scholar] [CrossRef]
- Hill, V.M.; O’Connor, R.M.; Sissoko, G.B.; Irobunda, I.S.; Leong, S.; Canman, J.C.; Stavropoulos, N.; Shirasu-Hiza, M. A bidirectional relationship between sleep and oxidative stress in Drosophila. PLoS Biol. 2018, 16, e2005206. [Google Scholar] [CrossRef]
- Moller, P.; Wallin, H.; Knudsen, L.E. Oxidative Stress Associated with Exercise, Psychological Stress and Life-Style Factors. Chem. Biol. Interact. 1996, 102, 17–36. Available online: http://biomednet.com/db/medline/96424598 (accessed on 3 May 2023). [CrossRef]
- Belaïdi, E. Oxidative Stress and Sleep Disorders. Available online: https://www.mdpi.com/journal/antioxidants/special_issues/Oxidative_Stress_and_Sleep_Disorders (accessed on 26 April 2023).
- Lauridsen, C.; Jensen, S.K. α-Tocopherol incorporation in mitochondria and microsomes upon supranutritional vitamin E supplementation. Genes Nutr. 2012, 7, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Roy, S. Tocotrienols in health and disease: The other half of the natural vitamin E family. Mol. Asp. Med. 2007, 28, 692–728. [Google Scholar] [CrossRef]
- Azzi, A.; Gysin, R.; Kempna, P.; Munteanu, A.; Negis, Y.; Villacorta, L.; Visarius, T.; Zingg, J.M. Vitamin E mediates cell signaling and regulation of gene expression. Ann. N. Y. Acad. Sci. 2004, 1031, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M. Vitamin E: A Role in Signal Transduction. Annu. Rev. Nutr. 2015, 35, 135–173. [Google Scholar] [CrossRef]
- Balbi, M.E.; Tonin, F.S.; Mendes, A.M.; Borba, H.H.; Wiens, A.; Fernandez-Llimos, F.; Pontarolo, R. Antioxidant effects of vitamins in type 2 diabetes: A meta-analysis of randomized controlled trials. Diabetol. Metab. Syndr. 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.K.; Ibrahim, W.; Wei, Z.; Chan, A.C. Vitamin E regulates mitochondrial hydrogen peroxide generation. Free Radic. Biol. Med. 1999, 27, 580–587. [Google Scholar] [CrossRef]
- El-Aal, A.A.; El-Ghffar, E.A.A.; Ghali, A.A.; Zughbur, M.R.; Sirdah, M.M. The effect of vitamin C and/or E supplementations on type 2 diabetic adult males under metformin treatment: A single-blinded randomized controlled clinical trial. Diabetes Metab. Syndr. 2018, 12, 483–489. [Google Scholar] [CrossRef]
- Vafa, M.; Haghighat, N.; Moslehi, N.; Eghtesadi, S.; Heydari, I. Effect of Tocotrienols enriched canola oil on glycemic control and oxidative status in patients with type 2 diabetes mellitus: A randomized double-blind placebo-controlled clinical trial. J. Res. Med. Sci. 2015, 20, 540–547. [Google Scholar] [CrossRef]
- Baliarsingh, S.; Beg, Z.H.; Ahmad, J. The therapeutic impacts of tocotrienols in type 2 diabetic patients with hyperlipidemia. Atherosclerosis 2005, 182, 367–374. [Google Scholar] [CrossRef]
- Ho, J.I.; Ng, E.Y.; Chiew, Y.; Koay, Y.Y.; Chuar, P.F.; Phang, S.C.W.; Ahmad, B.; Kadir, K.A. The effects of vitamin E on non-proliferative diabetic retinopathy in type 2 diabetes mellitus: Are they sustainable with 12 months of therapy. SAGE Open Med. 2022, 10, 20503121221095324. [Google Scholar] [CrossRef]
- Kalvaitus, K.; Portnoy, S.A. Experts Recommend Two-Pronged Approach to Treating Prediabetes. Available online: https://www.healio.com/news/endocrinology/20120325/experts-recommend-two-pronged-approach-to-treating-prediabetes (accessed on 11 January 2023).
- Suleman, F.; Khan, D.A.; Pervez, M.A.; Aamir, M. Effects of delta-tocotrienol supplementation on glycaemic control in individuals with prediabetes: A randomized controlled study. J. Pak. Med. Assoc. 2022, 72, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Kang, Z.; Wong, C. Vitamin E tocotrienols improve insulin sensitivity through activating peroxisome proliferator-activated receptors. Mol. Nutr. Food Res. 2010, 54, 345–352. [Google Scholar] [CrossRef]
- Mason, S.A.; Parker, L.; van der Pligt, P.; Wadley, G.D. Vitamin C supplementation for diabetes management: A comprehensive narrative review. Free Radic. Biol. Med. 2023, 194, 255–283. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Karp, J.; Sun, K.M.; Weaver, C.M. Decreasing Vitamin C Intake, Low Serum Vitamin C Level and Risk for US Adults with Diabetes. Nutrients 2022, 14, 3902. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Park, S. A Causal Relationship between Vitamin C Intake with Hyperglycemia and Metabolic Syndrome Risk: A Two-Sample Mendelian Randomization Study. Antioxidants 2022, 11, 857. [Google Scholar] [CrossRef]
- Ebenuwa, I.; Violet, P.C.; Padayatty, S.; Wang, Y.; Sun, H.; Adhikari, P.; Smith, S.; Tu, H.; Niyyati, M.; Wilkins, K.; et al. Abnormal urinary loss of vitamin C in diabetes: Prevalence and clinical characteristics of a vitamin C renal leak. Am. J. Clin. Nutr. 2022, 116, 274–284. [Google Scholar] [CrossRef]
- Shi, L.; Du, X.; Guo, P.; Huang, L.; Qi, P.; Gong, Q. Ascorbic acid supplementation in type 2 diabetes mellitus: A protocol for systematic review and meta-analysis. Medicine 2020, 99, e23125. [Google Scholar] [CrossRef]
- Mason, S.A.; Keske, M.A.; Wadley, G.D. Effects of Vitamin C Supplementation on Glycemic Control and Cardiovascular Risk Factors in People with Type 2 Diabetes: A GRADE-Assessed Systematic Review and Meta-analysis of Randomized Controlled Trials. Diabetes Care 2021, 44, 618–630. [Google Scholar] [CrossRef]
- Jiang, C.L.; Tsao, C.Y.; Lee, Y.C. Vitamin C attenuates predisposition to high-fat diet-induced metabolic dysregulation in GLUT10-deficient mouse model. Genes Nutr. 2022, 17, 10. [Google Scholar] [CrossRef]
- Wilson, R.; Willis, J.; Gearry, R.; Skidmore, P.; Fleming, E.; Frampton, C.; Carr, A. Inadequate Vitamin C Status in Prediabetes and Type 2 Diabetes Mellitus: Associations with Glycaemic Control, Obesity, and Smoking. Nutrients 2017, 9, 997. [Google Scholar] [CrossRef]
- Stanimirovic, J.; Radovanovic, J.; Banjac, K.; Obradovic, M.; Essack, M.; Zafirovic, S.; Gluvic, Z.; Gojobori, T.; Isenovic, E.R. Role of C-Reactive Protein in Diabetic Inflammation. Mediat. Inflamm 2022, 2022, 3706508. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Sagun, K.C.; Cárcamo, J.M.; Golde, D.W. Vitamin C enters mitochondria via facilitative glucose transporter 1 (Glut1) and confers mitochondrial protection against oxidative injury. FASEB J. 2005, 19, 1657–1667. [Google Scholar] [CrossRef]
- Bansal, A.; Hadimani, C.P. Low Plasma Ascorbate Levels in Type 2 Diabetic Patients With Adequate Dietary Vitamin C. J. Lab. Physicians 2021, 13, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J.; Taylor, P.B.; Lunec, J.; Girling, A.J.; Barnett, A.H. Low plasma ascorbate levels in patients with type 2 diabetes mellitus consuming adequate dietary vitamin C. Diabet. Med. 1994, 11, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Karne, R.J.; Hall, G.; Campia, U.; Panza, J.A.; Cannon, R.O.; Wang, Y.; Katz, A.; Levine, M.; Quon, M.J. High-dose oral vitamin C partially replenishes vitamin C levels in patients with Type 2 diabetes and low vitamin C levels but does not improve endothelial dysfunction or insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H137–H145. [Google Scholar] [CrossRef]
- Bonner, R.; Albajrami, O.; Hudspeth, J.; Upadhyay, A. Diabetic Kidney Disease. Prim. Care 2020, 47, 645–659. [Google Scholar] [CrossRef]
- Afkhami-Ardekani, M.; Shojaoddiny-Ardekani, A. Effect of vitamin C on blood glucose, serum lipids & serum insulin in type 2 diabetes patients. Indian J. Med. Res. 2007, 126, 471–474. [Google Scholar]
- Donin, A.S.; Dent, J.E.; Nightingale, C.M.; Sattar, N.; Owen, C.G.; Rudnicka, A.R.; Perkin, M.R.; Stephen, A.M.; Jebb, S.A.; Cook, D.G.; et al. Fruit, vegetable and vitamin C intakes and plasma vitamin C: Cross-sectional associations with insulin resistance and glycaemia in 9–10 year-old children. Diabet. Med. 2016, 33, 307–315. [Google Scholar] [CrossRef]
- Parajuli, S.; Jasmin, G.; Sirak, H.; Lee, A.F.; Nwosu, B.U. Prediabetes: Adherence to Nutrition Visits Decreases HbA1c in Children and Adolescents. Front. Endocrinol. 2022, 13, 916785. [Google Scholar] [CrossRef]
- Strain, J.J.; Mulholland, C.W. Vitamin C and vitamin E—Synergistic interactions in vivo? EXS 1992, 62, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A. Tocopherols, tocotrienols and tocomonoenols: Many similar molecules but only one vitamin E. Redox Biol. 2019, 26, 101259. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Natural Antioxidants: A Novel Therapeutic Approach to Autism Spectrum Disorders? Antioxidants 2020, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- de Souza Bastos, A.; Graves, D.T.; de Melo Loureiro, A.P.; Júnior, C.R.; Corbi, S.C.T.; Frizzera, F.; Scarel-Caminaga, R.M.; Câmara, N.O.; Andriankaja, O.M.; Hiyane, M.I.; et al. Diabetes and increased lipid peroxidation are associated with systemic inflammation even in well-controlled patients. J. Diabetes Complicat. 2016, 30, 1593–1599. [Google Scholar] [CrossRef]
- Augustine, J.; Troendle, E.P.; Barabas, P.; McAleese, C.A.; Friedel, T.; Stitt, A.W.; Curtis, T.M. The Role of Lipoxidation in the Pathogenesis of Diabetic Retinopathy. Front. Endocrinol. 2020, 11, 621938. [Google Scholar] [CrossRef]
- Sato, A.; Takino, Y.; Yano, T.; Fukui, K.; Ishigami, A. Determination of tissue-specific interaction between vitamin C and vitamin E. Br. J. Nutr. 2022, 128, 993–1003. [Google Scholar] [CrossRef]
- Porkkala-Sarataho, E.; Salonen, J.T.; Nyyssönen, K.; Kaikkonen, J.; Salonen, R.; Ristonmaa, U.; Diczfalusy, U.; Brigelius-Flohe, R.; Loft, S.; Poulsen, H.E. Long-term effects of vitamin E, vitamin C, and combined supplementation on urinary 7-hydro-8-oxo-2′-deoxyguanosine, serum cholesterol oxidation products, and oxidation resistance of lipids in nondepleted men. Arter. Thromb. Vasc. Biol. 2000, 20, 2087–2093. [Google Scholar] [CrossRef]
- Huang, H.Y.; Appel, L.J.; Croft, K.D.; Miller, E.R.; Mori, T.A.; Puddey, I.B. Effects of vitamin C and vitamin E on in vivo lipid peroxidation: Results of a randomized controlled trial. Am. J. Clin. Nutr. 2002, 76, 549–555. [Google Scholar] [CrossRef]
- Mazloom, Z.; Hejazi, N.; Dabbaghmanesh, M.H.; Tabatabaei, H.R.; Ahmadi, A.; Ansar, H. Effect of vitamin C supplementation on postprandial oxidative stress and lipid profile in type 2 diabetic patients. Pak. J. Biol. Sci. 2011, 14, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Calvagno, S.; Mauceri, B.; Misseri, M.; Tsami, A.; Vecchio, C.; Mastrosimone, G.; Di Pino, A.; Maiorca, D.; Judica, A.; et al. Effects of antioxidants on postprandial oxidative stress and endothelial dysfunction in subjects with impaired glucose tolerance and type 2 diabetes. Eur. J. Nutr. 2010, 49, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St. Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Jouihan, H.A.; Cooksey, R.C.; Jones, D.; Kim, H.J.; Winge, D.R.; McClain, D.A. Manganese supplementation protects against diet-induced diabetes in wild type mice by enhancing insulin secretion. Endocrinology 2013, 154, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Burlet, E.; Jain, S.K. Manganese supplementation reduces high glucose-induced monocyte adhesion to endothelial cells and endothelial dysfunction in Zucker diabetic fatty rats. J. Biol. Chem. 2013, 288, 6409–6416. [Google Scholar] [CrossRef]
- Du, S.; Wu, X.; Han, T.; Duan, W.; Liu, L.; Qi, J.; Niu, Y.; Na, L.; Sun, C. Dietary manganese and type 2 diabetes mellitus: Two prospective cohort studies in China. Diabetologia 2018, 61, 1985–1995. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, M.; Lui, G.; Chang, H.; Liu, W.; Li, Z.; Liu, Y.; Huang, G. Associations of Serum Manganese Levels with Prediabetes and Diabetes among ≥60-Year-Old Chinese Adults: A Population-Based Cross-Sectional Analysis. Nutrients 2016, 8, 497. [Google Scholar] [CrossRef]
- Chen, H.; Cui, Z.; Lu, W.; Wang, P.; Wang, J.; Zhou, Z.; Zhang, N.; Wang, Z.; Lin, T.; Song, Y.; et al. Association between serum manganese levels and diabetes in Chinese adults with hypertension. J. Clin. Hypertens 2022, 24, 918–927. [Google Scholar] [CrossRef]
- van der Schaft, N.; Schoufour, J.D.; Nano, J.; Kiefte-de Jong, J.C.; Muka, T.; Sijbrands, E.J.G.; Ikram, M.A.; Franco, O.H.; Voortman, T. Dietary antioxidant capacity and risk of type 2 diabetes mellitus, prediabetes and insulin resistance: The Rotterdam Study. Eur. J. Epidemiol. 2019, 34, 853–861. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Katz, M.L.; Stone, W.L.; Dratz, E.A. Fluorescent pigment accumulation in retinal pigment epithelium of antioxidant-deficient rats. Investig. Ophthalmol. Vis. Sci. 1978, 17, 1049–1058. [Google Scholar]
- Ogawa-Wong, A.N.; Berry, M.J.; Seale, L.A. Selenium and Metabolic Disorders: An Emphasis on Type 2 Diabetes Risk. Nutrients 2016, 8, 80. [Google Scholar] [CrossRef]
- Marcelino, G.; Machate, D.J.; Freitas, K.C.; Hiane, P.A.; Maldonade, I.R.; Pott, A.; Asato, M.A.; Candido, C.J.; Guimarães, R.C.A. β-Carotene: Preventive Role for Type 2 Diabetes Mellitus and Obesity: A Review. Molecules 2020, 25, 5803. [Google Scholar] [CrossRef]
- Jiang, Y.W.; Sun, Z.H.; Tong, W.W.; Yang, K.; Guo, K.Q.; Liu, G.; Pan, A. Dietary Intake and Circulating Concentrations of Carotenoids and Risk of Type 2 Diabetes: A Dose-Response Meta-Analysis of Prospective Observational Studies. Adv. Nutr. 2021, 12, 1723–1733. [Google Scholar] [CrossRef]
- Bright Futures Guidelines and Pocket Guide. Available online: https://www.aap.org/en/practice-management/bright-futures/bright-futures-materials-and-tools/bright-futures-guidelines-and-pocket-guide/ (accessed on 11 January 2023).
- Eddolls, W.T.B.; McNarry, M.A.; Stratton, G.; Winn, C.O.N.; Mackintosh, K.A. High-Intensity Interval Training Interventions in Children and Adolescents: A Systematic Review. Sport. Med. 2017, 47, 2363–2374. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuell, D.S.; Los, E.A.; Ford, G.A.; Stone, W.L. The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes. Antioxidants 2023, 12, 1139. https://doi.org/10.3390/antiox12061139
Tuell DS, Los EA, Ford GA, Stone WL. The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes. Antioxidants. 2023; 12(6):1139. https://doi.org/10.3390/antiox12061139
Chicago/Turabian StyleTuell, Dawn S., Evan A. Los, George A. Ford, and William L. Stone. 2023. "The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes" Antioxidants 12, no. 6: 1139. https://doi.org/10.3390/antiox12061139
APA StyleTuell, D. S., Los, E. A., Ford, G. A., & Stone, W. L. (2023). The Role of Natural Antioxidant Products That Optimize Redox Status in the Prevention and Management of Type 2 Diabetes. Antioxidants, 12(6), 1139. https://doi.org/10.3390/antiox12061139