
Targeting Oxidative Stress with Polyphenols to Fight Liver Diseases

 , , ,
, , , 
Abstract
:1. Introduction
2. Cellular Mechanisms of Redox Signaling
3. Intracellular Sources of ROS
3.1. Mitochondria
3.2. Cytochrome P450
4. Targeting Oxidative Stress with Natural Compounds in Liver Pathophysiology
4.1. Oxidative Stress in Liver Pathophysiology
4.1.1. Liver Ischemia/Reperfusion Injury
4.1.2. Non-Alcoholic Fatty Liver Disease
4.1.3. Hepatocellular Carcinoma
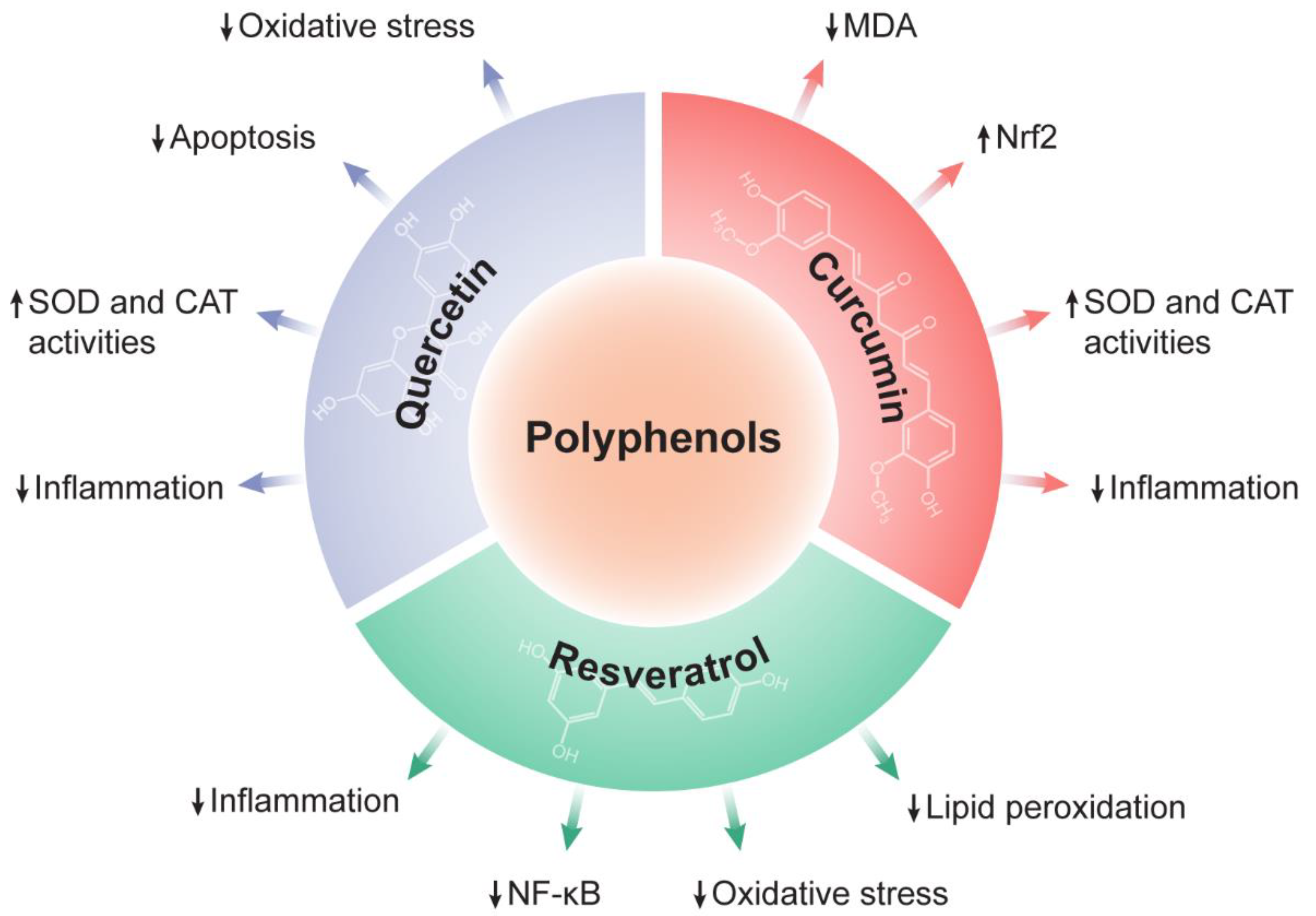
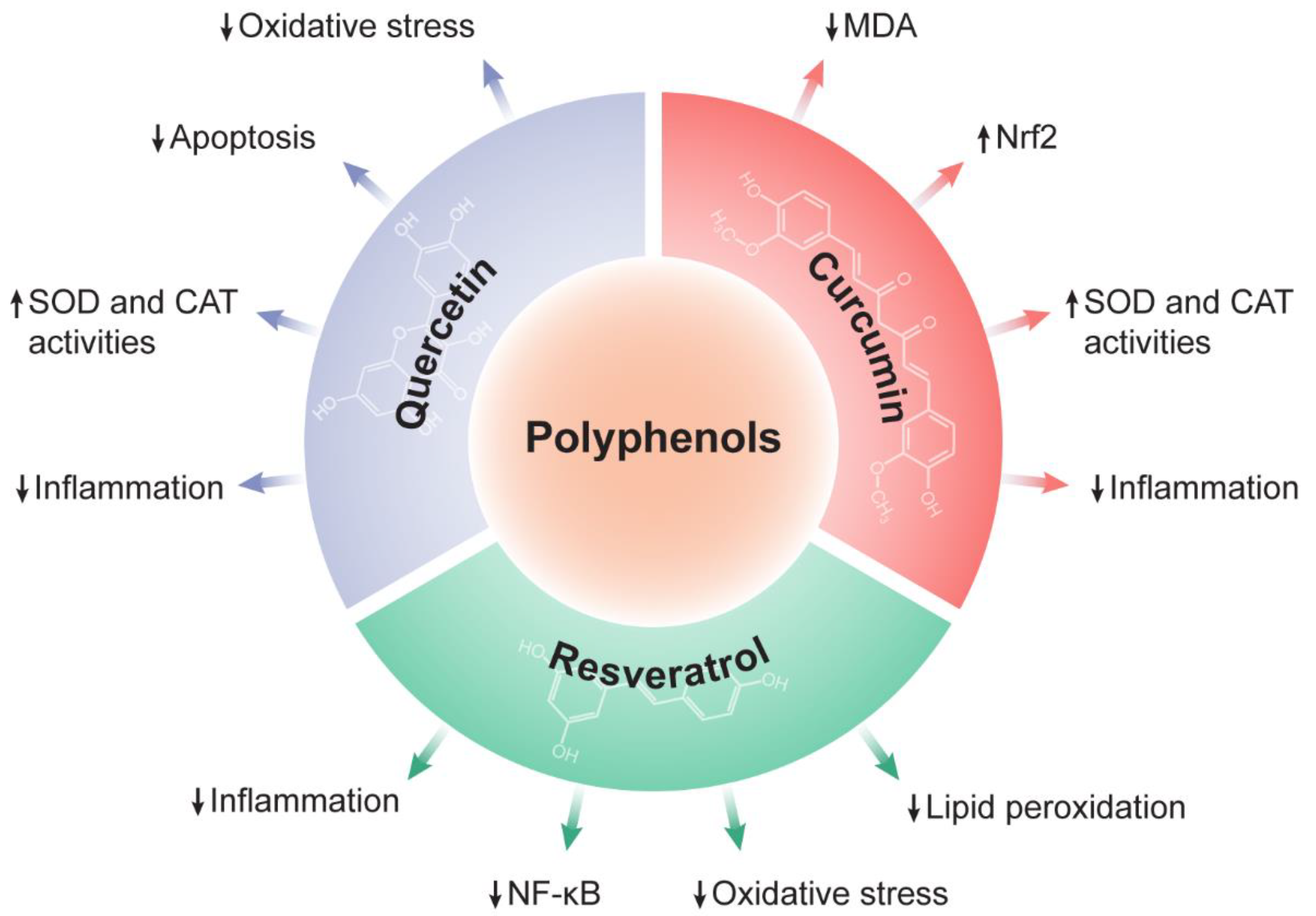
4.2. The Therapeutic Potential of Polyphenols against Liver Diseases
4.2.1. Quercetin
4.2.2. Resveratrol
4.2.3. Curcumin
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Hrycay, E.G.; Bandiera, S.M. Involvement of Cytochrome P450 in Reactive Oxygen Species Formation and Cancer, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 74, ISBN 9780128031193. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for Measuring Reactive Oxygen Species and Oxidative Damage in Cells and in Vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and Metabolic Health: The Interplay between ROS, CAMP and Sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Salzano, S.; Checconi, P.; Hanschmann, E.M.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of Inflammation and Oxidative Stress via Release of Glutathionylated Peroxiredoxin-2, Which Acts as a Danger Signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond Oxidative Stress: An Immunologist’s Guide to Reactive Oxygen Species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative Stress Induces Autophagic Cell Death Independent of Apoptosis in Transformed and Cancer Cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Datta, K.; Babbar, P.; Srivastava, T.; Sinha, S.; Chattopadhyay, P. P53 Dependent Apoptosis in Glioma Cell Lines in Response to Hydrogen Peroxide Induced Oxidative Stress. Int. J. Biochem. Cell Biol. 2002, 34, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.L.; Holbrook, N.J. Cellular Response to Oxidative Stress: Signaling for Suicide and Survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Frye, R.E.; Slattery, J.; Wynne, R.; Tippett, M.; Melnyk, S.; James, S.J. Oxidative Stress Induces Mitochondrial Dysfunction in a Subset of Autistic Lymphoblastoid Cell Lines. Transl. Psychiatry 2014, 4, e377. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Disease. Nat. Rev. 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of Oxidative Stress as an Anticancer Strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular Death, Reactive Oxygen Species (ROS) and Diabetic Complications Review-Article. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, Oxidative Stress and Biology of Ageing. Insight Rev. Artic. 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative Stress as a Crucial Factor in Liver Diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Ramachandran, A.; Jaeschke, H. Oxidative Stress and Acute Hepatic Injury. Curr. Opin. Toxicol. 2018, 7, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Huerta-Salgado, C.; Orozco-Aguilar, J.; Aguirre, F.; Tacchi, F.; Simon, F.; Cabello-Verrugio, C. Role of Oxidative Stress in Hepatic and Extrahepatic Dysfunctions during Nonalcoholic Fatty Liver Disease (NAFLD). Oxid. Med. Cell. Longev. 2020, 2020, 1617805. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxid. Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of Liver Diseases in the World. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Zhang, H.; Tsao, R. Dietary Polyphenols, Oxidative Stress and Antioxidant and Anti-Inflammatory Effects. Curr. Opin. Food Sci. 2016, 8, 33–42. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, R.H.; Gill, V.; Rice-Evans, C. Cell Proliferation and Oxidative Stress. Free Radic. Res. Commun. 1989, 7, 149–159. [Google Scholar] [CrossRef]
- Oka, S.; Tsuzuki, T.; Hidaka, M.; Ohno, M.; Nakatsu, Y.; Sekiguchi, M. Endogenous ROS Production in Early Differentiation State Suppresses Endoderm Differentiation via Transient FOXC1 Expression. Cell Death Discov. 2022, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A ROS-Dependent Mechanism Promotes CDK2 Phosphorylation to Drive Progression through S Phase. Dev. Cell 2022, 57, 1712–1727. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR Signalling Augments Macrophage Bactericidal Activity through Mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Dóka, É.; Van Laer, K.; Nagy, P.; Gräter, F.; Dick, T.P. A Proton Relay Enhances H2O2 Sensitivity of GAPDH to Facilitate Metabolic Adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef]
- Nayeem, M.A. Role of Oxylipins in Cardiovascular Diseases Review-Article. Acta Pharmacol. Sin. 2018, 39, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M. Unraveling the Truth about Antioxidants. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef]
- Nathan, C. Specificity of a Third Kind: Reactive Oxygen and Nitrogen Intermediates in Cell Signaling. J. Clin. Investig. 2003, 111, 769–778. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as Signalling Molecules: Mechanisms That Generate Specificity in ROS Homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Turpaev, K.T. Reactive Oxygen Species and Regulation of Gene Expression. Biochemistry 2002, 67, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen Peroxide Sensing, Signaling and Regulation of Transcription Factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Meng, T.; Fukada, T.; Tonks, N.K. Reversible Oxidation and Inactivation of Protein Tyrosine Phosphatases in Vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.S.; Machado, L.E.S.F. Preferential Redox Regulation of Cysteine-Based Protein Tyrosine Phosphatases: Structural and Biochemical Diversity. FEBS J. 2022, 289, 5480–5504. [Google Scholar] [CrossRef]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.-C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox Regulation of Protein Tyrosine Phosphatase 1B Involves a Sulphenyl-Amide Intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein Tyrosine Phosphatases: From Genes, to Function, to Disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.C.; et al. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-Stimulated Hydrogen Peroxide Reversibly Inhibits Protein-Tyrosine Phosphatase 1B in Vivo and Enhances the Early Insulin Action Cascade. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef] [Green Version]
- Londhe, A.D.; Bergeron, A.; Curley, S.M.; Zhang, F.; Rivera, K.D.; Kannan, A.; Coulis, G.; Rizvi, S.H.M.; Kim, S.J.; Pappin, D.J.; et al. Regulation of PTP1B Activation through Disruption of Redox-Complex Formation. Nat. Chem. Biol. 2020, 16, 122–125. [Google Scholar] [CrossRef]
- Mo, L.; Yang, C.; Gu, M.; Zheng, D.; Lin, L.; Wang, X.; Lan, A.; Hu, F.; Feng, J. PI3K/Akt Signaling Pathway-Induced Heme Oxygenase-1 Upregulation Mediates the Adaptive Cytoprotection of Hydrogen Peroxide Preconditioning against Oxidative Injury in PC12 Cells. Int. J. Mol. Med. 2012, 30, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox Regulation of PI 3-Kinase Signalling via Inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible Oxidation and Inactivation of the Tumor Suppressor PTEN in Cells Stimulated with Peptide Growth Factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase to Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive Oxygen Intermediates as Apparently Widely Used Messengers in the Activation of the NF-ΚB Transcription Factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Takada, Y.; Mukhopadhyay, A.; Kundu, G.C.; Mahabeleshwar, G.H.; Singh, S.; Aggarwal, B.B. Hydrogen Peroxide Activates NF-ΚB through Tyrosine Phosphorylation of IκBα and Serine Phosphorylation of P65. J. Biol. Chem. 2003, 278, 24233–24241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, H.; Manabe, T.; Oka, S.; Kamata, K.; Hirata, H. Hydrogen Peroxide Activates IκB Kinases through Phosphorylation of Serine Residues in the Activation Loops. FEBS Lett. 2002, 519, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halvey, P.J.; Hansen, J.M.; Johnson, J.M.; Go, Y.M.; Samali, A.; Jones, D.P. Selective Oxidative Stress in Cell Nuclei by Nuclear-Targeted D-Amino Acid Oxidase. Antioxid. Redox Signal. 2007, 9, 807–816. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Oka, S.I.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A Redox-Dependent Mechanism for Regulation of AMPK Activation by Thioredoxin1 during Energy Starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-Activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [Green Version]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-Derived ROS Activate AMP-Activated Protein Kinase (AMPK) Indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ighodaro, O.M.; Akinloye, O.A. First Line Defence Antioxidants-Superoxide Dismutase (SOD), Catalase (CAT) and Glutathione Peroxidase (GPX): Their Fundamental Role in the Entire Antioxidant Defence Grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Crapo, J.D.; Oury, T.; Rabouille, C.; Slot, J.W.; Chang, L.Y. Copper, Zinc Superoxide Dismutase Is Primarily a Cytosolic Protein in Human Cells. Proc. Natl. Acad. Sci. USA 1992, 89, 10405–10409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishigami, M. Superoxide Dismutase. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [CrossRef]
- Marklund, S.L. Extracellular Superoxide Dismutase and Other Superoxide Dismutase Isoenzymes in Tissues from Nine Mammalian Species. Biochem. J. 1984, 222, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [Green Version]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox Homeostasis Maintained by GPX4 Facilitates STING Activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Walczak, J.; Partyka, M.; Duszyński, J.; Szczepanowska, J. Implications of Mitochondrial Network Organization in Mitochondrial Stress Signalling in NARP Cybrid and Rho0 Cells. Sci. Rep. 2017, 7, 14864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial Morphology Transitions and Functions: Implications for Retrograde Signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulthuis, E.P.; Adjobo-Hermans, M.J.W.; Willems, P.H.G.M.; Koopman, W.J.H. Mitochondrial Morphofunction in Mammalian Cells. Antioxid. Redox Signal. 2019, 30, 2066–2109. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.I.; Alexeyev, M.F. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive Oxygen Species Generated from the Mitochondrial Electron Transport Chain Induce Cytochrome c Dissociation from Beef-Heart Submitochondrial Particles via Cardiolipin Peroxidation. Possible Role in the Apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive Oxygen Species (ROS)-Induced ROS Release: A New Phenomenon Accompanying Induction of the Mitochondrial Permeability Transition in Cardiac Myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1α Buffers ROS-Mediated Removal of Mitochondria during Myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.L.; Tan, E.K. Superoxide Drives Progression of Parkin/PINK1-Dependent Mitophagy Following Translocation of Parkin to Mitochondria. Cell Death Dis. 2017, 8, e3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy Is Triggered by Mild Oxidative Stress in a Mitochondrial Fission Dependent Manner. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Echtay, K.S.; Roussel, D.; St-Plerre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide Activates Mitochondrial Uncoupling Proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Rial, E. A History of the First Uncoupling Protein, UCP1. J. Bioenerg. Biomembr. 1999, 31, 399–406. [Google Scholar] [CrossRef]
- Bárcena, C.; Mayoral, P.; Quirós, P.M. Mitohormesis, an Antiaging Paradigm. In International Review of Cell and Molecular Biology; López-Otín, C., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 340, pp. 35–77. [Google Scholar]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants Prevent Health-Promoting Effects of Physical Exercise in Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose Restriction Extends Caenorhabditis Elegans Life Span by Inducing Mitochondrial Respiration and Increasing Oxidative Stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Zarse, K.; Schmeisser, S.; Groth, M.; Priebe, S.; Beuster, G.; Kuhlow, D.; Guthke, R.; Platzer, M.; Kahn, C.R.; Ristow, M. Impaired Insulin/IGF1 Signaling Extends Life Span by Promoting Mitochondrial L-Proline Catabolism to Induce a Transient ROS Signal. Cell Metab. 2012, 15, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Nebert, D.W.; Dalton, T.P. The Role of Cytochrome P450 Enzymes in Endogenous Signalling Pathways and Environmental Carcinogenesis. Nat. Rev. Cancer 2006, 6, 947–960. [Google Scholar] [CrossRef]
- Nebert, D.W.; Russell, D.W. Clinical Importance of the Cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Cytochrome P450 Substrate Oxidation: MiniReview. J. Biochem. Mol. Toxicol. 2007, 21, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Zangar, R.C.; Davydov, D.R.; Verma, S. Mechanisms That Regulate Production of Reactive Oxygen Species by Cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. [Google Scholar] [CrossRef]
- Morel, Y.; Barouki, R. Down-Regulation of Cytochrome P450 1A1 Gene Promoter by Oxidative Stress: Critical Contribution of Nuclear Factor 1. J. Biol. Chem. 1998, 273, 26969–26976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marí, M.; Cederbaum, A.I. Induction of Catalase, Alpha, and Microsomal Glutathione S-Transferase in CYP2E1 Overexpressing HepG2 Cells and Protection against Short-Term Oxidative Stress. Hepatology 2001, 33, 652–661. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, X. Recent Advances in Natural Products from Plants for Treatment of Liver Diseases. Eur. J. Med. Chem. 2013, 63, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Calixto, J.B. The Role of Natural Products in Modern Drug Discovery. An. Acad. Bras. Cienc. 2019, 91 (Suppl. 3), e20190105. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303. [Google Scholar] [CrossRef] [Green Version]
- Elkordy, A.A.; Haj-Ahmad, R.R.; Awaad, A.S.; Zaki, R.M. An Overview on Natural Product Drug Formulations from Conventional Medicines to Nanomedicines: Past, Present and Future. J. Drug Deliv. Sci. Technol. 2021, 63, 102459. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Buenz, E.J.; Verpoorte, R.; Bauer, B.A. The Ethnopharmacologic Contribution to Bioprospecting Natural Products. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 509–530. [Google Scholar] [CrossRef] [PubMed]
- Imran, I.B.; Engström, M.T.; Karonen, M.; Williams, A.R.; Salminen, J.-P. Alkaline Oxidization Can Increase the in Vitro Antiparasitic Activity of Proanthocyanidin-Rich Plant Extracts against Ascaris Suum. Exp. Parasitol. 2023, 248, 108493. [Google Scholar] [CrossRef]
- Bauermeister, A.; Calil, F.A.; Pinto, F.d.C.L.; Medeiros, T.C.T.; Almeida, L.C.; Silva, L.J.; de Melo, I.S.; Zucchi, T.D.; Costa-Lotufo, L.V.; Moraes, L.A.B. Pradimicin-IRD from Amycolatopsis Sp. IRD-009 and Its Antimicrobial and Cytotoxic Activities. Nat. Prod. Res. 2019, 33, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Banc, R.; Popa, D.S.; Cozma-Petruţ, A.; Filip, L.; Kiss, B.; Fărcaş, A.; Nagy, A.; Miere, D.; Loghin, F. Protective Effects of Wine Polyphenols on Oxidative Stress and Hepatotoxicity Induced by Acrylamide in Rats. Antioxidants 2022, 11, 1347. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Q.; Dai, W.; Li, S.; Feng, J.; Li, J.; Liu, T.; Xu, S.; Wang, W.; Lu, X.; et al. Quercetin Pretreatment Attenuates Hepatic Ischemia Reperfusion-Induced Apoptosis and Autophagy by Inhibiting ERK/NF-κ B Pathway. Gastroenterol. Res. Pract. 2017, 2017, 9724217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tan, H.Y.; Wang, N.; Cheung, F.; Hong, M.; Feng, Y. The Potential and Action Mechanism of Polyphenols in the Treatment of Liver Diseases. Oxid. Med. Cell. Longev. 2018, 2018, 8394818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.F.; Lin, C.H.; Lin, C.C.; Lin, Y.H.; Chen, C.F.; Lin, C.K.; Lin, S.C. Antioxidative Natural Product Protect against Econazole-Induced Liver Injuries. Toxicology 2004, 196, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.k.; Zhang, Y.f.; Xie, L.; Rong, F.; Zhu, X.y.; Xie, J.; Zhou, H.; Xu, T. Progress in the Treatment of Drug-Induced Liver Injury with Natural Products. Pharmacol. Res. 2022, 183, 106361. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef]
- Sgrò, P.; Ceci, R.; Lista, M.; Patrizio, F.; Sabatini, S.; Felici, F.; Sacchetti, M.; Bazzucchi, I.; Duranti, G.; Di Luigi, L. Quercetin Modulates IGF-I and IGF-II Levels after Eccentric Exercise-Induced Muscle-Damage: A Placebo-Controlled Study. Front. Endocrinol. 2021, 12, 745959. [Google Scholar] [CrossRef]
- Rasouli, H.; Farzaei, M.H.; Khodarahmi, R. Polyphenols and Their Benefits: A Review. Int. J. Food Prop. 2017, 20, 1700–1741. [Google Scholar] [CrossRef] [Green Version]
- Alexandrino, H.; Rolo, A.; Tralhão, J.G.; Castro e Sousa, F.; Palmeira, C. Mitochondria in Liver Regeneration: Energy Metabolism and Posthepatectomy Liver Dysfunction. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P.J., Ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 127–152. [Google Scholar]
- Bernal, W.; Lee, W.M.; Wendon, J.; Larsen, F.S.; Williams, R. Acute Liver Failure: A Curable Disease by 2024? J. Hepatol. 2015, 62, S112–S120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvo-Jave, E.E.; Escalante-Tattersfield, T.; Ortega-Salgado, J.A.; Piña, E.; Geller, D.A. Factors in the Pathophysiology of the Liver Ischemia-Reperfusion Injury. J. Surg. Res. 2008, 147, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver Ischaemia–Reperfusion Injury: A New Understanding of the Role of Innate Immunity. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 239–256. [Google Scholar] [CrossRef]
- Zhai, Y.; Petrowsky, H.; Hong, J.C.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Ischaemia-Reperfusion Injury in Liver Transplantation-from Bench to Bedside. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 79–89. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 Release Induced by Liver Ischemia Involves Toll-like Receptor 4-Dependent Reactive Oxygen Species Production and Calcium-Mediated Signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The Nuclear Factor HMGB1 Mediates Hepatic Injury after Murine Liver Ischemia-Reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Bamboat, Z.M.; Balachandran, V.P.; Ocuin, L.M.; Obaid, H.; Plitas, G.; DeMatteo, R.P. Toll-like Receptor 9 Inhibition Confers Protection from Liver Ischemia-Reperfusion Injury. Hepatology 2010, 51, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the Global Public Health Agenda for NAFLD: A Consensus Statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef]
- Stefan, N.; Häring, H.-U.; Cusi, K. Non-Alcoholic Fatty Liver Disease: Causes, Diagnosis, Cardiometabolic Consequences, and Treatment Strategies. Lancet Diabetes Endocrinol. 2018, 8587, 313–324. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic Fatty Liver Disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Neuschwander-tetri, B.A. Nonalcoholic Fatty Liver Disease: Clinical Features and Pathogenesis. Gastroenterol. Hepatol. 2006, 2, 282–291. [Google Scholar]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective Hepatic Mitochondrial Respiratory Chain in Patients with Nonalcoholic Steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial Dysfunction in Rat with Nonalcoholic Fatty Liver: Involvement of Complex I, Reactive Oxygen Species and Cardiolipin. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants 2021, 10, 174. [Google Scholar] [CrossRef]
- Koruk, M.; Taysi, S.; Savas, M.C.; Yilmaz, O.; Akcay, F.; Karakok, M. Oxidative Stress and Enzymatic Antioxidant Status in Patients with Nonalcoholic Steatohepatitis. Ann. Clin. Lab. Sci. 2004, 34, 57–62. [Google Scholar]
- Perlemuter, G.; Davit-Spraul, A.; Cosson, C.; Conti, M.; Bigorgne, A.; Paradis, V.; Corre, M.P.; Prat, L.; Kuoch, V.; Basdevant, A.; et al. Increase in Liver Antioxidant Enzyme Activities in Non-Alcoholic Fatty Liver Disease. Liver Int. 2005, 25, 946–953. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Shibata, T. Genomic Landscape of Hepatocarcinogenesis. J. Hum. Genet. 2021, 66, 845–851. [Google Scholar] [CrossRef]
- Li, S.; Wang, X.; Wu, Y.; Zhang, H.; Zhang, L.; Wang, C.; Zhang, R.; Guo, Z. 8-Hydroxy-2′-Deoxyguanosine Expression Predicts Hepatocellular Carcinoma Outcome. Oncol. Lett. 2012, 3, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, M.R.; Orlicky, D.J.; Prigge, J.R.; Krishna, P.; Talago, E.A.; Cavigli, I.R.; Eriksson, S.; Miller, C.G.; Kundert, J.A.; Sayin, V.I.; et al. TrxR1, Gsr, and Oxidative Stress Determine Hepatocellular Carcinoma Malignancy. Proc. Natl. Acad. Sci. USA 2019, 166, 11408–11417. [Google Scholar] [CrossRef] [Green Version]
- Scalbert, A.; Johnson, I.T.; Saltmarsh, M. Polyphenols: Antioxidants and Beyond. Am. J. Clin. Nutr. 2005, 81, 215–217. [Google Scholar] [CrossRef] [Green Version]
- Simón, J.; Casado-Andrés, M.; Goikoetxea-Usandizaga, N.; Serrano-Maciá, M.; Martínez-Chantar, M.L. Nutraceutical Properties of Polyphenols against Liver Diseases. Nutrients 2020, 12, 3517. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Tsatsakis, A.; Mamoulakis, C.; Teodoro, M.; Briguglio, G.; Caruso, E.; Tsoukalas, D.; Margina, D.; Dardiotis, E.; Kouretas, D.; et al. Current Evidence on the Effect of Dietary Polyphenols Intake on Chronic Diseases. Food Chem. Toxicol. 2017, 110, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A Concise Overview on the Chemistry, Occurrence, and Human Health. Phyther. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Villaescusa, B.; Rodriguez, E.S.; Rangel-Huerta, O.D. Polyphenols in Obesity and Metabolic Syndrome. In Obesity; del Moral, A.M., García, C.M.A., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 213–239. [Google Scholar]
- Zhang, Y.; Balasooriya, H.; Sirisena, S.; Ng, K. The Effectiveness of Dietary Polyphenols in Obesity Management: A Systematic Review and Meta-Analysis of Human Clinical Trials. Food Chem. 2023, 404, 134668. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Venkataraman, K.; Hollingsworth, A.; Piche, M.; Tai, T.C. Polyphenols: Benefits to the Cardiovascular System in Health and in Aging. Nutrients 2013, 5, 3779–3827. [Google Scholar] [CrossRef] [Green Version]
- Luca, S.V.; Macovei, I.; Bujor, A.; Miron, A.; Skalicka-Woźniak, K.; Aprotosoaie, A.C.; Trifan, A. Bioactivity of Dietary Polyphenols: The Role of Metabolites. Crit. Rev. Food Sci. Nutr. 2020, 60, 626–659. [Google Scholar] [CrossRef]
- Agati, G.; Azzarello, E.; Pollastri, S.; Tattini, M. Flavonoids as Antioxidants in Plants: Location and Functional Significance. Plant Sci. 2012, 196, 67–76. [Google Scholar] [CrossRef]
- Brunetti, C.; Di Ferdinando, M.; Fini, A.; Pollastri, S.; Tattini, M. Flavonoids as Antioxidants and Developmental Regulators: Relative Significance in Plants and Humans. Int. J. Mol. Sci. 2013, 14, 3540–3555. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Ramiro, I.; Vauzour, D.; Minihane, A.M. Polyphenols and Non-Alcoholic Fatty Liver Disease: Impact and Mechanisms. Proc. Nutr. Soc. 2016, 75, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, M.; Saeed, F.; Anjum, F.M.; Afzaal, M.; Tufail, T.; Bashir, M.S.; Ishtiaq, A.; Hussain, S.; Suleria, H.A.R. Natural Polyphenols: An Overview. Int. J. Food Prop. 2017, 20, 1689–1699. [Google Scholar] [CrossRef] [Green Version]
- Nijveldt, R.J.; Van Nood, E.; Van Hoorn, D.E.C.; Boelens, P.G.; Van Norren, K.; Van Leeuwen, P.A.M. Flavonoids: A Review of Probable Mechanisms of Action and Potential Applications. Am. J. Clin. Nutr. 2001, 74, 418–425. [Google Scholar] [CrossRef] [Green Version]
- Dorta, D.J.; Pigoso, A.A.; Mingatto, F.E.; Rodrigues, T.; Pestana, C.R.; Uyemura, S.A.; Santos, A.C.; Curti, C. Antioxidant Activity of Flavonoids in Isolated Mitochondria. Phyther. Res. 2008, 22, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hu, M.J.; Wang, Y.Q.; Cui, Y.L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Sharma, S.; Rath, S.K. A Versatile Flavonoid Quercetin: Study of Its Toxicity and Differential Gene Expression in the Liver of Mice. Phytomedicine Plus 2022, 2, 100148. [Google Scholar] [CrossRef]
- Chen, L.; Liu, J.; Mei, G.; Chen, H.; Peng, S.; Zhao, Y.; Yao, P.; Tang, Y. Quercetin and Non-Alcoholic Fatty Liver Disease: A Review Based on Experimental Data and Bioinformatic Analysis. Food Chem. Toxicol. 2021, 154, 112314. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Autophagy as a Molecular Target of Quercetin Underlying Its Protective Effects in Human Diseases. Arch. Physiol. Biochem. 2019, 128, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.M.M.; Bast, A. Health Effects of Quercetin: From Antioxidant to Nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef]
- Aytac, Z.; Kusku, S.I.; Durgun, E.; Uyar, T. Quercetin/β-Cyclodextrin Inclusion Complex Embedded Nanofibres: Slow Release and High Solubility. Food Chem. 2016, 197, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Shen, Y.J.; Chen, M.; Zhao, J.Y.; Chen, S.H.; Zhang, W.; Song, J.K.; Li, L.; Du, G.H. Quercetin Attenuates Ischemia Reperfusion Injury by Protecting the Blood-Brain Barrier through Sirt1 in MCAO Rats. J. Asian Nat. Prod. Res. 2021, 24, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Nisha, V.M.; Anusree, S.S.; Priyanka, A.; Raghu, K.G. Apigenin and Quercetin Ameliorate Mitochondrial Alterations by Tunicamycin-Induced ER Stress in 3T3-L1 Adipocytes. Appl. Biochem. Biotechnol. 2014, 174, 1365–1375. [Google Scholar] [CrossRef]
- Vidyashankar, S.; Sandeep Varma, R.; Patki, P.S. Quercetin Ameliorate Insulin Resistance and Up-Regulates Cellular Antioxidants during Oleic Acid Induced Hepatic Steatosis in HepG2 Cells. Toxicol. Vitr. 2013, 27, 945–953. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Heng, C.; Zhou, Y.; Jiang, Z.; Qian, X.; Du, L.; Mao, S.; Yin, X.; Lu, Q. Quercetin Improves Nonalcoholic Fatty Liver by Ameliorating Inflammation, Oxidative Stress, and Lipid Metabolism in Db/Db Mice. Phyther. Res. 2019, 33, 3140–3152. [Google Scholar] [CrossRef]
- Uylaş, M.U.; Şahin, A.; Şahintürk, V.; Alataş, İ.Ö. Quercetin Dose Affects the Fate of Hepatic Ischemia and Reperfusion Injury in Rats: An Experimental Research. Int. J. Surg. 2018, 53, 117–121. [Google Scholar] [CrossRef]
- Atef, Y.; El-Fayoumi, H.M.; Abdel-Mottaleb, Y.; Mahmoud, M.F. Quercetin and Tin Protoporphyrin Attenuate Hepatic Ischemia Reperfusion Injury: Role of HO-1. Naunyn. Schmiedebergs. Arch. Pharmacol. 2017, 390, 871–881. [Google Scholar] [CrossRef]
- Yamada, N.; Matsushima-Nishiwaki, R.; Kozawa, O. Quercetin Suppresses the Migration of Hepatocellular Carcinoma Cells Stimulated by Hepatocyte Growth Factor or Transforming Growth Factor-α: Attenuation of AKT Signaling Pathway. Arch. Biochem. Biophys. 2020, 682, 108296. [Google Scholar] [CrossRef]
- Jeon, J.S.; Kwon, S.; Ban, K.; Kwon Hong, Y.; Ahn, C.; Sung, J.S.; Choi, I. Regulation of the Intracellular ROS Level Is Critical for the Antiproliferative Effect of Quercetin in the Hepatocellular Carcinoma Cell Line HepG2. Nutr. Cancer 2019, 71, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, L.; Ma, Y.X.; Li, W.T.; Li, L.; Zhu, H.Z.; Wu, M.H.; Zhou, J.R. Quercetin Inhibits Growth of Hepatocellular Carcinoma by Apoptosis Induction in Part via Autophagy Stimulation in Mice. J. Nutr. Biochem. 2019, 69, 108–119. [Google Scholar] [CrossRef]
- Wu, R.; Zhou, T.; Xiong, J.; Zhang, Z.; Tian, S.; Wang, Y.; Chen, J.; Tian, X. Quercetin, the Ingredient of Xihuang Pills, Inhibits Hepatocellular Carcinoma by Regulating Autophagy and Macrophage Polarization. Front. Biosci. Landmark 2022, 27, 323. [Google Scholar] [CrossRef]
- Homayoonfal, M.; Gilasi, H.; Asemi, Z.; Khaksary Mahabady, M.; Asemi, R.; Yousefi, B. Quercetin Modulates Signal Transductions and Targets Non-Coding RNAs against Cancer Development. Cell. Signal. 2023, 107, 110667. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, J.; Peng, S.; Yang, G.; Cheng, X.; Chen, L.; Zhang, H.; Zhao, Y.; Yao, P.; Tang, Y. Autophagy and Exosomes Coordinately Mediate Quercetin’s Protective Effects on Alcoholic Liver Disease. J. Nutr. Biochem. 2023, 116, 109332. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Eo, S.K.; Lee, J.H.; Park, S.Y. Quercetin-Induced Autophagy Flux Enhances TRAIL-Mediated Tumor Cell Death. Oncol. Rep. 2015, 34, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, M.; Wang, J.; Guo, X.; Xiao, L.; Liu, P.; Liu, L.; Tang, Y.; Yao, P. Quercetin Ameliorates Autophagy in Alcohol Liver Disease Associated with Lysosome through MTOR-TFEB Pathway. J. Funct. Foods 2019, 52, 177–185. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.; Deng, Y.; Liao, L.; Zhou, M.; Peng, C.; Li, Y. Quercetin as a Protective Agent for Liver Diseases: A Comprehensive Descriptive Review of the Molecular Mechanism. Phyther. Res. 2021, 35, 4727–4747. [Google Scholar] [CrossRef]
- Ferreira-silva, M.; Faria-silva, C.; Carvalheiro, M.C.; Simões, S.; Marinho, H.S.; Marcelino, P.; Campos, M.C.; Metselaar, J.M.; Fernandes, E.; Baptista, P.V.; et al. Quercetin Liposomal Nanoformulation for Ischemia and Reperfusion Injury Treatment. Pharmaceutics 2022, 14, 104. [Google Scholar] [CrossRef]
- Briguglio, G.; Costa, C.; Pollicino, M.; Giambò, F.; Catania, S.; Fenga, C. Polyphenols in Cancer Prevention: New Insights (Review). Int. J. Funct. Nutr. 2020, 1, 9. [Google Scholar] [CrossRef]
- Granado-Serrano, A.B.; Martín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin Induces Apoptosis via Caspase Activation, Regulation of Bcl-2, and Inhibition of PI-3-Kinase/Akt and ERK Pathways in a Human Hepatoma Cell Line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar] [CrossRef] [Green Version]
- Momchilova, A.; Nikolaev, G.; Pankov, S.; Vassileva, E.; Krastev, N.; Robev, B.; Krastev, D.; Pinkas, A.; Pankov, R. Effect of Quercetin and Fingolimod, Alone or in Combination, on the Sphingolipid Metabolism in HepG2 Cells. Int. J. Mol. Sci. 2022, 23, 13916. [Google Scholar] [CrossRef]
- Su, X.; Zhou, D.; Li, N. Bioactive Stilbenes from Plants. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Cambridge, MA, USA, 2022; Volume 73, pp. 265–403. [Google Scholar]
- Wang, J.; Matsuzaki, K.; Kitanaka, S. Stilbene Derivatives from Pholidota Chinensis and Their Anti-Inflammatory Activity. Chem. Pharm. Bull. 2006, 54, 1216–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Chen, C.F.; Ma, X.P.; Wang, M.Y.; Wang, W.; Xia, Y.; Zhang, N.; Wu, M.K.; Pan, W.D. Antibacterial Stilbenes from the Tubers of Bletilla Striata. Fitoterapia 2019, 138, 104350. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Uno, T.; Ogino, F.; Ai, C.; Duo, J.; Sankawa, U. Antioxidant Constituents of Caragana Tibetica. Chem. Pharm. Bull. 2005, 53, 1204–1206. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Perris, A.; Jawed, J.J.; Hoda, M. Therapeutic Role of Resveratrol against Hepatocellular Carcinoma: A Review on Its Molecular Mechanisms of Action. Pharmacol. Res. Mod. Chin. Med. 2023, 6, 100233. [Google Scholar] [CrossRef]
- Gupta, S.C.; Kannappan, R.; Reuter, S.; Kim, J.H.; Aggarwal, B.B. Chemosensitization of Tumors by Resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 150–160. [Google Scholar] [CrossRef] [Green Version]
- De Mello Andrade, J.M.; Fasolo, D. Polyphenol Antioxidants from Natural Sources and Contribution to Health Promotion. In Polyphenols in Human Health and Disease; Watson, R.R., Preedy, V.R., Zibadi, S., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 1, pp. 253–265. [Google Scholar]
- Faghihzadeh, F.; Adibi, P.; Rafiei, R.; Hekmatdoost, A. Resveratrol Supplementation Improves Inflammatory Biomarkers in Patients with Nonalcoholic Fatty Liver Disease. Nutr. Res. 2014, 34, 837–843. [Google Scholar] [CrossRef]
- Farzin, L.; Asghari, S.; Rafraf, M.; Asghari-Jafarabadi, M.; Shirmohammadi, M. No Beneficial Effects of Resveratrol Supplementation on Atherogenic Risk Factors in Patients with Nonalcoholic Fatty Liver Disease. Int. J. Vitam. Nutr. Res. 2019, 90, 279–289. [Google Scholar] [CrossRef]
- He, D.; Guo, Z.; Pu, J.L.; Zheng, D.F.; Wei, X.F.; Liu, R.; Tang, C.Y.; Wu, Z.J. Resveratrol Preconditioning Protects Hepatocytes against Hepatic Ischemia Reperfusion Injury via Toll-like Receptor 4/Nuclear Factor-ΚB Signaling Pathway in Vitro and in Vivo. Int. Immunopharmacol. 2016, 35, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, X.; Han, X.; Sun, J.; Li, L.; Zhang, D.; Sun, G. Resveratrol Improves Hepatic Ischemia-Reperfusion Injury by Inhibiting Neutrophils via the ERK Signaling Pathway. Biomed. Pharmacother. 2023, 160, 114358. [Google Scholar] [CrossRef]
- Ismail, N.; Abdel–Mottaleb, Y.; Eissa Ahmed, A.A.; El-Maraghy, N.N. Novel Combination of Thymoquinone and Resveratrol Enhances Anticancer Effect on Hepatocellular Carcinoma Cell Line. Future J. Pharm. Sci. 2018, 4, 41–46. [Google Scholar] [CrossRef]
- Majdalawieh, A.F.; Fayyad, M.W. Recent Advances on the Anti-Cancer Properties of Nigella Sativa, a Widely Used Food Additive. J. Ayurveda Integr. Med. 2016, 7, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Wu, C.; Qiu, S.; Yuan, X.; Li, L. Effects of Resveratrol on Glucose Control and Insulin Sensitivity in Subjects with Type 2 Diabetes: Systematic Review and Meta-Analysis. Nutr. Metab. 2017, 14, 60. [Google Scholar] [CrossRef]
- Tabatabaie, M.; Abdollahi, S.; Salehi-Abargouei, A.; Clark, C.C.T.; Karimi-Nazari, E.; Fallahzadeh, H.; Rahmanian, M.; Mozaffari-Khosravi, H. The Effect of Resveratrol Supplementation on Serum Levels of Asymmetric De-Methyl-Arginine and Paraoxonase 1 Activity in Patients with Type 2 Diabetes: A Randomized, Double-Blind Controlled Trial. Phyther. Res. 2020, 34, 2023–2031. [Google Scholar] [CrossRef]
- Gómez-Zorita, S.; Fernández-Quintela, A.; MacArulla, M.T.; Aguirre, L.; Hijona, E.; Bujanda, L.; Milagro, F.; Martínez, J.A.; Portillo, M.P. Resveratrol Attenuates Steatosis in Obese Zucker Rats by Decreasing Fatty Acid Availability and Reducing Oxidative Stress. Br. J. Nutr. 2012, 107, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Sharma, N.K.; Thungapathra, M. Resveratrol Regulates Body Weight in Healthy and Ovariectomized Rats. Nutr. Metab. 2017, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Bujanda, L.; Hijona, E.; Larzabal, M.; Beraza, M.; Aldazabal, P.; García-Urkia, N.; Sarasqueta, C.; Cosme, A.; Irastorza, B.; González, A.; et al. Resveratrol Inhibits Nonalcoholic Fatty Liver Disease in Rats. BMC Gastroenterol. 2008, 8, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rašković, A.; Ćućuz, V.; Torović, L.; Tomas, A.; Gojković-Bukarica, L.; Ćebović, T.; Milijašević, B.; Stilinović, N.; Cvejić Hogervorst, J. Resveratrol Supplementation Improves Metabolic Control in Rats with Induced Hyperlipidemia and Type 2 Diabetes. Saudi Pharm. J. 2019, 27, 1036–1043. [Google Scholar] [CrossRef]
- Ali Sangouni, A.; Abdollahi, S.; Mozaffari-Khosravi, H. Effect of Resveratrol Supplementation on Hepatic Steatosis and Cardiovascular Indices in Overweight Subjects with Type 2 Diabetes: A Double-Blind, Randomized Controlled Trial. BMC Cardiovasc. Disord. 2022, 22, 212. [Google Scholar] [CrossRef]
- Faghihzadeh, F.; Adibi, P.; Hekmatdoost, A. The Effects of Resveratrol Supplementation on Cardiovascular Risk Factors in Patients with Non-Alcoholic Fatty Liver Disease: A Randomised, Double-Blind, Placebo-Controlled Study. Br. J. Nutr. 2015, 114, 796–803. [Google Scholar] [CrossRef]
- Akbari, M.; Tamtaji, O.R.; Lankarani, K.B.; Tabrizi, R.; Dadgostar, E.; Haghighat, N.; Kolahdooz, F.; Ghaderi, A.; Mansournia, M.A.; Asemi, Z. The Effects of Resveratrol on Lipid Profiles and Liver Enzymes in Patients with Metabolic Syndrome and Related Disorders: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Lipids Health Dis. 2020, 19, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, X.; Liu, Y.; Li, M. The Role of CD14 and Toll-like Receptor 4 of Kupffer Cells in Hepatic Ischemia-Reperfusion Injury in Rats. Transplant. Proc. 2012, 44, 937–941. [Google Scholar] [CrossRef]
- Hassan-Khabbar, S.; Cottart, C.H.; Wendum, D.; Vibert, F.; Clot, J.P.; Savouret, J.F.; Conti, M.; Nivet-Antoine, V. Postischemic Treatment by Trans-Resveratrol in Rat Liver Ischemia-Reperfusion: A Possible Strategy in Liver Surgery. Liver Transplant. 2008, 14, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.M.; Ebright, B.; Hua, B.; Slarve, I.; Zhou, Y.; Jia, Y.; Louie, S.G.; Stiles, B.L. Eicosanoids and Other Oxylipins in Liver Injury, Inflammation and Liver Cancer Development. Front. Physiol. 2023, 14, 1098467. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dudley, J.I.; Das, D.K. Dose-Dependency of Resveratrol in Providing Health Benefits. Dose-Response 2010, 8, 478–500. [Google Scholar] [CrossRef] [PubMed]
- Kaltenmeier, C.; Yazdani, H.O.; Handu, S.; Popp, B.; Geller, D.; Tohme, S. The Role of Neutrophils as a Driver in Hepatic Ischemia-Reperfusion Injury and Cancer Growth. Front. Immunol. 2022, 13, 3326. [Google Scholar] [CrossRef]
- De Oliveira, T.H.C.; Marques, P.E.; Proost, P.; Teixeira, M.M.M. Neutrophils: A Cornerstone of Liver Ischemia and Reperfusion Injury. Lab. Investig. 2018, 98, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amalraj, A.; Pius, A.; Gopi, S.; Gopi, S. Biological Activities of Curcuminoids, Other Biomolecules from Turmeric and Their Derivatives—A Review. J. Tradit. Complement. Med. 2017, 7, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soni, V.K.; Ratre, Y.K.; Mehta, A.; Dixit, A.K.; Dwivedi, M.; Shukla, D.; Kumar, A.; Vishvakarma, N.K. Curcumin: A Spice Pigment against Hepatic Cancer. In Theranostics and Precision Medicine for the Management of Hepatocellular Carcinoma; Nagaraju, G.P., Ahmad, S., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 3, pp. 141–159. [Google Scholar]
- Ming, T.; Tao, Q.; Tang, S.; Zhao, H.; Yang, H.; Liu, M.; Ren, S.; Xu, H. Curcumin: An Epigenetic Regulator and Its Application in Cancer. Biomed. Pharmacother. 2022, 156, 113956. [Google Scholar] [CrossRef]
- Huang, Y.; Zhan, Y.; Luo, G.; Zeng, Y.; McClements, D.J.; Hu, K. Curcumin Encapsulated Zein/Caseinate-Alginate Nanoparticles: Release and Antioxidant Activity under in Vitro Simulated Gastrointestinal Digestion. Curr. Res. Food Sci. 2023, 6, 100463. [Google Scholar] [CrossRef]
- Li, S.; You, J.; Wang, Z.; Liu, Y.; Wang, B.; Du, M.; Zou, T. Curcumin Alleviates High-Fat Diet-Induced Hepatic Steatosis and Obesity in Association with Modulation of Gut Microbiota in Mice. Food Res. Int. 2021, 143, 110270. [Google Scholar] [CrossRef]
- Ibrahim, S.G.; El-Emam, S.Z.; Mohamed, E.A.; Abd Ellah, M.F. Dimethyl Fumarate and Curcumin Attenuate Hepatic Ischemia/Reperfusion Injury via Nrf2/HO-1 Activation and Anti-Inflammatory Properties. Int. Immunopharmacol. 2020, 80, 106131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Shi, S.; Jiang, P.; Huang, X.; Zhao, J.; Jin, Y.; Shen, Y.; Zhou, X.; Liu, H.; Cai, J. Curcumin Alleviates Hepatic Ischemia-Reperfusion Injury by Inhibiting Neutrophil Extracellular Traps Formation. J. Investig. Surg. 2023, 36, 2164813. [Google Scholar] [CrossRef]
- Khan, H.; Ni, Z.; Feng, H.; Xing, Y.; Wu, X.; Huang, D.; Chen, L.; Niu, Y.; Shi, G. Combination of Curcumin with N-n-Butyl Haloperidol Iodide Inhibits Hepatocellular Carcinoma Malignant Proliferation by Downregulating Enhancer of Zeste Homolog 2 (EZH2)—LncRNA H19 to Silence Wnt/β-Catenin Signaling. Phytomedicine 2021, 91, 153706. [Google Scholar] [CrossRef]
- Ngu, M.H.; Norhayati, M.N.; Rosnani, Z.; Zulkifli, M.M. Curcumin as Adjuvant Treatment in Patients with Non-Alcoholic Fatty Liver (NAFLD) Disease: A Systematic Review and Meta-Analysis. Complement. Ther. Med. 2022, 68, 102843. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Morine, Y.; Xu, C.; Nakasu, C.; Wada, Y.; Teraoku, H.; Yamada, S.; Saito, Y.; Ikemoto, T.; Shimada, M.; et al. Curcumin-Mediated Resistance to Lenvatinib via EGFR Signaling Pathway in Hepatocellular Carcinoma. Cells 2023, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.S.; Srivastava, R.A.K. Curcumin and Quercetin Synergistically Inhibit Cancer Cell Proliferation in Multiple Cancer Cells and Modulate Wnt/β-Catenin Signaling and Apoptotic Pathways in A375 Cells. Phytomedicine 2019, 52, 117–128. [Google Scholar] [CrossRef]
- Arena, A.; Romeo, M.A.; Benedetti, R.; Masuelli, L.; Bei, R.; Saveria, M.; Montani, G.; Cirone, M. New Insights into Curcumin- and Resveratrol-Mediated Anti-Cancer Effects. Pharmaceuticals 2021, 14, 1068. [Google Scholar] [CrossRef]
- Patial, V.; Mahesh, S.; Sharma, S.; Pratap, K.; Singh, D.; Padwad, Y.S. Synergistic Effect of Curcumin and Piperine in Suppression of DENA-Induced Hepatocellular Carcinoma in Rats. Environ. Toxicol. Pharmacol. 2015, 40, 445–452. [Google Scholar] [CrossRef]
- Chung, S.S.; Vadgama, J.V. Curcumin and Epigallocatechin Gallate Inhibit the Cancer Stem Cell Phenotype via Down-Regulation of STAT3-NFκB Signaling. Anticancer Res. 2015, 35, 39–46. [Google Scholar]


{kind=link}
{kind=link}
| Model | Dosage | Mechanisms | Effects | Reference |
|---|---|---|---|---|
| NAFLD | ||||
| HepG2 cells | 10 µM (for 24 h) | ↓ TNFα ↑ Antioxidant defenses | Improved insulin-mediated glucose uptake Reduced inflammation | Vidyashankar et al. [158] |
| C57BLKS db/db mice | 100 mg/kg/day (for 8 weeks) | ↑ Antioxidant defenses ↑ FXR1/TGR5 signaling | Improved dyslipidemia Relieved liver swelling and liver enzymes Reduced lipid accumulation and hyperglycemia | Yang et al. [159] |
| HepG2 cells | 10 and 20 µM (for 24 h) | |||
| Liver I/R injury | ||||
| BALB/c mice | 100 and 200 mg/kg/day (for 5 days) | ↓ ALT and AST ↓TNFα and IL-6 ↑ p62 ↓ BECN1 and LC3 ↑ Bcl-2 ↓ Bax, CASP3 and CASP9 ↓ ERK/NF-κB pathway | Reduced serum liver enzymes Reduced histopathological liver damage Inhibited the release of proinflammatory cytokines Inhibited autophagy and alleviated apoptosis | Wu et al. [108] |
| Primary hepatocytes | 20 µM (for 24 h before I/R) | |||
| Spraque Dawley rats | 50 mg/kg (for 30 min before I/R) | ↓ AST and ALT ↓ MDA | Restored abnormal liver enzymes Caused liver histological improvement | Uylaş et al. [160] |
| Wistar rats | 50 mg/kg (before I/R) | ↑ GSH, SOD, and CAT ↓ MDA ↑ Bcl-2 ↓ TNFα, NF-κB, and HO-1 | Reduced oxidative stress Reduced hepatic degeneration Reduced inflammatory cytokines | Atef et al. [161] |
| Hepatocellular carcinoma | ||||
| HuH7 cells | 3 to 7 µM (for 1 h) | ↓ AKT signaling ↓ HGF ↓ TGFα | Suppressed the migration of HCC cells | Yamada et al. [162] |
| HepG2, HuH7, PLC/PRF-5 and Hep3B cells | 80 µM (for 24 and 48 h) | ↓ ROS ↓ Cyclin A and CHK1 ↑ HO-1 | Reduced proliferation of HCC cells | Jeon et al. [163] |
| BALB/c nude mice | 60 mg/kg/day | ↓ AKT and mTOR ↑ MAPK | Inhibited the growth of HCC Stimulated autophagy Induced apoptosis | Ji et al. [164] |
| SMMC7721, HepG2 and LO2 cells | 0 to 120 µM (for 24, 36, and 48 h) | |||
| Old BALB/c mice | 25, 50, and 100 mg/kg/day (for 21 days) | ↓ MMP-2 and MMP-6 ↓ NF-κB ↓ TNFα, IL-6, and IL-17A | Inhibited HCC proliferation and migration Promoted apoptosis Produced a reduction in the volume and weight of liver tumors | Wu et al. [165] |
| H22 and HepG2 cells | 25, 50, and 100 µM (for 24, 48, and 72 h) | |||
| Model | Dosage | Mechanisms | Effects | Reference |
|---|---|---|---|---|
| NAFLD | ||||
| Adult patients with hepatic steatosis | 500 mg/day (for 12 weeks) | ↓ ALT and AST ↓ NF-κB ↓ Cytokeratin-18 ↓ Bilirubin, HDL- cholesterol, and Apo a1 | Improved tissue response to insulin Alleviated oxidative stress and inflammation | Faghihzadeh et al. [182] Farzin et al. [183] |
| Liver I/R injury | ||||
| Sprague Dawley rats | 10 and 20 mg/kg (for 1 h before I/R) | ↓ TLR4 ↓ NF-κB ↓ p65 ↑ IκBα ↓ TNFα and IL-1β ↓ AST, ↓ ALT, and LDH | Decreased cell viability associated with hypoxia/reoxygenation Inhibited the activity of TLR4 receptors Reduced apoptosis | He et al. [184] |
| BRL-3A cells | 0 to 100 µM (for 2 and 18 h before I/R) | |||
| C57BL/6 mice | 25 mg/kg (for 1 week, 2 h, and 30 min before I/R) | ↓ NET release ↓ ROS production ↑ GSH and GPx activity ↓ ERK/c-Fos signaling | Ameliorated LIRI Inhibited the function of neutrophils Restrained neutrophils-mediated inflammatory response | Wang et al. [185] |
| Hepatocellular carcinoma | ||||
| HepG2 cells | 64.5 µM (for 24 h) | ↑ CASP3 ↓ GSH and MDA | Decreased cell viability in HepG2 Induced apoptosis | Ismail et al. [186] |
| Model | Dosage | Mechanisms | Effects | Reference |
|---|---|---|---|---|
| NAFLD | ||||
| Old C57BL/6 mice | Curcumin supplementation on HFD 0.2%(w/w) (for 10 weeks) | Gut microbiota modulation ↓ LPS levels | Reduced body weight gain and fat deposition Ameliorated insulin resistance and improved glucose tolerance Reduced hepatic steatosis | Li et al. [207] |
| Liver I/R injury | ||||
| Albino rats | Pretreatment with 400 mg/kg/day (for 14 days before I/R) | ↓ AST and ALT ↓ TNFα, iNOS, IL-1β and IL-6 ↑ Nrf2 and HO-1 | Protected the liver from I/R-induced injury Improved neutrophil infiltration and the inflammatory cascade | Ibrahim et al. [208] |
| Old C57BL/6 mice | Pretreatment with 100 mg/kg (for 3 h before I/R) | ↓ MEK/ERK pathway ↓ ROS production ↓ TNFα, IL-1β, and IL-6 | Alleviated hepatic I/R injury Reduced the production of NET Activated innate immune response | Zhu et al. [209] |
| Hepatocellular carcinoma | ||||
| Nude mice | 50 mg/kg/day (for 3 weeks) | ↓ EZH2 ↓ Wnt/β-catenin | Inhibited cell migration and invasion Inhibited tumorigenicity Induced apoptosis | Khan et al. [210] |
| Hep3B and SMMC-7721 cells | 15 µM (for 48 h) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, I.F.; Miranda, R.G.; Dorta, D.J.; Rolo, A.P.; Palmeira, C.M. Targeting Oxidative Stress with Polyphenols to Fight Liver Diseases. Antioxidants 2023, 12, 1212. https://doi.org/10.3390/antiox12061212
Machado IF, Miranda RG, Dorta DJ, Rolo AP, Palmeira CM. Targeting Oxidative Stress with Polyphenols to Fight Liver Diseases. Antioxidants. 2023; 12(6):1212. https://doi.org/10.3390/antiox12061212
Chicago/Turabian StyleMachado, Ivo F., Raul G. Miranda, Daniel J. Dorta, Anabela P. Rolo, and Carlos M. Palmeira. 2023. "Targeting Oxidative Stress with Polyphenols to Fight Liver Diseases" Antioxidants 12, no. 6: 1212. https://doi.org/10.3390/antiox12061212
APA StyleMachado, I. F., Miranda, R. G., Dorta, D. J., Rolo, A. P., & Palmeira, C. M. (2023). Targeting Oxidative Stress with Polyphenols to Fight Liver Diseases. Antioxidants, 12(6), 1212. https://doi.org/10.3390/antiox12061212