
Inclusion of a Catechol-Derived Hydrazinyl-Thiazole (CHT) in β-Cyclodextrin Nanocavity and Its Effect on Antioxidant Activity: A Calorimetric, Spectroscopic and Molecular Docking Approach

 , ,
, ,  , ,
, ,
Abstract
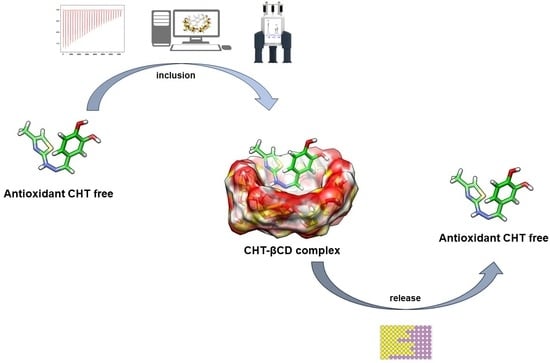
1. Introduction
2. Materials and Methods
2.1. Antioxidant Activity
2.2. Isothermal Titration Calorimetry (ITC)
2.3. 1H NMR Analysis
2.4. Molecular Docking
3. Results
3.1. Antioxidant Activity
3.2. Binding Affinity and Thermodynamics of CHT—β-CD Binding
3.3. Determination of the Stoichiometry by 1H NMR
3.4. ROESY Experiments
3.5. Evaluation of the Association Constant
3.6. Molecular Docking
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antony Muthu Prabhu, A.; Suresh Kumar, G.S.; Fatiha, M.; Sorimuthu, S.; Sundar Raj, M. Encapsulation of phenylalanine and 3,4-dihydroxyphenylalanine into β-cyclodextrin: Spectral and molecular modeling studies. J. Mol. Struct. 2015, 1079, 370–382. [Google Scholar] [CrossRef]
- Alper Öztürk, A.; Başaran, E.; Şenel, B.; Demirel, M.; Sarıca, Ş. Synthesis, characterization, antioxidant activity of Quercetin, Rutin and Quercetin-Rutin incorporated β-cyclodextrin inclusion complexes and determination of their activity in NIH-3T3, MDA-MB-231 and A549 cell lines. J. Mol. Struct. 2023, 1282, 135169. [Google Scholar] [CrossRef]
- Aree, T.; Jongrungruangchok, S. Crystallographic evidence for β-cyclodextrin inclusion complexation facilitating the improvement of antioxidant activity of tea (+)-catechin and (-)-epicatechin. Carbohydr. Polym. 2016, 140, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Kim, M.J.; Oh, W.Y.; Lee, J.H. Evaluation of deodorization techniques using cyclodextrins on the headspace volatiles and antioxidant properties of onion. Food Chem. 2023, 410, 135416. [Google Scholar] [CrossRef] [PubMed]
- Aguado, R.; Murtinho, D.; Valente, A.J.M. Association of antioxidant monophenolic compounds with β-cyclodextrin-functionalized cellulose and starch substrates. Carbohydr. Polym. 2021, 267, 118189. [Google Scholar] [CrossRef]
- Suresh Kumar, G.S.; Antony Muthu Prabhu, A.; Bhuvanesh, N. Studies on the self-catalyzed Knoevenagel condensation, characterization, DPPH radical scavenging activity, cytotoxicity, and molecular properties of 5-arylidene-2,2-dimethyl-1,3-dioxane-4,6-diones using single crystal XRD and DFT techniques. J. Mol. Struct. 2014, 1075, 166–177. [Google Scholar] [CrossRef]
- Liang, Y.; Hou, D.; Ni, Z.; Cao, M.; Cai, L. Preparation, characterization of naringenin, β-cyclodextrin and carbon quantum dot antioxidant nanocomposites. Food Chem. 2022, 375, 131646. [Google Scholar] [CrossRef]
- Mic, M.; Pîrnău, A.; Floare, C.G.; Miclăuș, M.; Kacso, I.; Palage, M.; Bogdan, M. Interaction of 1-methyl-1-({2-[4-(trifluoromethyl)phenyl]-1,3-thiazol-4-yl}methyl) piperidinium chloride with β-CD: Spectroscopic, calorimetric and molecular modeling approaches. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 195–204. [Google Scholar] [CrossRef]
- Maragos, S.; Archontaki, H.; Macheras, P.; Valsami, G. Effect of Cyclodextrin Complexation on the Aqueous Solubility and Solubility/Dose Ratio of Praziquantel. AAPS PharmSciTech 2009, 10, 1444. [Google Scholar] [CrossRef]
- Das, S.; Gazdag, Z.; Szente, L.; Meggyes, M.; Horváth, G.; Lemli, B.; Kunsági-Máté, S.; Kuzma, M.; Kőszegi, T. Antioxidant and antimicrobial properties of randomly methylated β cyclodextrin–captured essential oils. Food Chem. 2019, 278, 305–313. [Google Scholar] [CrossRef]
- Siva, S.; Li, C.; Cui, H.; Lin, L. Encompassment of isoeugenol in 2-hydroxypropyl-β-cyclodextrin using ultrasonication: Characterization, antioxidant and antibacterial activities. J. Mol. Liq. 2019, 296, 111777. [Google Scholar] [CrossRef]
- Navarro-Orcajada, S.; Conesa, I.; Matencio, A.; Rodríguez-Bonilla, P.; García-Carmona, F.; López-Nicolás, J.M. The use of cyclodextrins as solubility enhancers in the ORAC method may cause interference in the measurement of antioxidant activity. Talanta 2022, 243, e123336. [Google Scholar] [CrossRef] [PubMed]
- Junior, O.V.; Dantas, J.H.; Barão, C.E.; Zanoelo, E.F.; Cardozo-Filho, L.; de Moraes, F.F. Formation of inclusion compounds of (+)catechin with β-cyclodextrin in different complexation media: Spectral, thermal and antioxidant properties. J. Supercrit. Fluids 2017, 121, 10–18. [Google Scholar] [CrossRef]
- Yang, L.J.; Chang, Q.; Zhou, S.Y.; Yang, Y.H.; Xia, F.T.; Chen, W.; Li, M.; Yang, X.D. Host–guest interaction between brazilin and hydroxypropyl-β-cyclodextrin: Preparation, inclusion mode, molecular modelling and characterization. Dye. Pigment. 2018, 150, 193–201. [Google Scholar] [CrossRef]
- Lu, Z.; Cheng, B.; Hu, Y.; Zhang, Y.; Zou, G. Complexation of resveratrol with cyclodextrins: Solubility and antioxidant activity. Food Chem. 2009, 113, 17–20. [Google Scholar] [CrossRef]
- Lucas-Abellán, C.; Fortea, I.; López-Nicolás, J.M.; Núñez-Delicado, E. Cyclodextrins as resveratrol carrier system. Food Chem. 2007, 104, 39–44. [Google Scholar] [CrossRef]
- Mercader-Ros, M.T.; Lucas-Abellán, C.; Fortea, M.I.; Gabaldón, J.A.; Núñez-Delicado, E. Effect of HP-β-cyclodextrins complexation on the antioxidant activity of flavonols. Food Chem. 2010, 118, 769–773. [Google Scholar] [CrossRef]
- Hu, X.; Wang, X.; Han, L.; Li, S.; Zhou, W. Antioxidant and antimicrobial polyvinyl alcohol electrospun nanofibers containing baicalein-hydroxypropyl-β-cyclodextrin inclusion complex. Colloids Surfaces A Physicochem. Eng. Asp. 2021, 614, 126135. [Google Scholar] [CrossRef]
- Shiozawa, R.; Inoue, Y.; Murata, I.; Kanamoto, I. Effect of antioxidant activity of caffeic acid with cyclodextrins using ground mixture method. Asian J. Pharm. Sci. 2018, 13, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kalogeropoulos, N.; Yannakopoulou, K.; Gioxari, A.; Chiou, A.; Makris, D.P. Polyphenol characterization and encapsulation in β-cyclodextrin of a flavonoid-rich Hypericum perforatum (St John’s wort) extract. LWT Food Sci. Technol. 2010, 43, 882–889. [Google Scholar] [CrossRef]
- de Lima Paula, P.; de Lemos, A.S.O.; Queiroz, L.S.; Rocha, V.N.; Coimbra, E.S.; Fabri, R.L.; Denadai, Â.M.L. Supramolecular complexes between Plinia cauliflora (DC.) Kausel extracts and β-cyclodextrin: Physicochemical characterization and antioxidant and anti-inflammatory properties. J. Drug Deliv. Sci. Technol. 2023, 84, 104533. [Google Scholar] [CrossRef]
- Mic, M.; Pîrnău, A.; Floare, C.G.; Borlan, R.; Focsan, M.; Oniga, O.; Bogdan, M.; Vlase, L.; Oniga, I.; Marc, G. Antioxidant Activity Evaluation and Assessment of the Binding Affinity to HSA of a New Catechol Hydrazinyl-Thiazole Derivative. Antioxidants 2022, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.N.; Bristi, N.J.; Rafiquzzaman, M. Review on in vivo and in vitro methods evaluation of antioxidant activity. Saudi Pharm. J. 2013, 21, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Brand-Williams, W.; Cuvelier, M.E.; Berset, C. Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 1995, 28, 25–30. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Oniga, S.D.; Pîrnău, A.; Vlase, L.; Oniga, O. New Phenolic Derivatives of Thiazolidine-2,4-dione with Antioxidant and Antiradical Properties: Synthesis, Characterization, In Vitro Evaluation, and Quantum Studies. Molecules 2019, 24, 2060. [Google Scholar] [CrossRef]
- Marc, G.; Stana, A.; Franchini, A.H.; Vodnar, D.C.; Barta, G.; Tertiş, M.; Şanta, I.; Cristea, C.; Pîrnău, A.; Ciorîţă, A.; et al. Phenolic Thiazoles with Antioxidant and Antiradical Activity. Synthesis, In Vitro Evaluation, Toxicity, Electrochemical Behavior, Quantum Studies and Antimicrobial Screening. Antioxidants 2021, 10, 1707. [Google Scholar] [CrossRef]
- Da Ferreira, F.R.; Valentim, I.B.; Ramones, E.L.C.; Trevisan, M.T.S.; Olea-Azar, C.; Perez-Cruz, F.; de Abreu, F.C.; Goulart, M.O.F. Antioxidant activity of the mangiferin inclusion complex with β-cyclodextrin. LWT Food Sci. Technol. 2013, 51, 129–134. [Google Scholar] [CrossRef]
- Jullian, C.; Moyano, L.; Yañez, C.; Olea-Azar, C. Complexation of quercetin with three kinds of cyclodextrins: An antioxidant study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2007, 67, 230–234. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A 2013, 117, 12590–12600. [Google Scholar] [CrossRef]
- Bogdan, M.; Floare, C.G.; Buimaga-Iarinca, L.; Morari, C.; Pirnau, A. NMR study and computational assays of meclofenamic Na salt and β-cyclodextrin inclusion complex. J. Incl. Phenom. Macrocycl. Chem. 2016, 85, 111–120. [Google Scholar] [CrossRef]
- Floare, C.G.; Bogdan, M.; Tomoaia-Cotişel, M.; Mocanu, A. 1H NMR spectroscopic characterization of inclusion complex of desferrioxamine B chelator and β-cyclodextrin. J. Mol. Struct. 2022, 1248, 131477. [Google Scholar] [CrossRef]
- Caira, M.R.; Miller, J.L.; Nassimbeni, L.R. β-Cyclodextrin Inclusion Complexes of Mg2+ and Ca2+ Salts of Meclofenamic Acid: Preparation and Structural Characterisation. Supramol. Chem. 2006, 18, 553–559. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2010, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Stoica, C.I.; Marc, G.; Pîrnău, A.; Vlase, L.; Araniciu, C.; Oniga, S.; Palage, M.; Oniga, O. Thiazolyl-oxadiazole derivatives targeting lanosterol 14α-demethylase as potential antifungal agents: Design, synthesis and molecular docking studies. Farmacia 2016, 64, 390–397. [Google Scholar]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.-P.; Neumann, D. A new Lamarckian genetic algorithm for flexible ligand-receptor docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hazra, S.; Kumar, G.S. Physicochemical properties of inclusion complexes of sanguinarine with natural cyclodextrins: Spectroscopy, calorimetry and NMR studies. RSC Adv. 2015, 5, 1873–1882. [Google Scholar] [CrossRef]
- Mic, M.; Pîrnău, A.; Floare, C.G.; Marc, G.; Franchini, A.H.; Oniga, O.; Vlase, L.; Bogdan, M. Synthesis and molecular interaction study of a diphenolic hidrazinyl-thiazole compound with strong antioxidant and antiradical activity with HSA. J. Mol. Struct. 2021, 1244, 131278. [Google Scholar] [CrossRef]
- Mic, M.; Pîrnău, A.; Floare, C.G.; Bogdan, M. Study of the binding affinity between imatinib and α-1 glycoprotein using nuclear spin relaxation and isothermal titration calorimetry. Int. J. Biol. Macromol. 2020, 147, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Mic, M.; Pîrnău, A.; Neamţu, S.; Palage, M.; Bogdan, M. Molecular interaction of β-CD with 3-carboxy-1-[(2-phenyl-1,3-thiazol-4-yl) methyl]pyridin-1-ium iodide analyzed by isothermal titration calorimetry and NMR spectroscopy. J. Incl. Phenom. Macrocycl. Chem. 2015, 83, 257–265. [Google Scholar] [CrossRef]
- Pirnau, A.; Floare, C.G.; Bogdan, M. The complexation of flurbiprofen with β-cyclodextrin: A NMR study in aqueous solution. J. Incl. Phenom. Macrocycl. Chem. 2014, 78, 113–120. [Google Scholar] [CrossRef]
- Bogdan, M.; Caira, M.R.; Farcas, S.I. Inclusion of the Niflumic Acid Anion in β-cyclodextrin: A Solution NMR and X-ray Structural Investigation. Supramol. Chem. 2002, 14, 427–436. [Google Scholar] [CrossRef]
- Floare, C.G.; Balibanu, F.; Bogdan, M. CONSTEQ-A program for the calculation of the equilibrium constants using spectroscopic data. Stud. Univ. Babes-Bolyai Phys. 2005, 4, 451. [Google Scholar]
- Gu, A.; Wheate, N.J. Macrocycles as drug-enhancing excipients in pharmaceutical formulations. J. Incl. Phenom. Macrocycl. Chem. 2021, 100, 55–69. [Google Scholar] [CrossRef]
- Uekama, K.; Hirayama, F.; Irie, T. Cyclodextrin Drug Carrier Systems. Chem. Rev. 1998, 98, 2045–2076. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Ka (M−1) | ΔH (kJ mol−1) | TΔS (kJ mol−1) | ΔG (kJ mol−1) |
|---|---|---|---|---|
| 1.098 | 3.92 × 103 | −2.36 | 18.13 | −20.5 |
| Proton | Ha | Hb | Hc | Hd | He | Hf |
|---|---|---|---|---|---|---|
| δ | 7.029 | 6.801 | 7.137 | 7.843 | 6.387 | 2.145 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mic, M.; Pîrnău, A.; Floare, C.G.; Palage, M.D.; Oniga, O.; Marc, G. Inclusion of a Catechol-Derived Hydrazinyl-Thiazole (CHT) in β-Cyclodextrin Nanocavity and Its Effect on Antioxidant Activity: A Calorimetric, Spectroscopic and Molecular Docking Approach. Antioxidants 2023, 12, 1367. https://doi.org/10.3390/antiox12071367
Mic M, Pîrnău A, Floare CG, Palage MD, Oniga O, Marc G. Inclusion of a Catechol-Derived Hydrazinyl-Thiazole (CHT) in β-Cyclodextrin Nanocavity and Its Effect on Antioxidant Activity: A Calorimetric, Spectroscopic and Molecular Docking Approach. Antioxidants. 2023; 12(7):1367. https://doi.org/10.3390/antiox12071367
Chicago/Turabian StyleMic, Mihaela, Adrian Pîrnău, Călin G. Floare, Mariana Doina Palage, Ovidiu Oniga, and Gabriel Marc. 2023. "Inclusion of a Catechol-Derived Hydrazinyl-Thiazole (CHT) in β-Cyclodextrin Nanocavity and Its Effect on Antioxidant Activity: A Calorimetric, Spectroscopic and Molecular Docking Approach" Antioxidants 12, no. 7: 1367. https://doi.org/10.3390/antiox12071367
APA StyleMic, M., Pîrnău, A., Floare, C. G., Palage, M. D., Oniga, O., & Marc, G. (2023). Inclusion of a Catechol-Derived Hydrazinyl-Thiazole (CHT) in β-Cyclodextrin Nanocavity and Its Effect on Antioxidant Activity: A Calorimetric, Spectroscopic and Molecular Docking Approach. Antioxidants, 12(7), 1367. https://doi.org/10.3390/antiox12071367