

Oxidative Stress and Antioxidants in Age-Related Macular Degeneration


Abstract
:1. Introduction
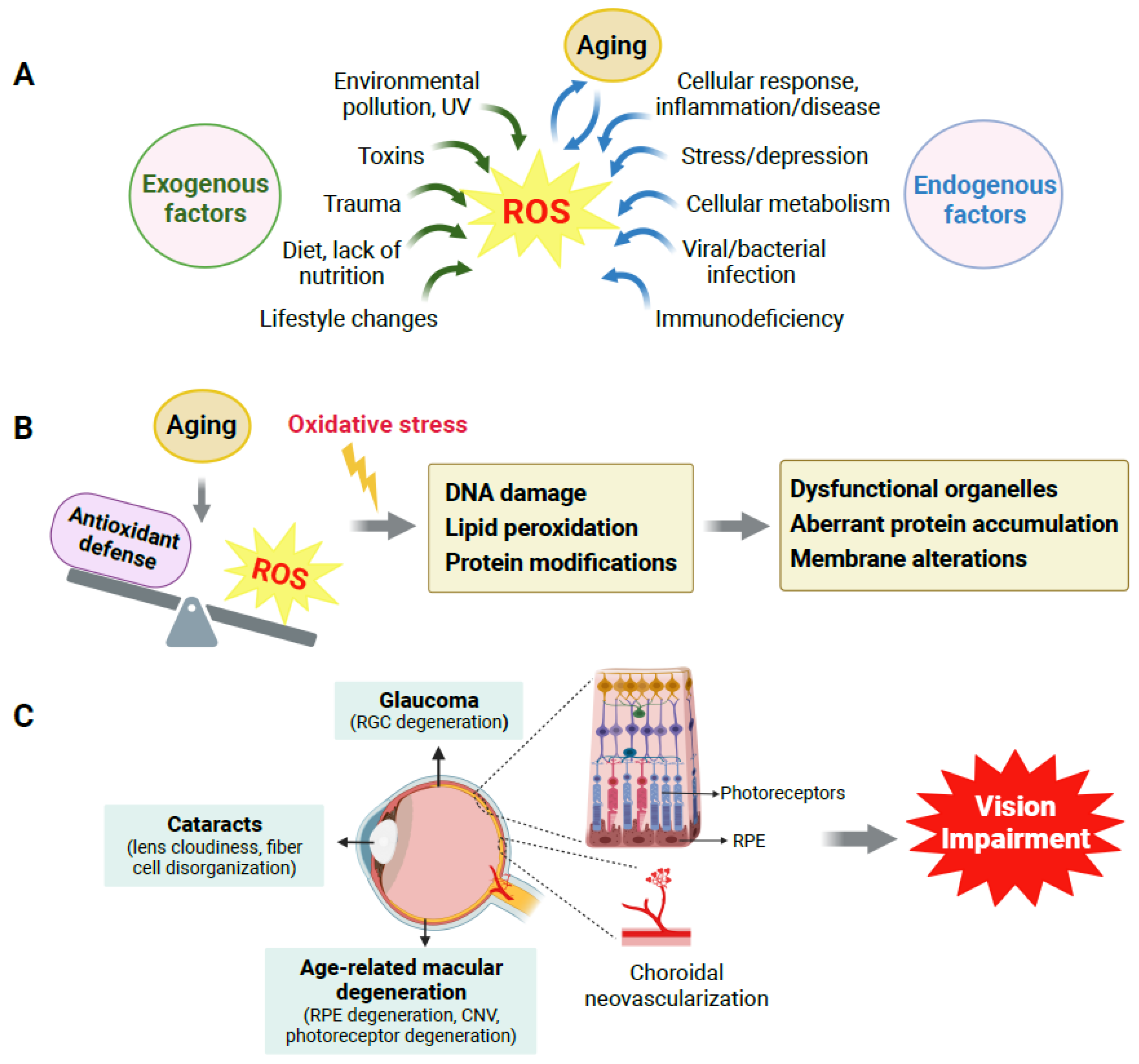
2. Production of ROS, Redox Homeostasis, and Their Impact on Aging
2.1. Imbalance of Redox Homeostasis Causes Oxidative Damage
2.2. ROS Accumulation Is One of Many Causes in Aging and Age-Related Disorders
3. Age-Related Eye Disorders
3.1. Overview of Age-Related Eye Disorders
3.2. Effects of Aging on the Eye, Retina and RPE
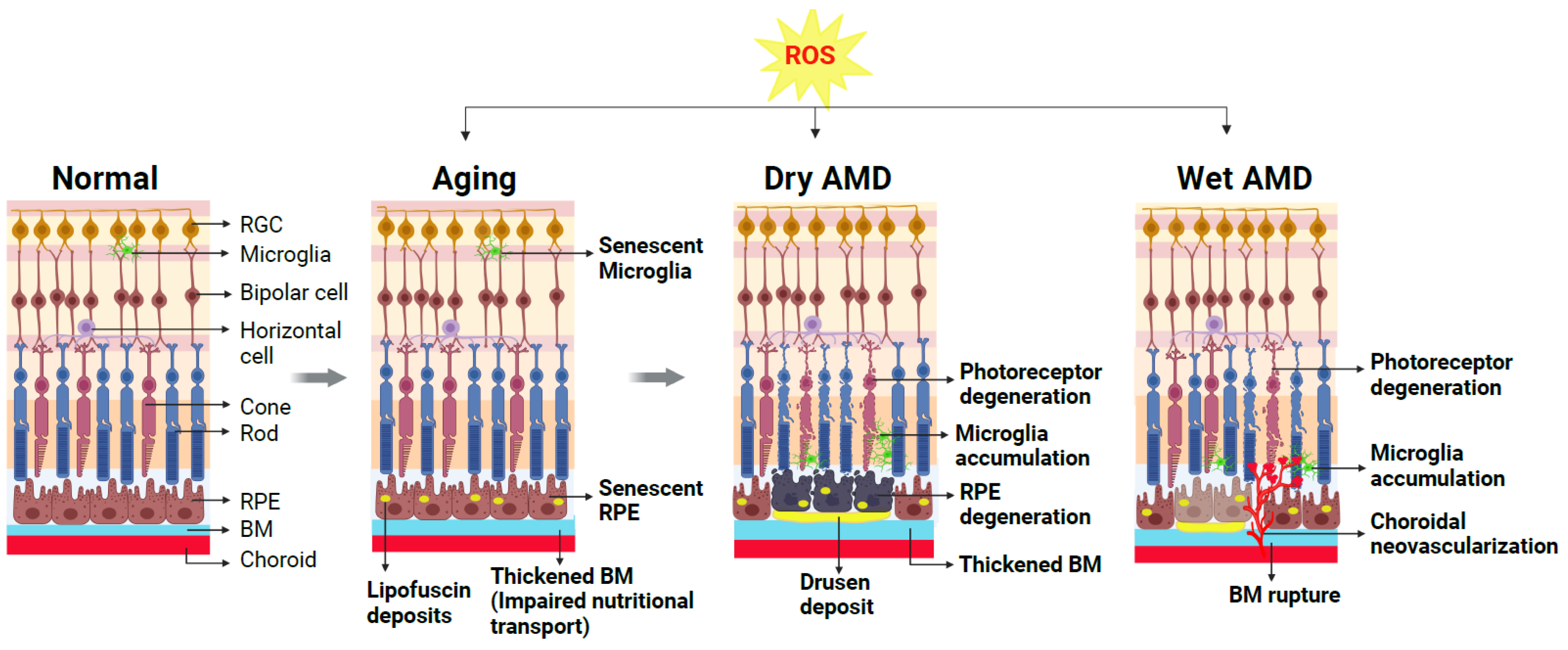
4. Age-Related Macular Degeneration: Classification, Pathophysiology, and Current Treatment
4.1. AMD: Clinical Classification
4.2. AMD: Dry and Wet Forms
4.3. AMD Pathogenesis: Genetic and Environmental Factors
4.4. Current Available Treatment for AMD
5. Presence of Oxidative Stress Markers in Clinical AMD
6. Experimental Studies of AMD Models Support the Pathogenic Role of Oxidative Stress in AMD
6.1. Genetic Models of Dry AMD Reflect Oxidative Stress and Oxidative Damage
6.2. Non-Genetic Models of Chemically Induced Oxidative RPE Damage Represent Features of Dry AMD
6.3. Recent Experimental Studies Have Established New Dry AMD Models with Identification of New Molecular Regulators of Oxidative Stress
6.4. Animal Models of Wet AMD: Roles of Oxidative Stress
6.5. In Vitro Models of RPE Oxidative Damage Further Support the Detrimental Role of Oxidative Stress
7. Therapeutic Roles of Antioxidants to Counter Oxidative Stress in AMD
7.1. Benefits of Antioxidant Intake in AMD Are Endorsed by AREDS I and II
7.2. Other Antioxidants in Protecting against AMD: Support from Additional Studies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosaka, S.; Obuki, M.; Nakajima, J.; Suzuki, M. Comparative study of antioxidants as quenchers or scavengers of reactive oxygen species based on quenching of MCLA-dependent chemiluminescence. Luminescence 2005, 20, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I. Oxidative Stress in Age-Related Macular Degeneration: Nrf2 as Therapeutic Target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Choo, P.P.; Woi, P.J.; Bastion, M.C.; Omar, R.; Mustapha, M.; Md Din, N. Review of Evidence for the Usage of Antioxidants for Eye Aging. Biomed. Res. Int. 2022, 2022, 5810373. [Google Scholar] [CrossRef]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [Green Version]
- Thorpe, G.W.; Fong, C.S.; Alic, N.; Higgins, V.J.; Dawes, I.W. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: Oxidative-stress-response genes. Proc. Natl. Acad. Sci. USA 2004, 101, 6564–6569. [Google Scholar] [CrossRef]
- Dogru, M.; Kojima, T.; Simsek, C.; Tsubota, K. Potential Role of Oxidative Stress in Ocular Surface Inflammation and Dry Eye Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES163–DES168. [Google Scholar] [CrossRef] [Green Version]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 10119. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Kregel, K.C.; Zhang, H.J. An integrated view of oxidative stress in aging: Basic mechanisms, functional effects, and pathological considerations. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R18–R36. [Google Scholar] [CrossRef]
- Yang, L.; Mih, N.; Anand, A.; Park, J.H.; Tan, J.; Yurkovich, J.T.; Monk, J.M.; Lloyd, C.J.; Sandberg, T.E.; Seo, S.W.; et al. Cellular responses to reactive oxygen species are predicted from molecular mechanisms. Proc. Natl. Acad. Sci. USA 2019, 116, 14368–14373. [Google Scholar] [CrossRef] [Green Version]
- Tsoukalas, D.; Fragkiadaki, P.; Docea, A.O.; Alegakis, A.K.; Sarandi, E.; Thanasoula, M.; Spandidos, D.A.; Tsatsakis, A.; Razgonova, M.P.; Calina, D. Discovery of potent telomerase activators: Unfolding new therapeutic and anti-aging perspectives. Mol. Med. Rep. 2019, 20, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The free radical theory of aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef]
- Gladyshev, V.N. The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal. 2014, 20, 727–731. [Google Scholar] [CrossRef]
- Smith, A.M.; Dragunow, M. Response to Watkins and Hutchinson. Trends Neurosci. 2014, 37, 190. [Google Scholar] [CrossRef]
- Hefendehl, J.K.; Neher, J.J.; Suhs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar] [PubMed]
- Chondrogianni, N.; Gonos, E.S. Overexpression of hUMP1/POMP proteasome accessory protein enhances proteasome-mediated antioxidant defence. Exp. Gerontol. 2007, 42, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. 2008, 4, 89–96. [Google Scholar] [PubMed]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Nashine, S. Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells 2021, 10, 2483. [Google Scholar] [CrossRef]
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar]
- Vyawahare, H.; Shinde, P. Age-Related Macular Degeneration: Epidemiology, Pathophysiology, Diagnosis, and Treatment. Cureus 2022, 14, e29583. [Google Scholar] [CrossRef]
- Caceres-Velez, P.R.; Hui, F.; Hercus, J.; Bui, B.; Jusuf, P.R. Restoring the oxidative balance in age-related diseases—An approach in glaucoma. Ageing Res. Rev. 2022, 75, 101572. [Google Scholar] [CrossRef]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wei, X. T Cell-Mediated Autoimmunity in Glaucoma Neurodegeneration. Front. Immunol. 2021, 12, 803485. [Google Scholar] [CrossRef]
- Alqawlaq, S.; Flanagan, J.G.; Sivak, J.M. All roads lead to glaucoma: Induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 2019, 183, 88–97. [Google Scholar] [CrossRef]
- Mozaffarieh, M.; Flammer, J. New insights in the pathogenesis and treatment of normal tension glaucoma. Curr. Opin. Pharmacol. 2013, 13, 43–49. [Google Scholar] [CrossRef]
- Medori, M.C.; Naureen, Z.; Dhuli, K.; Placidi, G.; Falsini, B.; Bertelli, M. Dietary supplements in retinal diseases, glaucoma, and other ocular conditions. J. Prev. Med. Hyg. 2022, 632 (Suppl. S3), E189–E199. [Google Scholar] [CrossRef]
- Chaudhry, S.; Dunn, H.; Carnt, N.; White, A. Nutritional supplementation in the prevention and treatment of glaucoma. Surv. Ophthalmol. 2022, 67, 1081–1098. [Google Scholar] [CrossRef]
- Lem, D.W.; Gierhart, D.L.; Davey, P.G. Carotenoids in the Management of Glaucoma: A Systematic Review of the Evidence. Nutrients 2021, 13, 1949. [Google Scholar] [CrossRef]
- Ramdas, W.D. The relation between dietary intake and glaucoma: A systematic review. Acta Ophthalmol. 2018, 96, 550–556. [Google Scholar] [CrossRef]
- Vision 2020: The cataract challenge. Community Eye Health 2000, 13, 17–19.
- Kaur, J.; Kukreja, S.; Kaur, A.; Malhotra, N.; Kaur, R. The oxidative stress in cataract patients. J. Clin. Diagn. Res. 2012, 6, 1629–1632. [Google Scholar] [CrossRef]
- Beebe, D.C.; Holekamp, N.M.; Shui, Y.B. Oxidative damage and the prevention of age-related cataracts. Ophthalmic Res. 2010, 44, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truscott, R.J. Age-related nuclear cataract-oxidation is the key. Exp. Eye Res. 2005, 80, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Braakhuis, A.J.; Donaldson, C.I.; Lim, J.C.; Donaldson, P.J. Nutritional Strategies to Prevent Lens Cataract: Current Status and Future Strategies. Nutrients 2019, 11, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiagarajan, R.; Manikandan, R. Antioxidants and cataract. Free Radic. Res. 2013, 47, 337–345. [Google Scholar] [CrossRef]
- Lim, J.C.; Caballero Arredondo, M.; Braakhuis, A.J.; Donaldson, P.J. Vitamin C and the Lens: New Insights into Delaying the Onset of Cataract. Nutrients 2020, 12, 3142. [Google Scholar] [CrossRef]
- Tanito, M. Reported evidence of vitamin E protection against cataract and glaucoma. Free Radic. Biol. Med. 2021, 177, 100–119. [Google Scholar] [CrossRef]
- Lin, J.B.; Tsubota, K.; Apte, R.S. A glimpse at the aging eye. NPJ Aging Mech. Dis. 2016, 2, 16003. [Google Scholar] [CrossRef] [Green Version]
- Sies, H.; Cadenas, E. Oxidative stress: Damage to intact cells and organs. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1985, 311, 617–631. [Google Scholar] [CrossRef]
- Samuel, M.A.; Zhang, Y.; Meister, M.; Sanes, J.R. Age-related alterations in neurons of the mouse retina. J. Neurosci. 2011, 31, 16033–16044. [Google Scholar] [CrossRef] [Green Version]
- Harwerth, R.S.; Wheat, J.L.; Rangaswamy, N.V. Age-related losses of retinal ganglion cells and axons. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4437–4443. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, P.; Nag, T.C.; Wadhwa, S. Age-related decrease in rod bipolar cell density of the human retina: An immunohistochemical study. J. Biosci. 2007, 32, 293–298. [Google Scholar] [CrossRef]
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056. [Google Scholar] [CrossRef]
- Lee, K.S.; Lin, S.; Copland, D.A.; Dick, A.D.; Liu, J. Cellular senescence in the aging retina and developments of senotherapies for age-related macular degeneration. J. Neuroinflamm. 2021, 18, 32. [Google Scholar] [CrossRef]
- Gu, X.; Neric, N.J.; Crabb, J.S.; Crabb, J.W.; Bhattacharya, S.K.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related changes in the retinal pigment epithelium (RPE). PLoS ONE 2012, 7, e38673. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Banerjee, K.; Lehmann, G.L.; Almeida, D.; Hajjar, K.A.; Benedicto, I.; Jiang, Z.; Radu, R.A.; Thompson, D.H.; Rodriguez-Boulan, E.; et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc. Natl. Acad. Sci. USA 2021, 118, e2100122118. [Google Scholar] [CrossRef]
- Boulton, M.E. Studying melanin and lipofuscin in RPE cell culture models. Exp. Eye Res. 2014, 126, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ach, T.; Huisingh, C.; McGwin, G., Jr.; Messinger, J.D.; Zhang, T.; Bentley, M.J.; Gutierrez, D.B.; Ablonczy, Z.; Smith, R.T.; Sloan, K.R.; et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4832–4841. [Google Scholar] [CrossRef] [Green Version]
- Soundara Pandi, S.P.; Rajendran, A.; Radha Krishnan, S.; Anto, M.J.; Gardiner, T.; Chakravarthy, U.; Veerappan, M. Characterization of age-related macular degeneration in Indian donor eyes. Indian J. Ophthalmol. 2021, 69, 642–646. [Google Scholar] [CrossRef]
- Steinberg, J.S.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S. Foveal Sparing of Reticular Drusen in Eyes With Early and Intermediate Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4267–4274. [Google Scholar] [CrossRef] [Green Version]
- Elsner, A.E.; Berk, L.; Burns, S.A.; Rosenberg, P.R. Aging and human cone photopigments. J. Opt. Soc. Am. A 1988, 5, 2106–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keunen, J.E.; van Norren, D.; van Meel, G.J. Density of foveal cone pigments at older age. Investig. Ophthalmol. Vis. Sci. 1987, 28, 985–991. [Google Scholar]
- Jackson, G.R.; Owsley, C.; Curcio, C.A. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res. Rev. 2002, 1, 381–396. [Google Scholar] [CrossRef]
- Chirco, K.R.; Sohn, E.H.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Structural and molecular changes in the aging choroid: Implications for age-related macular degeneration. Eye 2017, 31, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Fields, M.A.; Hoshino, R.; Priore, L.V. Effects of aging and anatomic location on gene expression in human retina. Front. Aging Neurosci. 2012, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, F.L., III; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, Y.J.; Won, J.Y. Molecular Mechanisms of Retinal Pigment Epithelium Dysfunction in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 12298. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Hicks, D.; Hamel, C.P. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Maurya, M.; Bora, K.; Blomfield, A.K.; Pavlovich, M.C.; Huang, S.; Liu, C.-H.; Chen, J. Oxidative stress in retinal pigment epithelium degeneration: From pathogenesis to therapeutic targets in dry age-related macular degeneration. Neural Regen. Res. 2023, 18, 2173–2181. [Google Scholar] [CrossRef]
- Wong, J.H.C.; Ma, J.Y.W.; Jobling, A.I.; Brandli, A.; Greferath, U.; Fletcher, E.L.; Vessey, K.A. Exploring the pathogenesis of age-related macular degeneration: A review of the interplay between retinal pigment epithelium dysfunction and the innate immune system. Front. Neurosci. 2022, 16, 1009599. [Google Scholar] [CrossRef]
- Cabral, T.; Mello, L.G.M.; Lima, L.H.; Polido, J.; Regatieri, C.V.; Belfort, R., Jr.; Mahajan, V.B. Retinal and choroidal angiogenesis: A review of new targets. Int. J. Retina Vitreous 2017, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Yeo, N.J.Y.; Chan, E.J.J.; Cheung, C. Choroidal Neovascularization: Mechanisms of Endothelial Dysfunction. Front. Pharmacol. 2019, 10, 1363. [Google Scholar] [CrossRef] [Green Version]
- Farecki, M.-L.; Gutfleisch, M.; Faatz, H.; Rothaus, K.; Heimes, B.; Spital, G.; Lommatzsch, A.; Pauleikhoff, D. Characteristics of type 1 and 2 CNV in exudative AMD in OCT-Angiography. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 913–921. [Google Scholar] [CrossRef]
- Yannuzzi, L.A.; Freund, K.B.; Takahashi, B.S. Review of retinal angiomatous proliferation or type 3 neovascularization. Retina 2008, 28, 375–384. [Google Scholar] [CrossRef]
- Cooke Bailey, J.N.; Pericak-Vance, M.A.; Haines, J.L. Genome-wide association studies: Getting to pathogenesis, the role of inflammation/complement in age-related macular degeneration. Cold Spring Harb. Perspect. Med. 2014, 4, a017186. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.V.; Leitner, W.P.; Staples, M.K.; Anderson, D.H. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp. Eye Res. 2001, 73, 887–896. [Google Scholar] [CrossRef]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Francis, P.J.; Hamon, S.C.; Ott, J.; Weleber, R.G.; Klein, M.L. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J. Med. Genet. 2009, 46, 300–307. [Google Scholar] [CrossRef]
- Spencer, K.L.; Hauser, M.A.; Olson, L.M.; Schmidt, S.; Scott, W.K.; Gallins, P.; Agarwal, A.; Postel, E.A.; Pericak-Vance, M.A.; Haines, J.L. Deletion of CFHR3 and CFHR1 genes in age-related macular degeneration. Hum. Mol. Genet. 2008, 17, 971–977. [Google Scholar] [CrossRef] [Green Version]
- Mohamad, N.A.; Ramachandran, V.; Mohd Isa, H.; Chan, Y.M.; Ngah, N.F.; Ching, S.M.; Hoo, F.K.; Wan Sulaiman, W.A.; Inche Mat, L.N.; Mohamed, M.H. Association of HTRA1 and ARMS2 gene polymorphisms with response to intravitreal ranibizumab among neovascular age-related macular degenerative subjects. Hum. Genom. 2019, 13, 13. [Google Scholar] [CrossRef]
- Tosi, G.M.; Caldi, E.; Neri, G.; Nuti, E.; Marigliani, D.; Baiocchi, S.; Traversi, C.; Cevenini, G.; Tarantello, A.; Fusco, F.; et al. HTRA1 and TGF-β1 Concentrations in the Aqueous Humor of Patients With Neovascular Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Kanda, A.; Chen, W.; Othman, M.; Branham, K.E.; Brooks, M.; Khanna, R.; He, S.; Lyons, R.; Abecasis, G.R.; Swaroop, A. A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 16227–16232. [Google Scholar] [CrossRef] [PubMed]
- Micklisch, S.; Lin, Y.; Jacob, S.; Karlstetter, M.; Dannhausen, K.; Dasari, P.; von der Heide, M.; Dahse, H.M.; Schmölz, L.; Grassmann, F.; et al. Age-related macular degeneration associated polymorphism rs10490924 in ARMS2 results in deficiency of a complement activator. J. Neuroinflamm. 2017, 14, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velilla, S.; García-Medina, J.J.; García-Layana, A.; Dolz-Marco, R.; Pons-Vázquez, S.; Pinazo-Durán, M.D.; Gómez-Ulla, F.; Arévalo, J.F.; Díaz-Llopis, M.; Gallego-Pinazo, R. Smoking and age-related macular degeneration: Review and update. J. Ophthalmol. 2013, 2013, 895147. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Cote, J.; Rosner, B. Progression of age-related macular degeneration: Association with dietary fat, transunsaturated fat, nuts, and fish intake. Arch. Ophthalmol. 2003, 121, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mitsuhashi, T.; Matsuo, T.; Yorifuji, T.; Hamada, J.; Liu, Y. Alcohol Consumption and Age-related Macular Degeneration: A Systematic Review and Dose-response Meta-analysis. Curr. Eye Res. 2021, 46, 1900–1907. [Google Scholar] [CrossRef]
- Zhang, Q.Y.; Tie, L.J.; Wu, S.S.; Lv, P.L.; Huang, H.W.; Wang, W.Q.; Wang, H.; Ma, L. Overweight, Obesity, and Risk of Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1276–1283. [Google Scholar] [CrossRef] [Green Version]
- Meyers, K.J.; Liu, Z.; Millen, A.E.; Iyengar, S.K.; Blodi, B.A.; Johnson, E.; Snodderly, D.M.; Klein, M.L.; Gehrs, K.M.; Tinker, L.; et al. Joint Associations of Diet, Lifestyle, and Genes with Age-Related Macular Degeneration. Ophthalmology 2015, 122, 2286–2294. [Google Scholar] [CrossRef] [Green Version]
- Schick, T.; Ersoy, L.; Lechanteur, Y.T.; Saksens, N.T.; Hoyng, C.B.; den Hollander, A.I.; Kirchhof, B.; Fauser, S. History Of Sunlight Exposure Is a Risk Factor for Age-Related Macular Degeneration. Retina 2016, 36, 787–790. [Google Scholar] [CrossRef]
- Millen, A.E.; Meyers, K.J.; Liu, Z.; Engelman, C.D.; Wallace, R.B.; LeBlanc, E.S.; Tinker, L.F.; Iyengar, S.K.; Robinson, J.G.; Sarto, G.E.; et al. Association between vitamin D status and age-related macular degeneration by genetic risk. JAMA Ophthalmol. 2015, 133, 1171–1179. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wong, W.T. Innate Immunity in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2021, 1256, 121–141. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.; Charbel Issa, P.; Cano, M.; Brandstatter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Alic, L.; Papac-Milicevic, N.; Czamara, D.; Rudnick, R.B.; Ozsvar-Kozma, M.; Hartmann, A.; Gurbisz, M.; Hoermann, G.; Haslinger-Hutter, S.; Zipfel, P.F.; et al. A genome-wide association study identifies key modulators of complement factor H binding to malondialdehyde-epitopes. Proc. Natl. Acad. Sci. USA 2020, 117, 9942–9951. [Google Scholar] [CrossRef] [Green Version]
- Parravano, M.; Costanzo, E.; Scondotto, G.; Trifirò, G.; Virgili, G. Anti-VEGF and Other Novel Therapies for Neovascular Age-Related Macular Degeneration: An Update. BioDrugs 2021, 35, 673–692. [Google Scholar] [CrossRef]
- Kaiser, S.M.; Arepalli, S.; Ehlers, J.P. Current and Future Anti-VEGF Agents for Neovascular Age-Related Macular Degeneration. J. Exp. Pharmacol. 2021, 13, 905–912. [Google Scholar] [CrossRef]
- Modi, Y.S.; Tanchon, C.; Ehlers, J.P. Comparative safety and tolerability of anti-VEGF therapy in age-related macular degeneration. Drug. Saf. 2015, 38, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Sun, D.; Lu, H.; Dai, R.; Xing, L.; Dong, H.; Wang, L.; Wei, D.; Jiang, B.; Jiao, Y.; et al. Comparison of effectiveness and safety between conbercept and ranibizumab for treatment of neovascular age-related macular degeneration. A retrospective case-controlled non-inferiority multiple center study. Eye 2018, 32, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Al-Khersan, H.; Hussain, R.M.; Ciulla, T.A.; Dugel, P.U. Innovative therapies for neovascular age-related macular degeneration. Expert. Opin. Pharmacother. 2019, 20, 1879–1891. [Google Scholar] [CrossRef]
- Cabral de Guimaraes, T.A.; Daich Varela, M.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: Therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef]
- Schmidt-Erfurth, U.; Kaiser, P.K.; Korobelnik, J.F.; Brown, D.M.; Chong, V.; Nguyen, Q.D.; Ho, A.C.; Ogura, Y.; Simader, C.; Jaffe, G.J.; et al. Intravitreal aflibercept injection for neovascular age-related macular degeneration: Ninety-six-week results of the VIEW studies. Ophthalmology 2014, 121, 193–201. [Google Scholar] [CrossRef]
- Ozveren, M. Results of Intravitreal Anti-VEGF Injection in Choroidal Neovascularization Caused by Pathologies Other Than Age-Related Macular Degeneration. Beyoglu Eye J. 2020, 5, 129–134. [Google Scholar] [CrossRef]
- Miller, J.W. Beyond VEGF—The Weisenfeld Lecture. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6911–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to Anti-VEGF therapy in neovascular AMD: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem. Pharmacol. 2019, 168, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Uemura, A.; Fruttiger, M.; D’Amore, P.A.; De Falco, S.; Joussen, A.M.; Sennlaub, F.; Brunck, L.R.; Johnson, K.T.; Lambrou, G.N.; Rittenhouse, K.D.; et al. VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog. Retin. Eye Res. 2021, 84, 100954. [Google Scholar] [CrossRef]
- Tzoumas, N.; Riding, G.; Williams, M.A.; Steel, D.H. Complement inhibitors for age-related macular degeneration. Cochrane Database Syst. Rev. 2023, 6, CD009300. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Hanus, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell. Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, M.; Milliner, C.; Bell, B.A.; Bonilha, V.L. Oxidative stress in the retina and retinal pigment epithelium (RPE): Role of aging, and DJ-1. Redox Biol. 2020, 37, 101623. [Google Scholar] [CrossRef]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Lau, L.I.; Liu, C.J.; Wei, Y.H. Increase of 8-hydroxy-2′-deoxyguanosine in aqueous humor of patients with exudative age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5486–5490. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.K.; Dong, A.; Hackett, S.F.; Bell, W.R.; Green, W.R.; Campochiaro, P.A. Oxidative damage in age-related macular degeneration. Histol. Histopathol. 2007, 22, 1301–1308. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Lu, L.; Gu, X.; Hong, L.; Laird, J.; Jaffe, K.; Choi, J.; Crabb, J.; Salomon, R.G. Synthesis and structural characterization of carboxyethylpyrrole-modified proteins: Mediators of age-related macular degeneration. Bioorg. Med. Chem. 2009, 17, 7548–7561. [Google Scholar] [CrossRef]
- Jeffrey, B.G.; Weisinger, H.S.; Neuringer, M.; Mitchell, D.C. The role of docosahexaenoic acid in retinal function. Lipids 2001, 36, 859–871. [Google Scholar] [CrossRef]
- Gu, X.; Meer, S.G.; Miyagi, M.; Rayborn, M.E.; Hollyfield, J.G.; Crabb, J.W.; Salomon, R.G. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J. Biol. Chem. 2003, 278, 42027–42035. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Pauer, G.J.; Yue, X.; Narendra, U.; Sturgill, G.M.; Bena, J.; Gu, X.; Peachey, N.S.; Salomon, R.G.; Hagstrom, S.A.; et al. Assessing susceptibility to age-related macular degeneration with proteomic and genomic biomarkers. Mol. Cell. Proteom. 2009, 8, 1338–1349. [Google Scholar] [CrossRef] [Green Version]
- Ethen, C.M.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Age-related macular degeneration and retinal protein modification by 4-hydroxy-2-nonenal. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3469–3479. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Pauer, G.J.; Yue, X.; Narendra, U.; Sturgill, G.M.; Bena, J.; Gu, X.; Peachey, N.S.; Salomon, R.G.; Hagstrom, S.A.; et al. Proteomic and genomic biomarkers for age-related macular degeneration. Adv. Exp. Med. Biol. 2010, 664, 411–417. [Google Scholar] [CrossRef]
- Totan, Y.; Yağci, R.; Bardak, Y.; Ozyurt, H.; Kendir, F.; Yilmaz, G.; Sahin, S.; Sahin Tiğ, U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009, 34, 1089–1093. [Google Scholar] [CrossRef]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, J.; Rowe, G.C.; Khadka, A.; Diaz-Aguilar, D.; Spencer, C.; Arany, Z.; Saint-Geniez, M. PGC-1α Induces Human RPE Oxidative Metabolism and Antioxidant Capacity. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1038–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decanini, A.; Nordgaard, C.L.; Feng, X.; Ferrington, D.A.; Olsen, T.W. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 2007, 143, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Elbay, A.; Ozer, O.F.; Akkan, J.C.U.; Celik, U.; Kutlutürk, I.; Koytak, A.; Ozdemir, H. Comparison of serum thiol-disulphide homeostasis and total antioxidant-oxidant levels between exudative age-related macular degeneration patients and healthy subjects. Int. Ophthalmol. 2017, 37, 1095–1101. [Google Scholar] [CrossRef]
- Ramazan, Z.K.; Sarı, İ.; Yıldırım, B.G.; Güntürk, İ.; Küçük, E.; Erşan, S.; Seydel, G. Evaluation of oxidative stress, 3-Nitrotyrosine, and HMGB-1 levels in patients with wet type Age-Related Macular Degeneration. J. Med. Biochem. 2022, 41, 275–281. [Google Scholar] [CrossRef]
- Zafrilla, P.; Losada, M.; Perez, A.; Caravaca, G.; Mulero, J. Biomarkers of oxidative stress in patients with wet age related macular degeneration. J. Nutr. Health Aging 2013, 17, 219–222. [Google Scholar] [CrossRef]
- Mrowicka, M.; Mrowicki, J.; Szaflik, J.P.; Szaflik, M.; Ulinska, M.; Szaflik, J.; Majsterek, I. Analysis of antioxidative factors related to AMD risk development in the polish patients. Acta Ophthalmol. 2017, 95, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Dong, Y.; Yang, H.; Pan, X.; Fan, R.; Zhai, L. Serum superoxide dismutase and malondialdehyde levels in a group of Chinese patients with age-related macular degeneration. Aging Clin. Exp. Res. 2011, 23, 264–267. [Google Scholar] [CrossRef]
- Anand, A.; Sharma, N.K.; Gupta, A.; Prabhakar, S.; Sharma, S.K.; Singh, R. Superoxide dismutase1 levels in North Indian population with age-related macular degeneration. Oxid. Med. Cell. Longev. 2013, 2013, 365046. [Google Scholar] [CrossRef] [Green Version]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [Green Version]
- Green, W.R. Histopathology of age-related macular degeneration. Mol. Vis. 1999, 5, 27. [Google Scholar]
- Chee, R.I.; Mahrous, A.; Koenig, L.; Mandel, L.S.; Yazdanie, F.; Chan, C.C.; Gupta, M.P. Histopathology of Age-Related Macular Degeneration and Implications for Pathogenesis and Therapy. Adv. Exp. Med. Biol. 2021, 1256, 67–88. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Cai, B.; Fishkin, N.; Jang, Y.P.; Krane, S.; Vollmer, H.R.; Zhou, J.; Nakanishi, K. A2E, a fluorophore of RPE lipofuscin: Can it cause RPE degeneration? Adv. Exp. Med. Biol. 2003, 533, 205–211. [Google Scholar] [CrossRef]
- Gerth, C. The role of the ERG in the diagnosis and treatment of Age-Related Macular Degeneration. Doc. Ophthalmol. 2009, 118, 63–68. [Google Scholar] [CrossRef]
- Chuang, J.-Z.; Yang, N.; Nakajima, N.; Otsu, W.; Fu, C.; Yang, H.H.; Lee, M.P.; Akbar, A.F.; Badea, T.C.; Guo, Z.; et al. Retinal pigment epithelium-specific CLIC4 mutant is a mouse model of dry age-related macular degeneration. Nat. Commun. 2022, 13, 374. [Google Scholar] [CrossRef]
- Seminotti, B.; Grings, M.; Tucci, P.; Leipnitz, G.; Saso, L. Nuclear Factor Erythroid-2-Related Factor 2 Signaling in the Neuropathophysiology of Inherited Metabolic Disorders. Front. Cell. Neurosci. 2021, 15, 785057. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, M.; Lester, K.L.; Han, Z. Light-induced Nrf2(−/−) mice as atrophic age-related macular degeneration model and treatment with nanoceria laden injectable hydrogel. Sci. Rep. 2019, 9, 14573. [Google Scholar] [CrossRef] [Green Version]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res. 2014, 119, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Behndig, A.; Svensson, B.; Marklund, S.L.; Karlsson, K. Superoxide dismutase isoenzymes in the human eye. Investig. Ophthalmol. Vis. Sci. 1998, 39, 471–475. [Google Scholar]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Pang, J.J.; Renganathan, K.; Zhan, X.; Crabb, J.W.; Kim, S.R.; Sparrow, J.R.; Hauswirth, W.W.; Lewin, A.S. SOD2 knockdown mouse model of early AMD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunaief, J.L. Iron induced oxidative damage as a potential factor in age-related macular degeneration: The Cogan Lecture. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4660–4664. [Google Scholar] [CrossRef] [Green Version]
- Hadziahmetovic, M.; Dentchev, T.; Song, Y.; Haddad, N.; He, X.; Hahn, P.; Pratico, D.; Wen, R.; Harris, Z.L.; Lambris, J.D.; et al. Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2728–2736. [Google Scholar] [CrossRef]
- Sivapathasuntharam, C.; Hayes, M.J.; Shinhmar, H.; Kam, J.H.; Sivaprasad, S.; Jeffery, G. Complement factor H regulates retinal development and its absence may establish a footprint for age related macular degeneration. Sci. Rep. 2019, 9, 1082. [Google Scholar] [CrossRef] [Green Version]
- Coffey, P.J.; Gias, C.; McDermott, C.J.; Lundh, P.; Pickering, M.C.; Sethi, C.; Bird, A.; Fitzke, F.W.; Maass, A.; Chen, L.L.; et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. USA 2007, 104, 16651–16656. [Google Scholar] [CrossRef]
- Shaw, P.X.; Zhang, L.; Zhang, M.; Du, H.; Zhao, L.; Lee, C.; Grob, S.; Lim, S.L.; Hughes, G.; Lee, J.; et al. Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2012, 109, 13757–13762. [Google Scholar] [CrossRef]
- Ufret-Vincenty, R.L.; Aredo, B.; Liu, X.; McMahon, A.; Chen, P.W.; Sun, H.; Niederkorn, J.Y.; Kedzierski, W. Transgenic mice expressing variants of complement factor H develop AMD-like retinal findings. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5878–5887. [Google Scholar] [CrossRef] [Green Version]
- Landowski, M.; Kelly, U.; Klingeborn, M.; Groelle, M.; Ding, J.D.; Grigsby, D.; Bowes Rickman, C. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 3703–3711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.H.; Reddick, R.L.; Piedrahita, J.A.; Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef]
- Saadane, A.; Petrov, A.; Mast, N.; El-Darzi, N.; Dao, T.; Alnemri, A.; Song, Y.; Dunaief, J.L.; Pikuleva, I.A. Mechanisms that minimize retinal impact of apolipoprotein E absence. J. Lipid Res. 2018, 59, 2368–2382. [Google Scholar] [CrossRef] [Green Version]
- Dithmar, S.; Curcio, C.A.; Le, N.A.; Brown, S.; Grossniklaus, H.E. Ultrastructural changes in Bruch’s membrane of apolipoprotein E-deficient mice. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2035–2042. [Google Scholar]
- Fernández-Robredo, P.; Rodríguez, J.A.; Sádaba, L.M.; Recalde, S.; García-Layana, A. Egg yolk improves lipid profile, lipid peroxidation and retinal abnormalities in a murine model of genetic hypercholesterolemia. J. Nutr. Biochem. 2008, 19, 40–48. [Google Scholar] [CrossRef]
- Levy, O.; Lavalette, S.; Hu, S.J.; Housset, M.; Raoul, W.; Eandi, C.; Sahel, J.A.; Sullivan, P.M.; Guillonneau, X.; Sennlaub, F. APOE Isoforms Control Pathogenic Subretinal Inflammation in Age-Related Macular Degeneration. J. Neurosci. 2015, 35, 13568–13576. [Google Scholar] [CrossRef] [Green Version]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Iacovelli, J.; Spencer, C.; Saint-Geniez, M. Direct effect of sodium iodate on neurosensory retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, M.; Nakamura, S.; Inoue, Y.; Nishinaka, A.; Nakamura, M.; Shimazawa, M.; Hara, H. Irreversible Photoreceptors and RPE Cells Damage by Intravenous Sodium Iodate in Mice Is Related to Macrophage Accumulation. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3476–3487. [Google Scholar] [CrossRef] [Green Version]
- Enzmann, V.; Row, B.W.; Yamauchi, Y.; Kheirandish, L.; Gozal, D.; Kaplan, H.J.; McCall, M.A. Behavioral and anatomical abnormalities in a sodium iodate-induced model of retinal pigment epithelium degeneration. Exp. Eye Res. 2006, 82, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Chowers, G.; Cohen, M.; Marks-Ohana, D.; Stika, S.; Eijzenberg, A.; Banin, E.; Obolensky, A. Course of Sodium Iodate-Induced Retinal Degeneration in Albino and Pigmented Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.E.; Alsaeedi, H.A.; Rashid, M.B.A.; Lam, C.; Harun, M.H.N.; Saleh, M.; Luu, C.D.; Kumar, S.S.; Ng, M.H.; Isa, H.M.; et al. Retinal degeneration rat model: A study on the structural and functional changes in the retina following injection of sodium iodate. J. Photochem. Photobiol. B Biol. 2019, 196, 111514. [Google Scholar] [CrossRef] [PubMed]
- Monés, J.; Leiva, M.; Peña, T.; Martínez, G.; Biarnés, M.; Garcia, M.; Serrano, A.; Fernandez, E. A Swine Model of Selective Geographic Atrophy of Outer Retinal Layers Mimicking Atrophic AMD: A Phase I Escalating Dose of Subretinal Sodium Iodate. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3974–3983. [Google Scholar] [CrossRef] [Green Version]
- Govindaraju, V.K.; Bodas, M.; Vij, N. Cigarette smoke induced autophagy-impairment regulates AMD pathogenesis mechanisms in ARPE-19 cells. PLoS ONE 2017, 12, e0182420. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, M.; Nagai, N.; Sussan, T.E.; Biswal, S.; Handa, J.T. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE 2008, 3, e3119. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Hadzic, S.; Roxlau, E.T.; Fuehler, B.; Janise-Libawski, A.; Wimmer, T.; Lei, B.; Li, S.W.; Weissmann, N.; Stieger, K. Retinal tissue develops an inflammatory reaction to tobacco smoke and electronic cigarette vapor in mice. J. Mol. Med. 2021, 99, 1459–1469. [Google Scholar] [CrossRef]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24 Pt B, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Huang, S.; Liu, C.H.; Wang, Z.; Fu, Z.; Britton, W.R.; Blomfield, A.K.; Kamenecka, T.M.; Dunaief, J.L.; Solt, L.A.; Chen, J. REV-ERBα regulates age-related and oxidative stress-induced degeneration in retinal pigment epithelium via NRF2. Redox Biol. 2022, 51, 102261. [Google Scholar] [CrossRef]
- Gupta, U.; Ghosh, S.; Wallace, C.T.; Shang, P.; Xin, Y.; Nair, A.P.; Yazdankhah, M.; Strizhakova, A.; Ross, M.A.; Liu, H.; et al. Increased LCN2 (lipocalin 2) in the RPE decreases autophagy and activates inflammasome-ferroptosis processes in a mouse model of dry AMD. Autophagy 2023, 19, 92–111. [Google Scholar] [CrossRef]
- Zhong, Y.; Li, J.; Wang, J.J.; Chen, C.; Tran, J.T.; Saadi, A.; Yu, Q.; Le, Y.Z.; Mandal, M.N.; Anderson, R.E.; et al. X-box binding protein 1 is essential for the anti-oxidant defense and cell survival in the retinal pigment epithelium. PLoS ONE 2012, 7, e38616. [Google Scholar] [CrossRef] [Green Version]
- Lambert, V.; Lecomte, J.; Hansen, S.; Blacher, S.; Gonzalez, M.-L.A.; Struman, I.; Sounni, N.E.; Rozet, E.; de Tullio, P.; Foidart, J.M.; et al. Laser-induced choroidal neovascularization model to study age-related macular degeneration in mice. Nat. Protoc. 2013, 8, 2197–2211. [Google Scholar] [CrossRef]
- Gong, Y.; Li, J.; Sun, Y.; Fu, Z.; Liu, C.H.; Evans, L.; Tian, K.; Saba, N.; Fredrick, T.; Morss, P.; et al. Optimization of an Image-Guided Laser-Induced Choroidal Neovascularization Model in Mice. PLoS ONE 2015, 10, e0132643. [Google Scholar] [CrossRef] [Green Version]
- Dobi, E.T.; Puliafito, C.A.; Destro, M. A new model of experimental choroidal neovascularization in the rat. Arch. Ophthalmol. 1989, 107, 264–269. [Google Scholar] [CrossRef]
- Tobe, T.; Ortega, S.; Luna, J.D.; Ozaki, H.; Okamoto, N.; Derevjanik, N.L.; Vinores, S.A.; Basilico, C.; Campochiaro, P.A. Targeted disruption of the FGF2 gene does not prevent choroidal neovascularization in a murine model. Am. J. Pathol. 1998, 153, 1641–1646. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Zhao, L.; Li, Y.; Liu, Y.; Xiao, W.; Song, Y.; Luo, L.; Huang, D.; Yancopoulos, G.D.; Wiegand, S.J.; et al. A subretinal matrigel rat choroidal neovascularization (CNV) model and inhibition of CNV and associated inflammation and fibrosis by VEGF trap. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6009–6017. [Google Scholar] [CrossRef]
- Qiu, G.; Stewart, J.M.; Sadda, S.; Freda, R.; Lee, S.; Guven, D.; de Juan, E., Jr.; Varner, S.E. A new model of experimental subretinal neovascularization in the rabbit. Exp. Eye Res. 2006, 83, 141–152. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Ling, J.X.; Wallace, T.M.; Dithmar, S.; Lawson, D.H.; Cohen, C.; Elner, V.M.; Elner, S.G.; Sternberg, P., Jr. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis. 2002, 8, 119–126. [Google Scholar]
- Oh, H.; Takagi, H.; Takagi, C.; Suzuma, K.; Otani, A.; Ishida, K.; Matsumura, M.; Ogura, Y.; Honda, Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1891–1898. [Google Scholar]
- Jo, Y.J.; Sonoda, K.H.; Oshima, Y.; Takeda, A.; Kohno, R.; Yamada, J.; Hamuro, J.; Yang, Y.; Notomi, S.; Hisatomi, T.; et al. Establishment of a new animal model of focal subretinal fibrosis that resembles disciform lesion in advanced age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6089–6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, K.; Spaide, R.F.; Ellis, E.A.; Iwabuchi, S.; Ogura, Y.; Armstrong, D. Lipid hydroperoxide stimulates subretinal choroidal neovascularization in the rabbit. Exp. Eye Res. 2002, 74, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kambhampati, S.P.; Bhutto, I.A.; McLeod, D.S.; Lutty, G.A.; Kannan, R.M. Evolution of oxidative stress, inflammation and neovascularization in the choroid and retina in a subretinal lipid induced age-related macular degeneration model. Exp. Eye Res. 2021, 203, 108391. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Bhutto, I.A.; Merges, C.; Grebe, R.; Emmert, D.; McLeod, D.S.; Armstrong, D.; Lutty, G.A. A rat model for choroidal neovascularization using subretinal lipid hydroperoxide injection. Am. J. Pathol. 2010, 176, 3085–3097. [Google Scholar] [CrossRef]
- Spilsbury, K.; Garrett, K.L.; Shen, W.Y.; Constable, I.J.; Rakoczy, P.E. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am. J. Pathol. 2000, 157, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Blaauwgeers, H.G.; Holtkamp, G.M.; Rutten, H.; Witmer, A.N.; Koolwijk, P.; Partanen, T.A.; Alitalo, K.; Kroon, M.E.; Kijlstra, A.; van Hinsbergh, V.W.; et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris. Evidence for a trophic paracrine relation. Am. J. Pathol. 1999, 155, 421–428. [Google Scholar] [CrossRef]
- Okamoto, N.; Tobe, T.; Hackett, S.F.; Ozaki, H.; Vinores, M.A.; LaRochelle, W.; Zack, D.J.; Campochiaro, P.A. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: A new model of intraretinal and subretinal neovascularization. Am. J. Pathol. 1997, 151, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hu, Y.; Lu, K.; Flannery, J.G.; Ma, J.X. Very low density lipoprotein receptor, a negative regulator of the wnt signaling pathway and choroidal neovascularization. J. Biol. Chem. 2007, 282, 34420–34428. [Google Scholar] [CrossRef] [Green Version]
- Heckenlively, J.R.; Hawes, N.L.; Friedlander, M.; Nusinowitz, S.; Hurd, R.; Davisson, M.; Chang, B. Mouse model of subretinal neovascularization with choroidal anastomosis. Retina 2003, 23, 518–522. [Google Scholar] [CrossRef]
- Joyal, J.S.; Sun, Y.; Gantner, M.L.; Shao, Z.; Evans, L.P.; Saba, N.; Fredrick, T.; Burnim, S.; Kim, J.S.; Patel, G.; et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat. Med. 2016, 22, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Nagai, N.; Lundh von Leithner, P.; Izumi-Nagai, K.; Hosking, B.; Chang, B.; Hurd, R.; Adamson, P.; Adamis, A.P.; Foxton, R.H.; Ng, Y.S.; et al. Spontaneous CNV in a novel mutant mouse is associated with early VEGF-A-driven angiogenesis and late-stage focal edema, neural cell loss, and dysfunction. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3709–3719. [Google Scholar] [CrossRef]
- Lu, Z.; Lin, V.; May, A.; Che, B.; Xiao, X.; Shaw, D.H.; Su, F.; Wang, Z.; Du, H.; Shaw, P.X. HTRA1 synergizes with oxidized phospholipids in promoting inflammation and macrophage infiltration essential for ocular VEGF expression. PLoS ONE 2019, 14, e0216808. [Google Scholar] [CrossRef]
- Abokyi, S.; To, C.H.; Lam, T.T.; Tse, D.Y. Central Role of Oxidative Stress in Age-Related Macular Degeneration: Evidence from a Review of the Molecular Mechanisms and Animal Models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [Green Version]
- Parsons, N.; Annamalai, B.; Obert, E.; Schnabolk, G.; Tomlinson, S.; Rohrer, B. Inhibition of the alternative complement pathway accelerates repair processes in the murine model of choroidal neovascularization. Mol. Immunol. 2019, 108, 8–12. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef]
- Zurawa-Janicka, D.; Kobiela, J.; Stefaniak, T.; Wozniak, A.; Narkiewicz, J.; Wozniak, M.; Limon, J.; Lipinska, B. Changes in expression of serine proteases HtrA1 and HtrA2 during estrogen-induced oxidative stress and nephrocarcinogenesis in male Syrian hamster. Acta Biochim. Pol. 2008, 55, 9–19. [Google Scholar] [CrossRef]
- Supanji; Shimomachi, M.; Hasan, M.Z.; Kawaichi, M.; Oka, C. HtrA1 is induced by oxidative stress and enhances cell senescence through p38 MAPK pathway. Exp. Eye Res. 2013, 112, 79–92. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, M.; Liang, J.; Chen, Y.; Wang, Y.; Wang, Y.; Xiao, Y.; Zhao, Z.; Wan, X.; Jiang, M.; et al. SLC7A11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting RPE Ferroptosis and VEGF Production. Front. Cell Dev. Biol. 2021, 9, 639851. [Google Scholar] [CrossRef]
- Nakamura, S.; Noguchi, T.; Inoue, Y.; Sakurai, S.; Nishinaka, A.; Hida, Y.; Masuda, T.; Nakagami, Y.; Horai, N.; Tsusaki, H.; et al. Nrf2 Activator RS9 Suppresses Pathological Ocular Angiogenesis and Hyperpermeability. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Totan, Y.; Cekiç, O.; Borazan, M.; Uz, E.; Sögüt, S.; Akyol, O. Plasma malondialdehyde and nitric oxide levels in age related macular degeneration. Br. J. Ophthalmol. 2001, 85, 1426–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, O.; Ateş, N.A.; Tamer, L.; Muşlu, N.; Ercan, B.; Atik, U.; Kanik, A. Changes in antioxidant enzyme activity and malondialdehyde level in patients with age-related macular degeneration. Ophthalmologica 2004, 218, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Kaneko, H.; Hayashi, Y.; Takayama, K.; Hwang, S.J.; Nishizawa, Y.; Kimoto, R.; Nagasaka, Y.; Tsunekawa, T.; Matsuura, T.; et al. Malondialdehyde induces autophagy dysfunction and VEGF secretion in the retinal pigment epithelium in age-related macular degeneration. Free Radic. Biol. Med. 2016, 94, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Delaplane, S.; Jiang, A.; Reed, A.; He, Y.; Fishel, M.; Nyland, R.L., 2nd; Borch, R.F.; Qiao, X.; Georgiadis, M.M.; et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: Small-molecule inhibition of the redox function of Ape1. Antioxid. Redox Signal. 2008, 10, 1853–1867. [Google Scholar] [CrossRef]
- Jiang, A.; Gao, H.; Kelley, M.R.; Qiao, X. Inhibition of APE1/Ref-1 redox activity with APX3330 blocks retinal angiogenesis in vitro and in vivo. Vision. Res. 2011, 51, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Liu, M.; He, Y.; Yang, B. Quercetin protect cigarette smoke extracts induced inflammation and apoptosis in RPE cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2010–2015. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.M.; Huang, D.Y.; Sekar, P.; Hsu, S.H.; Lin, W.W. Correction to: Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J. Biomed. Sci. 2019, 26, 66. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.M.; Zheng, W. Artemisinin protects human retinal pigment epithelial cells from hydrogen peroxide-induced oxidative damage through activation of ERK/CREB signaling. Redox Biol. 2016, 9, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Ni, T.; Yang, W.; Xing, Y. Protective effects of delphinidin against H(2)O(2)-induced oxidative injuries in human retinal pigment epithelial cells. Biosci. Rep. 2019, 39, BSR20190689. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shen, H.; Shi, J.; Tang, L. The effects of alpha lipoic acid in preventing oxidative stress-induced retinal pigment epithelial cell injury. Can. J. Physiol. Pharmacol. 2014, 92, 765–772. [Google Scholar] [CrossRef]
- Savion, N.; Dahamshi, S.; Morein, M.; Kotev-Emeth, S. S-Allylmercapro-N-Acetylcysteine Attenuates the Oxidation-Induced Lens Opacification and Retinal Pigment Epithelial Cell Death In Vitro. Antioxidants 2019, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Kanzaki, Y.; Fujita, H.; Sato, K.; Hosokawa, M.; Matsumae, H.; Morizane, Y.; Ohuchi, H. Protrusion of KCNJ13 Gene Knockout Retinal Pigment Epithelium Due to Oxidative Stress-Induced Cell Death. Investig. Ophthalmol. Vis. Sci. 2022, 63, 29. [Google Scholar] [CrossRef]
- Ma, X.; Chen, H.; Jian, S.; He, J.; Liu, Y.; Han, S.; Chang, L.; Li, P.; Chen, Y.A.; Liu, X.; et al. DAPL1 deficiency in mice impairs antioxidant defenses in the RPE and leads to retinal degeneration with AMD-like features. Redox Biol. 2023, 62, 102675. [Google Scholar] [CrossRef]
- Blanco-Ayala, T.; Andérica-Romero, A.C.; Pedraza-Chaverri, J. New insights into antioxidant strategies against paraquat toxicity. Free Radic. Res. 2014, 48, 623–640. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Ryhänen, T.; Karjalainen, H.M.; Lammi, M.J.; Suuronen, T.; Huhtala, A.; Kontkanen, M.; Teräsvirta, M.; Uusitalo, H.; Salminen, A. Geldanamycin increases 4-hydroxynonenal (HNE)-induced cell death in human retinal pigment epithelial cells. Neurosci. Lett. 2005, 382, 185–190. [Google Scholar] [CrossRef]
- Grassmann, F.; Friedrich, U.; Fauser, S.; Schick, T.; Milenkovic, A.; Schulz, H.L.; von Strachwitz, C.N.; Bettecken, T.; Lichtner, P.; Meitinger, T.; et al. A Candidate Gene Association Study Identifies DAPL1 as a Female-Specific Susceptibility Locus for Age-Related Macular Degeneration (AMD). Neuromol. Med. 2015, 17, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Y.; Wang, J.N.; Cao, Q.C.; Sun, R.X.; Zhu, H.J.; Zhang, Y.R.; Ji, J.D.; Liu, Q.H. m(6)A modification of circSPECC1 suppresses RPE oxidative damage and maintains retinal homeostasis. Cell Rep. 2022, 41, 111671. [Google Scholar] [CrossRef]
- Armento, A.; Honisch, S.; Panagiotakopoulou, V.; Sonntag, I.; Jacob, A.; Bolz, S.; Kilger, E.; Deleidi, M.; Clark, S.; Ueffing, M. Loss of Complement Factor H impairs antioxidant capacity and energy metabolism of human RPE cells. Sci. Rep. 2020, 10, 10320. [Google Scholar] [CrossRef]
- Trakkides, T.O.; Schäfer, N.; Reichenthaler, M.; Kühn, K.; Brandwijk, R.; Toonen, E.J.M.; Urban, F.; Wegener, J.; Enzmann, V.; Pauly, D. Oxidative Stress Increases Endogenous Complement-Dependent Inflammatory and Angiogenic Responses in Retinal Pigment Epithelial Cells Independently of Exogenous Complement Sources. Antioxidants 2019, 8, 548. [Google Scholar] [CrossRef] [Green Version]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age-Related Eye Disease Study 2 Research Group. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013, 309, 2005–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Medicine. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2001. [Google Scholar] [CrossRef] [Green Version]
- Gorusupudi, A.; Nelson, K.; Bernstein, P.S. The Age-Related Eye Disease 2 Study: Micronutrients in the Treatment of Macular Degeneration. Adv. Nutr. 2017, 8, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krinsky, N.I. Possible biologic mechanisms for a protective role of xanthophylls. J. Nutr. 2002, 132, 540S–542S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seddon, J.M.; Ajani, U.A.; Sperduto, R.D.; Hiller, R.; Blair, N.; Burton, T.C.; Farber, M.D.; Gragoudas, E.S.; Haller, J.; Miller, D.T.; et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA 1994, 272, 1413–1420. [Google Scholar] [CrossRef]
- Fu, Z.; Liegl, R.; Wang, Z.; Gong, Y.; Liu, C.H.; Sun, Y.; Cakir, B.; Burnim, S.B.; Meng, S.S.; Löfqvist, C.; et al. Adiponectin Mediates Dietary Omega-3 Long-Chain Polyunsaturated Fatty Acid Protection Against Choroidal Neovascularization in Mice. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3862–3870. [Google Scholar] [CrossRef] [Green Version]
- Rezende, F.A.; Lapalme, E.; Qian, C.X.; Smith, L.E.; SanGiovanni, J.P.; Sapieha, P. Omega-3 supplementation combined with anti-vascular endothelial growth factor lowers vitreal levels of vascular endothelial growth factor in wet age-related macular degeneration. Am. J. Ophthalmol. 2014, 158, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.; Chawla, R.; Kumar, A. Antioxidant supplements in age-related macular degeneration: Are they actually beneficial? Ther. Adv. Ophthalmol. 2021, 13, 25158414211030418. [Google Scholar] [CrossRef]
- Manresa, N.; Mulero, J.; Losada, M.; Zafrilla, P. Routes of Oxidative Stress in Age-Related Macular Degeneration. Int. J. Ophthalmol. Clin. Res. 2016, 3, 49. [Google Scholar] [CrossRef]
- Bungau, S.; Abdel-Daim, M.M.; Tit, D.M.; Ghanem, E.; Sato, S.; Maruyama-Inoue, M.; Yamane, S.; Kadonosono, K. Health Benefits of Polyphenols and Carotenoids in Age-Related Eye Diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9783429. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Aggarwal, B.B. Resveratrol: A multitargeted agent for age-associated chronic diseases. Cell Cycle 2008, 7, 1020–1035. [Google Scholar] [CrossRef] [Green Version]
- Dugas, B.; Charbonnier, S.; Baarine, M.; Ragot, K.; Delmas, D.; Ménétrier, F.; Lherminier, J.; Malvitte, L.; Khalfaoui, T.; Bron, A.; et al. Effects of oxysterols on cell viability, inflammatory cytokines, VEGF, and reactive oxygen species production on human retinal cells: Cytoprotective effects and prevention of VEGF secretion by resveratrol. Eur. J. Nutr. 2010, 49, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Nagineni, C.N.; Raju, R.; Nagineni, K.K.; Kommineni, V.K.; Cherukuri, A.; Kutty, R.K.; Hooks, J.J.; Detrick, B. Resveratrol Suppresses Expression of VEGF by Human Retinal Pigment Epithelial Cells: Potential Nutraceutical for Age-related Macular Degeneration. Aging Dis. 2014, 5, 88–100. [Google Scholar] [CrossRef]
- Kang, J.H.; Choung, S.Y. Protective effects of resveratrol and its analogs on age-related macular degeneration in vitro. Arch. Pharm. Res. 2016, 39, 1703–1715. [Google Scholar] [CrossRef]
- Khan, A.A.; Dace, D.S.; Ryazanov, A.G.; Kelly, J.; Apte, R.S. Resveratrol regulates pathologic angiogenesis by a eukaryotic elongation factor-2 kinase-regulated pathway. Am. J. Pathol. 2010, 177, 481–492. [Google Scholar] [CrossRef]
- Nagai, N.; Kubota, S.; Tsubota, K.; Ozawa, Y. Resveratrol prevents the development of choroidal neovascularization by modulating AMP-activated protein kinase in macrophages and other cell types. J. Nutr. Biochem. 2014, 25, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Koto, T.; Nagai, N.; Mochimaru, H.; Kurihara, T.; Izumi-Nagai, K.; Satofuka, S.; Shinoda, H.; Noda, K.; Ozawa, Y.; Inoue, M.; et al. Eicosapentaenoic acid is anti-inflammatory in preventing choroidal neovascularization in mice. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4328–4334. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Fu, Z.; Edin, M.L.; Liu, C.H.; Wang, Z.; Shao, Z.; Fredrick, T.W.; Saba, N.J.; Morss, P.C.; Burnim, S.B.; et al. Cytochrome P450 Oxidase 2C Inhibition Adds to ω-3 Long-Chain Polyunsaturated Fatty Acids Protection Against Retinal and Choroidal Neovascularization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1919–1927. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Tomita, Y.; Edin, M.L.; Ren, A.; Ko, M.; Yang, J.; Bull, E.; Zeldin, D.C.; Hellström, A.; Fu, Z.; et al. Cytochrome P450 oxidase 2J inhibition suppresses choroidal neovascularization in mice. Metabolism 2022, 134, 155266. [Google Scholar] [CrossRef]
- Courtaut, F.; Aires, V.; Acar, N.; Bretillon, L.; Guerrera, I.C.; Chhuon, C.; Pais de Barros, J.P.; Olmiere, C.; Delmas, D. RESVEGA, a Nutraceutical Omega-3/Resveratrol Supplementation, Reduces Angiogenesis in a Preclinical Mouse Model of Choroidal Neovascularization. Int. J. Mol. Sci. 2021, 22, 11023. [Google Scholar] [CrossRef]
- Cai, X.; McGinnis, J.F. Nanoceria: A Potential Therapeutic for Dry AMD. Adv. Exp. Med. Biol. 2016, 854, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tisi, A.; Pulcini, F.; Carozza, G.; Mattei, V.; Flati, V.; Passacantando, M.; Antognelli, C.; Maccarone, R.; Delle Monache, S. Antioxidant Properties of Cerium Oxide Nanoparticles Prevent Retinal Neovascular Alterations In Vitro and In Vivo. Antioxidants 2022, 11, 1133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wong, L.L.; Karakoti, A.S.; Seal, S.; McGinnis, J.F. Nanoceria inhibit the development and promote the regression of pathologic retinal neovascularization in the Vldlr knockout mouse. PLoS ONE 2011, 6, e16733. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Seal, S.; McGinnis, J.F. Sustained inhibition of neovascularization in vldlr−/− mice following intravitreal injection of cerium oxide nanoparticles and the role of the ASK1-P38/JNK-NF-kappaB pathway. Biomaterials 2014, 35, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisi, A.; Flati, V.; Delle Monache, S.; Lozzi, L.; Passacantando, M.; Maccarone, R. Nanoceria Particles Are an Eligible Candidate to Prevent Age-Related Macular Degeneration by Inhibiting Retinal Pigment Epithelium Cell Death and Autophagy Alterations. Cells 2020, 9, 1617. [Google Scholar] [CrossRef]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef] [Green Version]
- Winkler, B.S.; Boulton, M.E.; Gottsch, J.D.; Sternberg, P. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32. [Google Scholar]
- Qin, L.; Bartlett, H.; Griffiths, H.R.; Eperjesi, F.; Armstrong, R.A.; Gherghel, D. Macular pigment optical density is related to blood glutathione levels in healthy individuals. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5029–5033. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Terluk, M.R.; Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Yuan, C.; Kartha, R.V.; Montezuma, S.R.; Ferrington, D.A. N-Acetyl-L-cysteine Protects Human Retinal Pigment Epithelial Cells from Oxidative Damage: Implications for Age-Related Macular Degeneration. Oxid. Med. Cell. Longev. 2019, 2019, 5174957. [Google Scholar] [CrossRef] [Green Version]
- Hara, R.; Inomata, Y.; Kawaji, T.; Sagara, N.; Inatani, M.; Fukushima, M.; Tanihara, H. Suppression of choroidal neovascularization by N-acetyl-cysteine in mice. Curr. Eye Res. 2010, 35, 1012–1020. [Google Scholar] [CrossRef]
- Eye Disease Case-Control Study Group. Antioxidant status and neovascular age-related macular degeneration. Arch. Ophthalmol. 1993, 111, 104–109. [Google Scholar] [CrossRef]
- Aoki, A.; Inoue, M.; Nguyen, E.; Obata, R.; Kadonosono, K.; Shinkai, S.; Hashimoto, H.; Sasaki, S.; Yanagi, Y. Dietary n-3 Fatty Acid, α-Tocopherol, Zinc, vitamin D, vitamin C, and β-carotene are Associated with Age-Related Macular Degeneration in Japan. Sci. Rep. 2016, 6, 20723. [Google Scholar] [CrossRef] [Green Version]
- Edwards, G.; Olson, C.G.; Euritt, C.P.; Koulen, P. Molecular Mechanisms Underlying the Therapeutic Role of Vitamin E in Age-Related Macular Degeneration. Front. Neurosci. 2022, 16, 890021. [Google Scholar] [CrossRef]
- Snodderly, D.M. Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitamins. Am. J. Clin. Nutr. 1995, 62 (Suppl. S6), 1448S–1461S. [Google Scholar] [CrossRef]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for preventing age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 7, Cd000253. [Google Scholar] [CrossRef]
- Cho, E.; Seddon, J.M.; Rosner, B.; Willett, W.C.; Hankinson, S.E. Prospective study of intake of fruits, vegetables, vitamins, and carotenoids and risk of age-related maculopathy. Arch. Ophthalmol. 2004, 122, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Olza, J.; Aranceta-Bartrina, J.; González-Gross, M.; Ortega, R.M.; Serra-Majem, L.; Varela-Moreiras, G.; Gil, Á. Reported Dietary Intake and Food Sources of Zinc, Selenium, and Vitamins A, E and C in the Spanish Population: Findings from the ANIBES Study. Nutrients 2017, 9, 697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrowicka, M.; Mrowicki, J.; Kucharska, E.; Majsterek, I. Lutein and Zeaxanthin and Their Roles in Age-Related Macular Degeneration-Neurodegenerative Disease. Nutrients 2022, 14, 827. [Google Scholar] [CrossRef] [PubMed]
- Korobelnik, J.F.; Rougier, M.B.; Delyfer, M.N.; Bron, A.; Merle, B.M.J.; Savel, H.; Chêne, G.; Delcourt, C.; Creuzot-Garcher, C. Effect of Dietary Supplementation With Lutein, Zeaxanthin, and ω-3 on Macular Pigment: A Randomized Clinical Trial. JAMA Ophthalmol. 2017, 135, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, E.-S.M.; Akhtar, H.; Zaheer, K.; Ali, R. Dietary sources of lutein and zeaxanthin carotenoids and their role in eye health. Nutrients 2013, 5, 1169–1185. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Liu, R.; Du, J.H.; Liu, T.; Wu, S.S.; Liu, X.H. Lutein, Zeaxanthin and Meso-zeaxanthin Supplementation Associated with Macular Pigment Optical Density. Nutrients 2016, 8, 426. [Google Scholar] [CrossRef]
- Chew, E.Y.; Clemons, T.; SanGiovanni, J.P.; Danis, R.; Domalpally, A.; McBee, W.; Sperduto, R.; Ferris, F.L. The Age-Related Eye Disease Study 2 (AREDS2): Study design and baseline characteristics (AREDS2 report number 1). Ophthalmology 2012, 119, 2282–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christen, W.G.; Schaumberg, D.A.; Glynn, R.J.; Buring, J.E. Dietary ω-3 fatty acid and fish intake and incident age-related macular degeneration in women. Arch. Ophthalmol. 2011, 129, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Mikołajczak, N.; Sobiechowska, D.A.; Tańska, M. Edible flowers as a new source of natural antioxidants for oxidative protection of cold-pressed oils rich in omega-3 fatty acids. Food Res. Int. 2020, 134, 109216. [Google Scholar] [CrossRef]
- Fan, H.; Song, J.T. Potential mechanisms of macular degeneration protection by fatty fish consumption. Curr. Opin. Pharmacol. 2022, 63, 102186. [Google Scholar] [CrossRef]
- Leung, H.H.; Ng, A.L.K.; Durand, T.; Kawasaki, R.; Oger, C.; Balas, L.; Galano, J.-M.; Wong, I.Y.H.; Lee, J.C.-Y. Increase in omega-6 and decrease in omega-3 polyunsaturated fatty acid oxidation elevates the risk of exudative AMD development in adults with Chinese diet. Free Radic. Biol. Med. 2019, 145, 349–356. [Google Scholar] [CrossRef]
- Clinical Trial. Resveratrol for Exudative Age-Related Macular Degeneration (AGED). Identifier: NCT02625376. Available online: https://clinicaltrials.gov/ct2/show/NCT02625376 (accessed on 1 April 2023).
- Garcia-Layana, A.; Recalde, S.; Hernandez, M.; Abraldes, M.J.; Nascimento, J.; Hernandez-Galilea, E.; Olmedilla-Alonso, B.; Escobar-Barranco, J.J.; Zapata, M.A.; Silva, R.; et al. A Randomized Study of Nutritional Supplementation in Patients with Unilateral Wet Age-Related Macular Degeneration. Nutrients 2021, 13, 1253. [Google Scholar] [CrossRef]
- Kowalski, M.; Bielecka-Kowalska, A.; Oszajca, K.; Eusebio, M.; Jaworski, P.; Bartkowiak, J.; Szemraj, J. Manganese superoxide dismutase (MnSOD) gene (Ala-9Val, Ile58Thr) polymorphism in patients with age-related macular degeneration (AMD). Med. Sci. Monit. 2010, 16, Cr190–Cr196. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Antioxidants | Chemical Nature | Dietary Sources | Functions | Effects in AMD | References |
|---|---|---|---|---|---|
| Clinical Studies | |||||
| Vitamins C and E | Vitamin C as ascorbic acid; Vitamins E as tocopherol and tocotrienol. | Vitamin C: citrus fruits, berries, and red pepper Vitamin E: nuts, seeds, greens, whole grains. | Vitamin C reduces oxidative stress and promotes immune protection. Vitamin E maintains retinal structure and function and scavenges peroxyl radicals. | Low intake was associated with neovascular AMD. Protective against AMD progression as a component of AREDS and AREDS2 formula. | [221,222,253,254,255,256] |
| Vitamin A, β-carotene | Pure form of vitamin A as retinol; β-carotene as precursor of vitamin A. | Green leafy vegetables, fruits, dairy products, fish, and eggs. | Vitamin A aids formation of photopigment to support vision, also quenches free radicals. | β-carotene is protective against AMD progression as a component of AREDS formula. | [221,222,229,256,257,258] |
| Zinc | Essential trace mineral, and micronutrients | Oysters, meat, seafood, fish, poultry, cereals, and grains. | Functions as a co-factor of antioxidant enzymes and protects against oxidative damage. | Significantly reduced the risk of developing advanced AMD, as a component of AREDS and AREDS2 formula. | [221,222,259] |
| Lutein and zeaxanthin. | Xanthophyll family of carotenoids | Green leafy vegetables, egg yolks, yellow-orange fruits. | Functions as components of macular pigment, filters blue light and quenches single oxygen and free radicals. | Reduces the risk of AMD progression. Replaces β-carotene in AREDS2 formula to reduce cancer risk. | [222,260,261,262,263] |
| ω-3 long-chain polyunsaturated fatty acid (LCPUFA) | Mainly as eicosapentaenoic (EPA, 20:5 ω-3), and docosahexaenoic (DHA, 22:6 ω-3) acids | Nuts and seeds, plant oils, nuts, fish, and seafood. | Supports photoreceptor membrane structure, reduces inflammation, and limits oxidative stress. | Lower intake increases the risk of neovascular AMD, and higher intake was associated with decreased risk of AMD in prospective studies. Yet addition to AREDS2 did not further reduce risk of AMD progression. | [222,261,264,265,266,267,268] |
| Resveratrol | Polyphenols | Grape juices, wines, berries, and peanuts. | Protects against oxidative stress, anti-inflammatory. | Addition to AREDS EU formula did not have significant effects in wet AMD. Another clinical trial result pending. | [269,270] |
| Cu/Zn superoxide dismutase (SOD1) | Antioxidant enzymes | Supplementing copper may increase SOD levels in endogenous synthesis. | Scavenges superoxide anion and protects against oxidative stress. | Decreased in erythrocytes and serum of AMD patients in a few studies yet increased in other studies reflecting potential compensation. Increased SOD protein observed in AMD doner eyes. | [125,128,129,130,131] |
| MnSOD (SOD2) | Antioxidant enzymes | Manganese is required for endogenous synthesis. | A mitochondrial enzyme detoxifies free radicals from mitochondrial respiration. | Lowest expression was observed in wet and dry form of AMD, with genetic correlation. Increased protein levels in AMD donor eyes. | [125,271] |
| Experimental Studies | |||||
| ω-3 long-chain polyunsaturated fatty acid (LCPUFA) | Mainly as eicosapentaenoic (EPA, 20:5 ω-3), and docosahexaenoic (DHA, 22:6 ω-3) acids | Nuts and seeds, plant oils, nuts, fish, and seafood. | Supports photoreceptor membrane structure, reduces inflammation, and limits oxidative stress. | Inhibits laser-induced CNV in mouse models. | [227,238,239,240,241] |
| Resveratrol | Polyphenols | Grape juices, wines, berries, peanuts. | Protects against oxidative stress, anti-inflammatory. | Suppresses laser-induced CNV. Prevents excessive VEGF production. | [233,234,235,236,237] |
| SOD1 and SOD2 | Antioxidant enzymes | Endogenous synthesis. Requires trace minerals Cu, Zn, Mn. | Scavenges superoxide anion in cytosol and in mitochondria and protects against oxidative stress. | SOD1 knockout and SOD2 down lead to AMD-like features in mice with RPE degeneration. A small fraction of SOD1 knockout mice develop CNV. | [142,143,144] |
| Glutathione (GSH) | Tripeptide consisting of three amino acids: glutamic acid, cysteine, and glycine | Produced endogenously in the liver. Adding sulfur- and selenium-rich food helps. | Scavenges electrophilic and oxidant species to reduced oxidative stress. | Protects RPE cells from oxidative damage in ARPE19 and primary RPE isolated from AMD donors. | [248,249,250,251] |
| N-acetyl-cysteine (NAC) | A precursor of L-cysteine, aids synthesis of glutathione | Seafood, chicken, turkey, fish, protein-rich foods. | Antioxidant and cytoprotectant. | Reduces oxidative damage in RPE and suppresses laser-induced CNV in mice. | [251,252] |
| Cerium oxide nanoparticles (CeO2-NPs, nanoceria) | Nanomaterial | Not applicable. Synthetic material. | Inorganic antioxidants mimicking SOD and catalase. | Prevents retinal neovascularization in vitro and in Vldlr−/− mice. Protects RPE from oxidative damage. | [242,243,244,245,246] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kushwah, N.; Bora, K.; Maurya, M.; Pavlovich, M.C.; Chen, J. Oxidative Stress and Antioxidants in Age-Related Macular Degeneration. Antioxidants 2023, 12, 1379. https://doi.org/10.3390/antiox12071379
Kushwah N, Bora K, Maurya M, Pavlovich MC, Chen J. Oxidative Stress and Antioxidants in Age-Related Macular Degeneration. Antioxidants. 2023; 12(7):1379. https://doi.org/10.3390/antiox12071379
Chicago/Turabian StyleKushwah, Neetu, Kiran Bora, Meenakshi Maurya, Madeline C. Pavlovich, and Jing Chen. 2023. "Oxidative Stress and Antioxidants in Age-Related Macular Degeneration" Antioxidants 12, no. 7: 1379. https://doi.org/10.3390/antiox12071379
APA StyleKushwah, N., Bora, K., Maurya, M., Pavlovich, M. C., & Chen, J. (2023). Oxidative Stress and Antioxidants in Age-Related Macular Degeneration. Antioxidants, 12(7), 1379. https://doi.org/10.3390/antiox12071379