
Aging, NRF2, and TAU: A Perfect Match for Neurodegeneration?



Abstract
: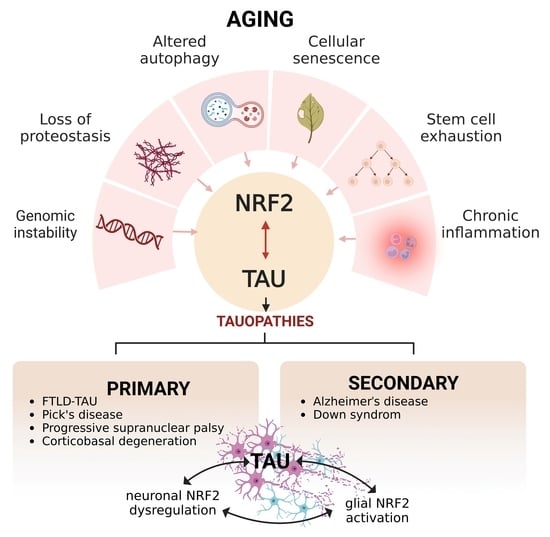
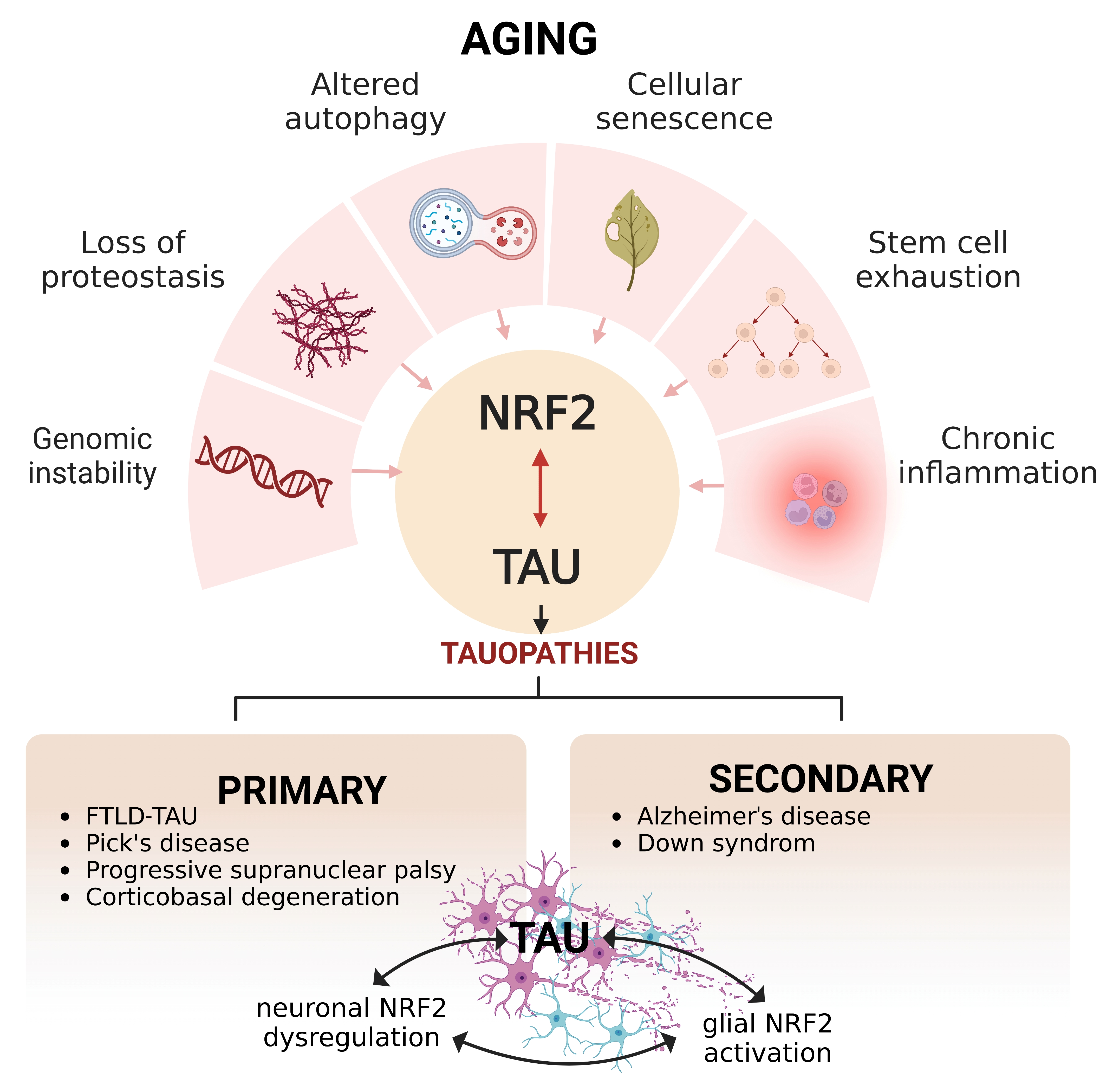{kind=link}
{kind=link}
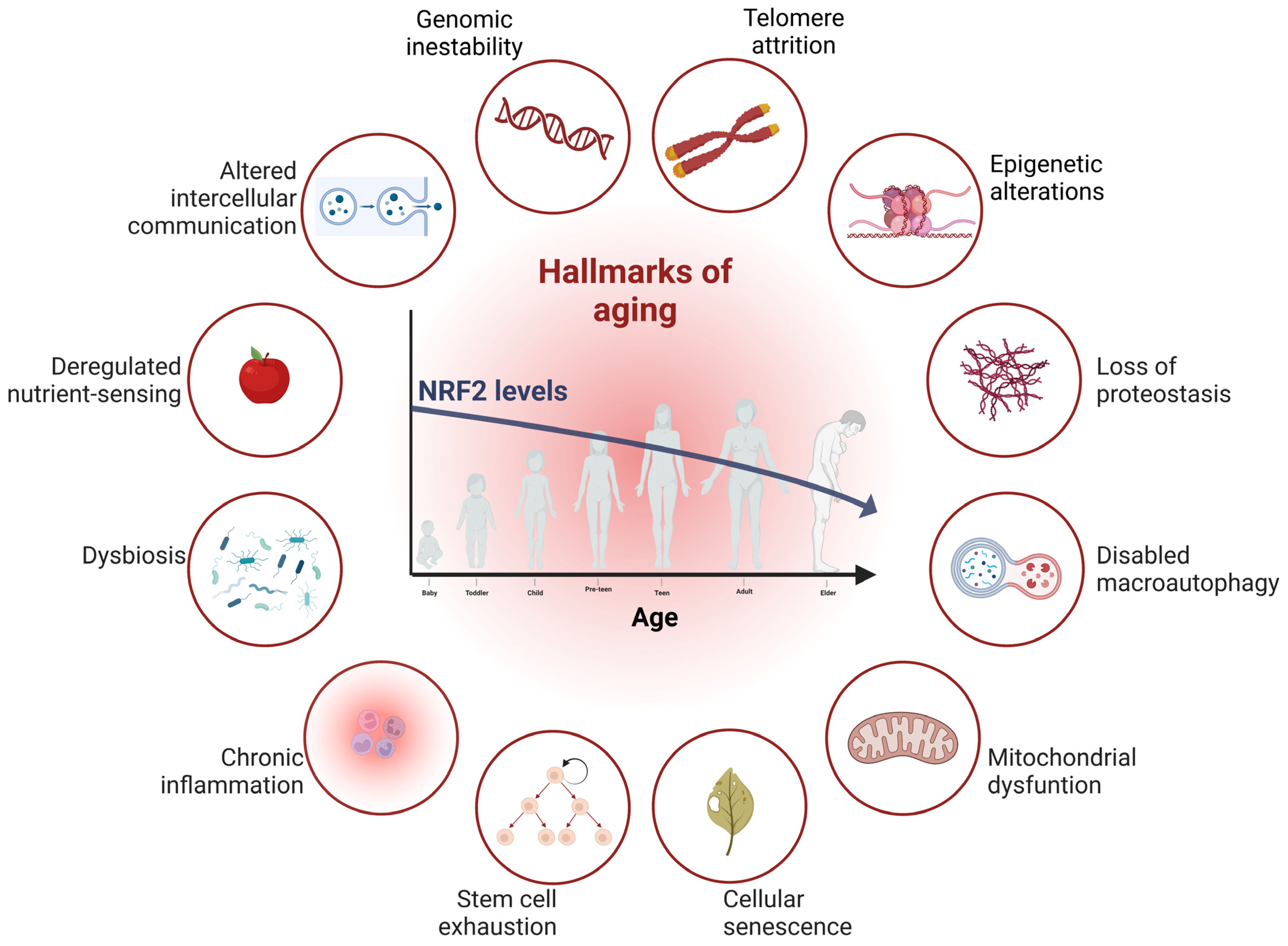{kind=link}
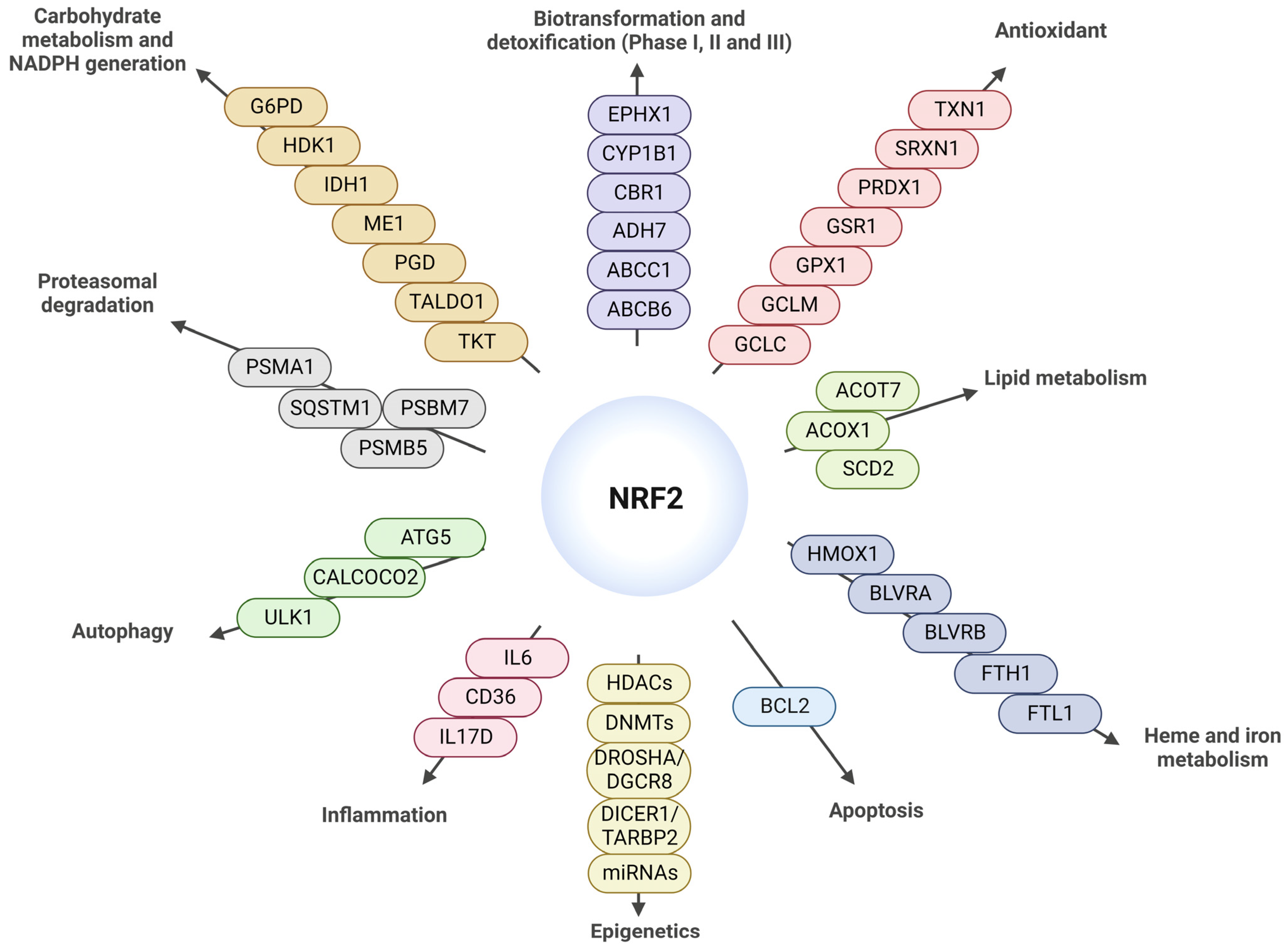{kind=link}
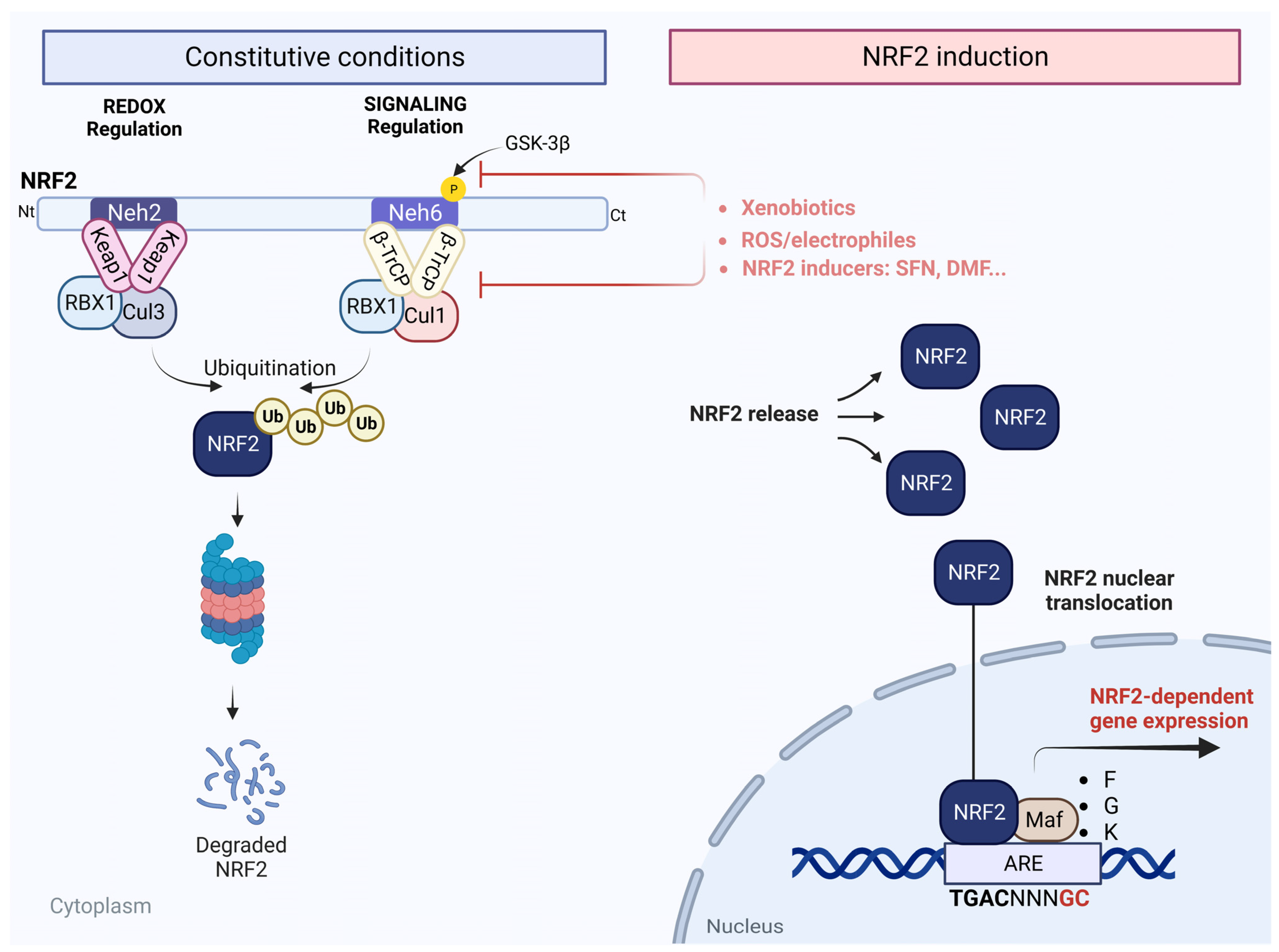
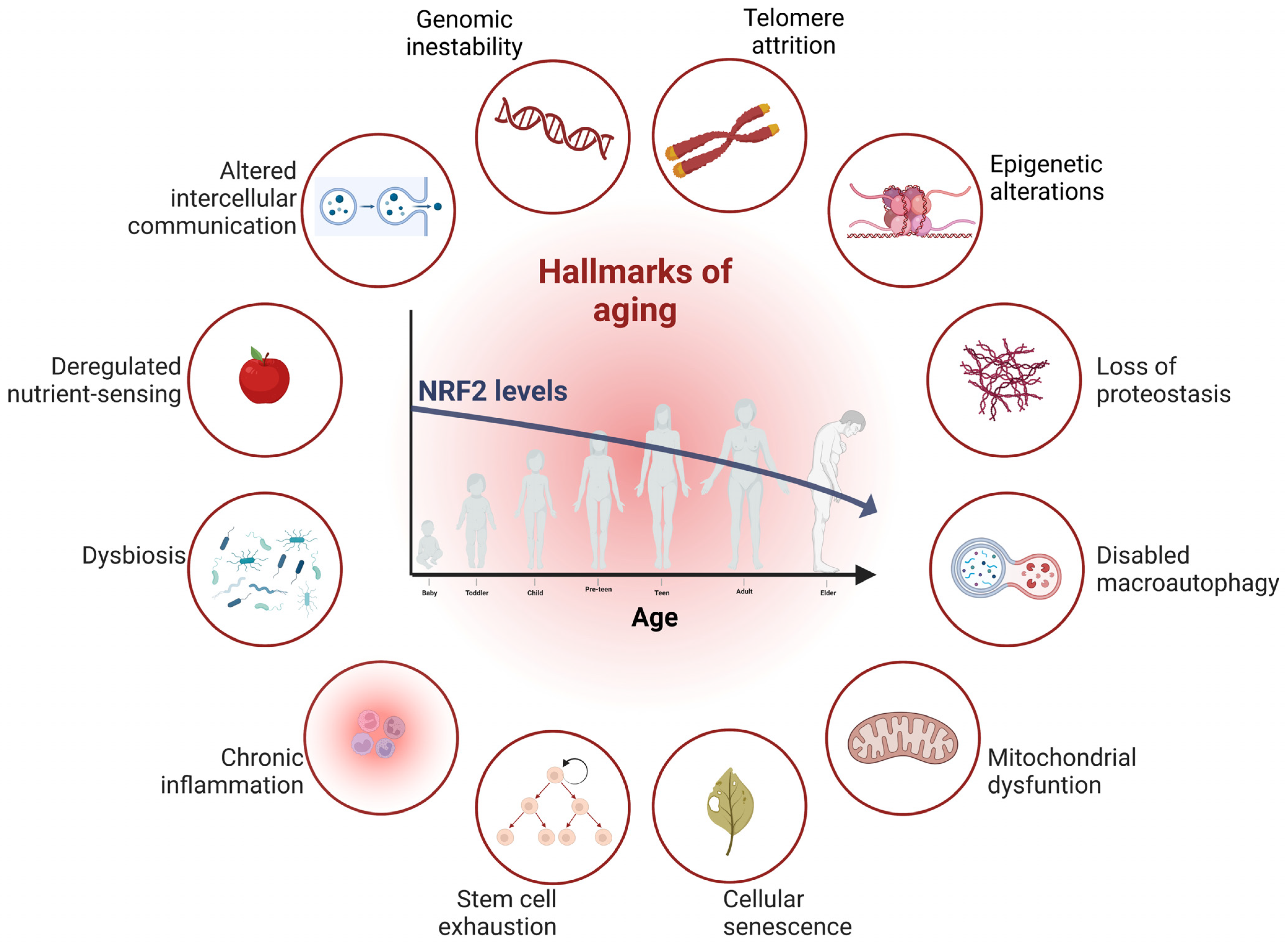
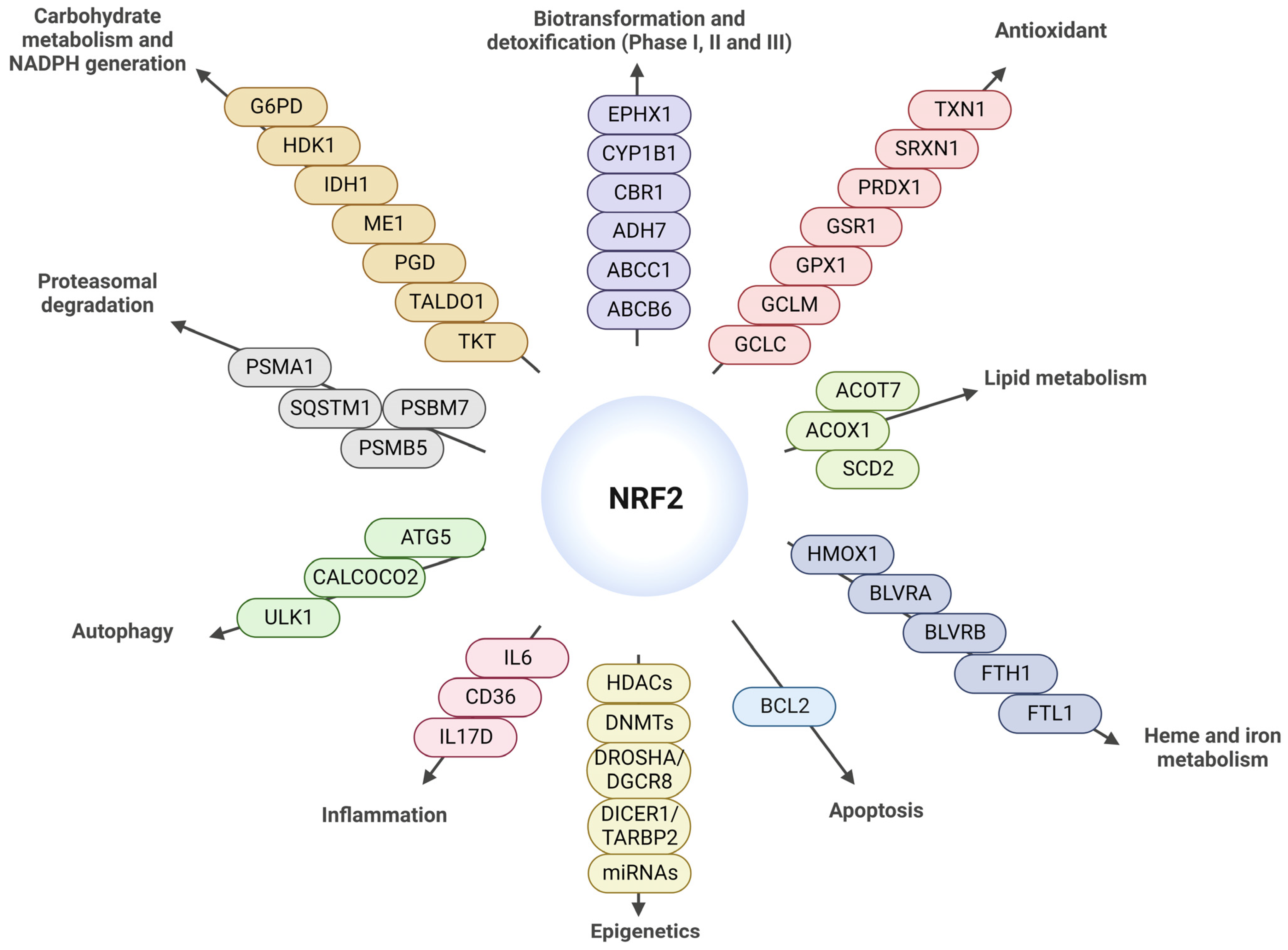
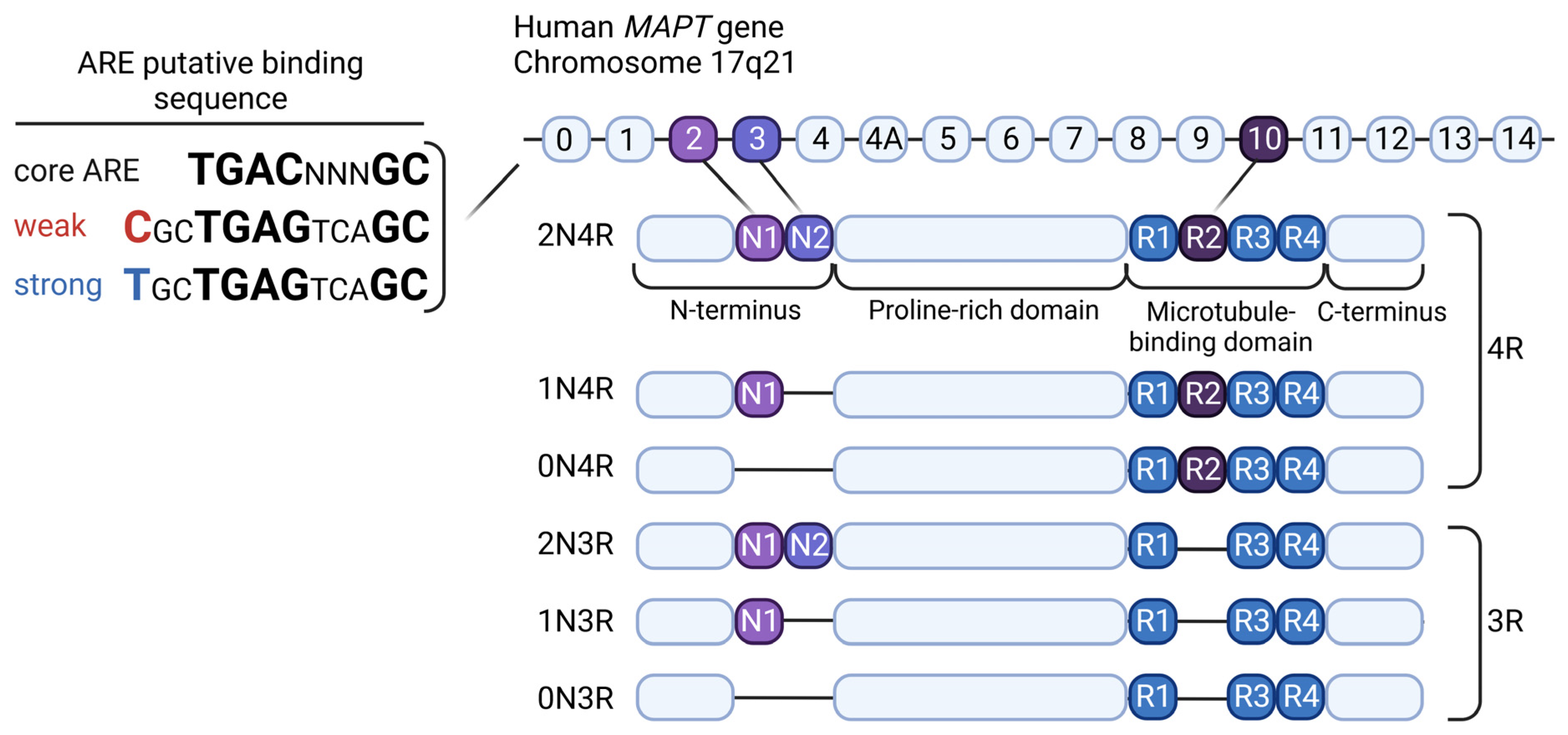{kind=link}
{kind=link}
1. Introduction
2. The NRF2 Pathway
3. Aging and Its Connection to NRF2
3.1. Genomic Instability and NRF2
3.2. NRF2 Is a Regulator of Protein Degradation Systems, Proteasomes, and Autophagy
3.3. Implication of NRF2 in Cellular Senescence and Stem Cell Exhaustion
3.4. NRF2 as an Inflammation Modulator
4. TAU and Its Modulation during Aging
5. Aging, Mitochondrial Dysfunction, and Ferroptosis: Role of NRF2 and TAU
6. NRF2 and TAU Interconnection
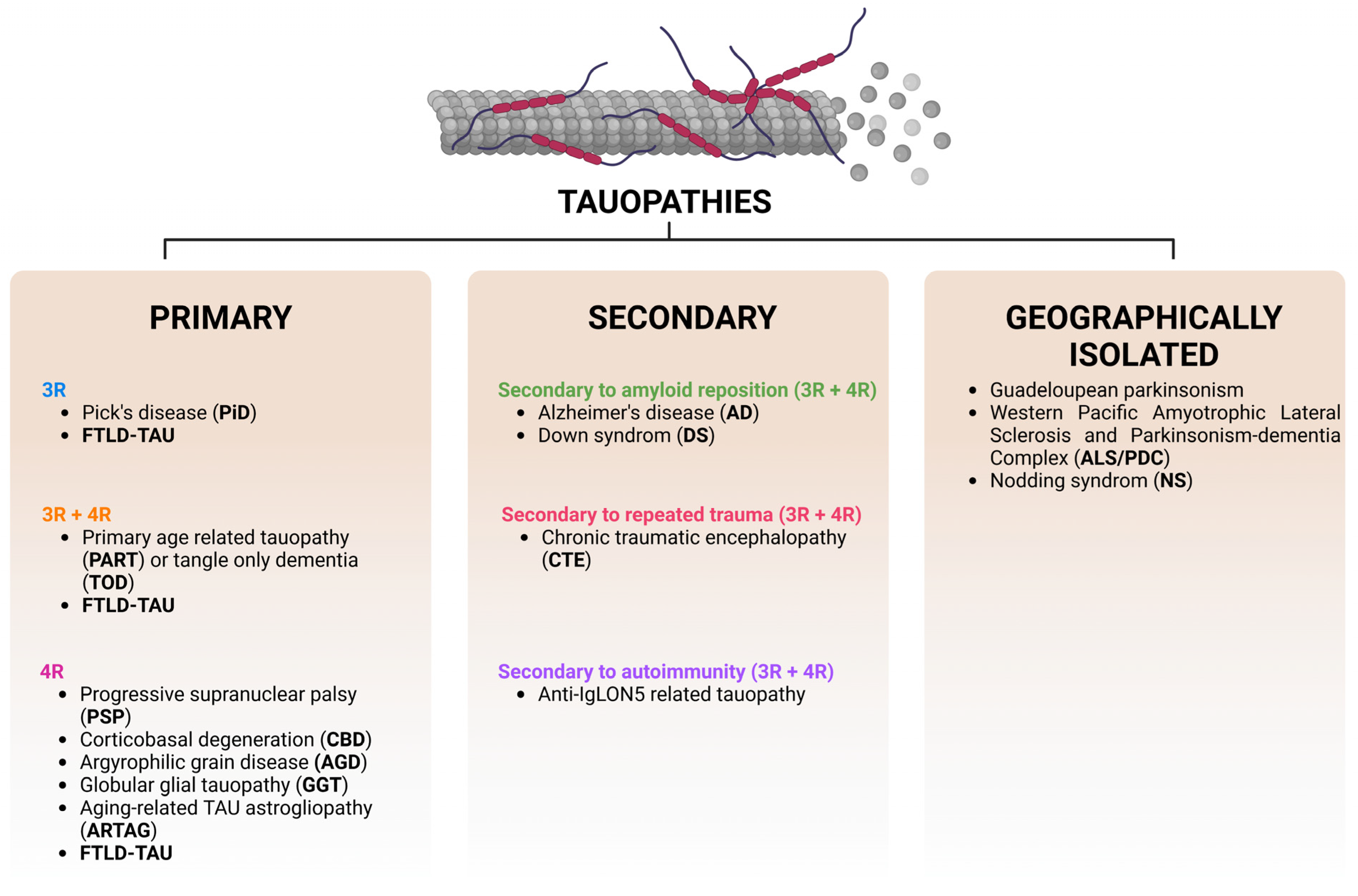
7. Types of Tauopathies and Their Main Molecular Characteristics
8. Primary Tauopathies and Their Connection to NRF2
8.1. Relevance of NRF2 in FTLD
8.2. Involvement of NRF2 in Pick’s Disease
8.3. Role of NRF2 in PSP and CBD
9. Secondary Tauopathies and Their Link to NRF2
9.1. Implication of NRF2 in AD
9.2. Role of NRF2 in DS
9.3. Role of NRF2 in CTE
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Itoh, K.; Igarashi, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Cloning and Characterization of a Novel Erythroid Cell-Derived CNC Family Transcription Factor Heterodimerizing with the Small Maf Family Proteins. Mol. Cell Biol. 1995, 15, 4184–4193. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP Promotes Glycogen Synthase Kinase 3-Dependent Degradation of the Nrf2 Transcription Factor in a Keap1-Independent Manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobón-Velasco, J.C.; Devijver, H.; García-Mayoral, M.F.; van Leuven, F.; et al. Structural and Functional Characterization of Nrf2 Degradation by the Glycogen Synthase Kinase 3/β-TrCP Axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/β-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lv, Y.-F.; Zhao, J.-L.; You, Q.-D.; Jiang, Z.-Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci. USA 2000, 97, 12475–12480. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by Protein Kinase C Regulates Antioxidant Response Element-mediated Transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, S. Upregulation of heme oxygenase-1 expression by dehydrodiconiferyl alcohol (DHCA) through the AMPK-Nrf2 dependent pathway. Toxicol. Appl. Pharmacol. 2014, 281, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [Green Version]
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol. Cell Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Blasco, D.; Santofimia-Castaño, P.; Gonzalez, A.; Almeida, A.; Bolaños, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Vrba, J.; Gažák, R.; Kuzma, M.; Papoušková, B.; Vacek, J.; Weiszenstein, M.; Křen, V.; Ulrichová, J. A Novel Semisynthetic Flavonoid 7-O-Galloyltaxifolin Upregulates Heme Oxygenase-1 in RAW264.7 Cells via MAPK/Nrf2 Pathway. J. Med. Chem. 2013, 56, 856–866. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [Green Version]
- Matsumaru, D.; Motohashi, H. The KEAP1-NRF2 System in Healthy Aging and Longevity. Antioxidants 2021, 10, 1929. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B.; Madhavan, L.; Zhang, D.D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [Green Version]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L.; Zou, Y.; Hu, M.; Lee, J.; Nambiar, S.M.; Garcia, V.; Bao, Q.; et al. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Liver Physiol. 2010, 299, G1211–G1221. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Lastres-Becker, I.; Innamorato, N.G.; Jaworski, T.; Rábano, A.; Kügler, S.; van Leuven, F.; Cuadrado, A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 2014, 137, 78–91. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Ulusoy, A.; Innamorato, N.G.; Sahin, G.; Rábano, A.; Kirik, D.; Cuadrado, A.I. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar] [CrossRef] [Green Version]
- Bourgonje, A.R.; Kloska, D.; Grochot-Przęczek, A.; Feelisch, M.; Cuadrado, A.; van Goor, H. Personalized redox medicine in inflammatory bowel diseases: An emerging role for HIF-1α and NRF2 as therapeutic targets. Redox Biol. 2023, 60, 102603. [Google Scholar] [CrossRef] [PubMed]
- Silva-Llanes, I.; Shin, C.H.; Jiménez-Villegas, J.; Gorospe, M.; Lastres-Becker, I. The Transcription Factor NRF2 Has Epigenetic Regulatory Functions Modulating HDACs, DNMTs, and miRNA Biogenesis. Antioxidants 2023, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; García-Yagüe, Á.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Rojo, A.I.; Arias, E.; Díaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Jiang, C.; Gao, J. Transcriptional factor Nrf2 is essential for aggresome formation during proteasome inhibition. Biomed. Rep. 2019, 11, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Tsakiri, E.N.; Sykiotis, G.P.; Papassideri, I.S.; Terpos, E.; Dimopoulos, M.A.; Gorgoulis, V.G.; Bohmann, D.; Trougakos, I.P. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 2013, 12, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Kensler, T.W. Induction of 26S proteasome subunit PSMB5 by the bifunctional inducer 3-methylcholanthrene through the Nrf2-ARE, but not the AhR/Arnt-XRE, pathway. Biochem. Biophys. Res. Commun. 2006, 345, 1350–1357. [Google Scholar] [CrossRef]
- Lodato, M.A.; Ziegenfuss, J.S. The two faces of DNA oxidation in genomic and functional mosaicism during aging in human neurons. Front. Aging 2022, 3, 991460. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, J.; Schwer, B.; El-Khamisy, S.F. Editorial: Genomic Instability and Neurodegeneration. Front. Aging Neurosci. 2022, 14, 940459. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, Y.; Ji, K.; Liu, Y.; Kong, Y.; Nie, S.; Na Li, N.; Hao, J.; Xie, Y.; Xu, C.; et al. NRF2 preserves genomic integrity by facilitating ATR activation and G2 cell cycle arrest. Nucleic Acids Res. 2020, 48, 9109–9123. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, C.; Liu, Q. Roles of NRF2 in DNA damage repair. Cell. Oncol. 2023. [Google Scholar] [CrossRef]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Frankowska, N.; Lisowska, K.; Witkowski, J.M. Proteolysis dysfunction in the process of aging and age-related diseases. Front. Aging 2022, 3, 927630. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Tabibzadeh, S. Role of autophagy in aging: The good, the bad, and the ugly. Aging Cell 2023, 22, e13753. [Google Scholar] [CrossRef]
- Koizumi, S.; Hamazaki, J.; Murata, S. Transcriptional regulation of the 26S proteasome by Nrf1. Proc. Jpn. Acad. Ser. B 2018, 94, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, M.-K.; Cho, J.-M.; Huang, B.; Shin, S.; Kensler, T.W. Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic. Biol. Med. 2007, 43, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants Enhance Mammalian Proteasome Expression through the Keap1-Nrf2 Signaling Pathway. Mol. Cell Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.-K.; Wakabayashi, N.; Itoh, K.; Motohashi, H.; Yamamoto, M.; Kensler, T.W. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J. Biol. Chem. 2003, 278, 8135–8145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death Dis. 2019, 10, 510. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ma, S.; Zhao, X.; Wen, B.; Sun, P.; Fu, Z. Upregulation of antioxidant and autophagy pathways via NRF2 activation protects spinal cord neurons from ozone damage. Mol. Med. Rep. 2021, 23, 12067. [Google Scholar] [CrossRef]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.-M.; Mandelkow, E.; Cuervo, A.M. Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 2018, 17, e12692. [Google Scholar] [CrossRef] [Green Version]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.-J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct Interaction between Nrf2 and p21Cip1/WAF1 Upregulates the Nrf2-Mediated Antioxidant Response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Xu, Y.; Luo, Y.; Wang, N.-X.; Xiao, J.-H. Role of Nrf2 in cell senescence regulation. Mol. Cell Biochem. 2020, 476, 247–259. [Google Scholar] [CrossRef]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.-D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef] [Green Version]
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, dev156059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicaise, A.M.; Willis, C.M.; Crocker, S.J.; Pluchino, S. Stem Cells of the Aging Brain. Front. Aging Neurosci. 2020, 12, 247. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, D.M.; Brown, D.R. Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J. Neurochem. 2019, 151, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.; Wang, Y.; Kim, H.-S.; Lalli, M.A.; Kosik, K.S. Nrf2, a Regulator of the Proteasome, Controls Self-Renewal and Pluripotency in Human Embryonic Stem Cells. Stem Cells 2014, 32, 2616–2625. [Google Scholar] [CrossRef] [Green Version]
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robledinos-Antón, N.; Rojo, A.I.; Ferreiro, E.; Núñez, Á.; Krause, K.-H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Anandhan, A.; Zhang, D.D.; Madhavan, L. An NRF2 Perspective on Stem Cells and Ageing. Front. Aging 2021, 2, 690686. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Yan, X.; Wintergerst, K.A.; Cai, L.; Keller, B.B.; Tan, Y. Nrf2: Redox and Metabolic Regulator of Stem Cell State and Function. Trends Mol. Med. 2019, 26, 185–200. [Google Scholar] [CrossRef] [Green Version]
- Kahroba, H.; Ramezani, B.; Maadi, H.; Sadeghi, M.R.; Jaberie, H.; Ramezani, F. The role of Nrf2 in neural stem/progenitors cells: From maintaining stemness and self-renewal to promoting differentiation capability and facilitating therapeutic application in neurodegenerative disease. Ageing Res. Rev. 2021, 65, 101211. [Google Scholar] [CrossRef]
- Meldolesi, J. Role of Senescent Astrocytes in Health and Disease. Int. J. Mol. Sci. 2023, 24, 8498. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System during Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Antignano, I.; Liu, Y.; Offermann, N.; Capasso, M. Aging microglia. Cell. Mol. Life Sci. 2023, 80, 126. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Vicente-Gutierrez, C.; Bonora, N.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Bates, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Abrogating mitochondrial ROS in neurons or astrocytes reveals cell-specific impact on mouse behaviour. Redox Biol. 2021, 41, 101917. [Google Scholar] [CrossRef]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription Factors NRF2 and NF-κB Are Coordinated Effectors of the Rho Family, GTP-binding Protein RAC1 during Inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [Green Version]
- Lan, T.; Li, Y.; Fan, C.; Wang, L.; Wang, W.; Chen, S.; Yu, S.Y. MicroRNA-204-5p reduction in rat hippocampus contributes to stress-induced pathology via targeting RGS12 signaling pathway. J. Neuroinflamm. 2021, 18, 243. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.K.; Datusalia, A.K.; Sharma, S.S. Posttranscriptional regulation of Nrf2 through miRNAs and their role in Alzheimer’s disease. Pharmacol. Res. 2022, 175, 106018. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Boyarko, B.; Hook, V. Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neurodegeneration. Front. Neurosci. 2021, 15, 702788. [Google Scholar] [CrossRef]
- Avila, J.; de Barreda, E.G.; Pallas-Bazarra, N.; Hernandez, F. Tau and neuron aging. Aging Dis. 2013, 4, 23–28. [Google Scholar]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Höglinger, G.U.; et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol. 2017, 133, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Juan, P.; Moreno, S.; de Rojas, I.; Hernández, I.; Valero, S.; Alegret, M.; Montrreal, L.; González, P.G.; Lage, C.; López-García, S.; et al. The MAPT H1 Haplotype Is a Risk Factor for Alzheimer’s Disease in APOE ε4 Non-carriers. Front. Aging Neurosci. 2019, 11, 327. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Siahaan, V.; Tan, R.; Humhalova, T.; Libusova, L.; Lacey, S.E.; Tan, T.; Dacy, M.; Ori-McKenney, K.M.; McKenney, R.J.; Braun, M.; et al. Microtubule lattice spacing governs cohesive envelope formation of tau family proteins. Nat. Chem. Biol. 2022, 18, 1224–1235. [Google Scholar] [CrossRef]
- Nunez, W.A.; Combs, B.; Gamblin, T.C.; Ackley, B.D. Age-dependent accumulation of tau aggregation in Caenorhabditis elegans. Front. Aging 2022, 3, 928574. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Sealey, M.; Ruiz, E.; Pegasiou, C.M.; Brookes, K.; Green, S.; Crisford, A.; Duque-Vasquez, M.; Luckett, E.; Robertson, R.; et al. Age-related changes in tau and autophagy in human brain in the absence of neurodegeneration. PLoS ONE 2023, 18, e0262792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 9, 707268. [Google Scholar] [CrossRef]
- Wegmann, S.; Bennett, R.E.; Delorme, L.; Robbins, A.B.; Hu, M.; MacKenzie, D.; Kirk, M.J.; Schiantarelli, J.; Tunio, N.; Amaral, A.C.; et al. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019, 5, eaaw6404. [Google Scholar] [CrossRef] [Green Version]
- Park, S.A.; Ahn, S.I.; Gallo, J.-M. Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep. 2016, 49, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Campbell, M.R.; Lacher, S.E.; Cho, H.-Y.; Wan, M.; Crowl, C.L.; Chorley, B.N.; Bond, G.L.; Kleeberger, S.R.; Slattery, M.; et al. A Polymorphic Antioxidant Response Element Links NRF2/sMAF Binding to Enhanced MAPT Expression and Reduced Risk of Parkinsonian Disorders. Cell Rep. 2016, 15, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Mitochondria in neurodegeneration. Curr. Opin. Physiol. 2022, 26, 100532. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Zhang, Y.; Abramov, A.Y.; Dinkova-Kostova, A.T. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem. J. 2014, 457, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Impacts Cellular Bioenergetics by Controlling Substrate Availability for Mitochondrial Respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Cvetko, F.; Caldwell, S.T.; Higgins, M.; Suzuki, T.; Yamamoto, M.; Prag, H.A.; Hartley, R.C.; Dinkova-Kostova, A.T.; Murphy, M.P. Nrf2 is activated by disruption of mitochondrial thiol homeostasis but not by enhanced mitochondrial superoxide production. J. Biol. Chem. 2021, 296, 100169. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Shilovsky, G.A.; Ashapkin, V.V. Transcription Factor Nrf2 and Mitochondria—Friends or Foes in the Regulation of Aging Rate. Biochemistry 2022, 87, 1477–1486. [Google Scholar] [CrossRef]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef]
- Eckert, A.; Nisbet, R.; Grimm, A.; Götz, J. March separate, strike together—Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau Accumulation Causes Mitochondrial Distribution Deficits in Neurons in a Mouse Model of Tauopathy and in Human Alzheimer’s Disease Brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; de Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. An Update on Mitochondrial Reactive Oxygen Species Production. Antioxidants 2020, 9, 472. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Carter, B.Z.; Andreeff, M.; Ishizawa, J. Molecular Mechanisms of Ferroptosis and Updates of Ferroptosis Studies in Cancers and Leukemia. Cells 2023, 12, 1128. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.; Liu, P.; Wei, Y.; Tan, H.; Wang, Q.; Jiang, Z.; Yang, K.; et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, eade9585. [Google Scholar] [CrossRef]
- Anandhan, A.; Dodson, M.; Schmidlin, C.J.; Liu, P.; Zhang, D.D. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 2020, 27, 436–447. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, Y.; Liu, Y.; Liu, Q.; Sun, H.; Mei, M.; Liao, X. Ferroptosis promotes microtubule-associated protein tau aggregation via GSK-3β activation and proteasome inhibition. Mol. Neurobiol. 2022, 59, 1486–1501. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Martinez-Valbuena, I.; de Andrea, C.E.; Villalba-Esparza, M.; Ilaalagan, S.; Couto, B.; Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Cell-Specific Dysregulation of Iron and Oxygen Homeostasis as a Novel Pathophysiology in PSP. Ann. Neurol. 2022, 93, 431–445. [Google Scholar] [CrossRef]
- Odetti, P.; Garibaldi, S.; Norese, R.; Angelini, G.; Marinelli, L.; Valentini, S.; Menini, S.; Traverso, N.; Zaccheo, D.; Siedlak, S.; et al. Lipoperoxidation Is Selectively Involved in Progressive Supranuclear Palsy. J. Neuropathol. Exp. Neurol. 2000, 59, 393–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenough, M.A.; Lane, D.J.R.; Balez, R.; Anastacio, H.T.D.; Zeng, Z.; Ganio, K.; McDevitt, C.A.; Acevedo, K.; Belaidi, A.A.; Koistinaho, J.; et al. Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differ. 2022, 29, 2123–2136. [Google Scholar] [CrossRef]
- Derry, P.J.; Hegde, M.L.; Jackson, G.R.; Kayed, R.; Tour, J.M.; Tsai, A.-L.; Kent, T.A. Revisiting the intersection of amyloid, pathologically modified tau and iron in Alzheimer’s disease from a ferroptosis perspective. Prog. Neurobiol. 2020, 184, 101716. [Google Scholar] [CrossRef]
- Jakaria, M.; Belaidi, A.A.; Bush, A.I.; Ayton, S. Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J. Neurochem. 2021, 159, 804–825. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.-F.; Zou, T.; Tuo, Q.-Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Caffrey, T.M.; Joachim, C.; Wade-Martins, R. Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol. Aging 2008, 29, 1923–1929. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, T.M.; Wade-Martins, R. The role of MAPT sequence variation in mechanisms of disease susceptibility. Biochem. Soc. Trans. 2012, 40, 687–692. [Google Scholar] [CrossRef]
- Lai, M.C.; Bechy, A.-L.; Denk, F.; Collins, E.; Gavriliouk, M.; Zaugg, J.B.; Ryan, B.J.; Wade-Martins, R.; Caffrey, T.M. Haplotype-specific MAPT exon 3 expression regulated by common intronic polymorphisms associated with Parkinsonian disorders. Mol. Neurodegener. 2017, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V.W. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 4496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhang, S.; Zheng, H. The cargo receptor SQSTM1 ameliorates neurofibrillary tangle pathology and spreading through selective targeting of pathological MAPT (microtubule associated protein tau). Autophagy 2019, 15, 583–598. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Ji, C.; Pallo, S.; Rahman, I.; Johnson, G.V. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age-dependent manner. Neurobiol. Aging 2018, 63, 128–139. [Google Scholar] [CrossRef]
- Chung, D.-E.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Natl. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Holper, S.; Watson, R.; Yassi, N. Tau as a Biomarker of Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 7307. [Google Scholar] [CrossRef]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Ganguly, J.; Jog, M. Tauopathy and Movement Disorders—Unveiling the Chameleons and Mimics. Front. Neurol. 2020, 11, 599384. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.; Falcon, B.; Kotecha, A.; van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-based Classification of Tauopathies. Nature 2021, 598, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Ruiz, A.; Sánchez-Juan, P. Genetic Architecture of Primary Tauopathies. Neuroscience 2023, 518, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, K.-M.; Yang, L.; Dong, Q.; Yu, J.-T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 2022, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Newcombe, E.A.; Camats-Perna, J.; Silva, M.L.; Valmas, N.; Huat, T.J.; Medeiros, R. Inflammation: The link between comorbidities, genetics, and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langworth-Green, C.; Patel, S.; Jaunmuktane, Z.; Jabbari, E.; Morris, H.; Thom, M.; Lees, A.; Hardy, J.; Zandi, M.; Duff, K. Chronic effects of inflammation on tauopathies. Lancet Neurol. 2023, 22, 430–442. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Esteras, N.; Rohrer, J.D.; Hardy, J.; Wray, S.; Abramov, A.Y. Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10 + 16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 2017, 12, 410–422. [Google Scholar] [CrossRef]
- Albers, D.S.; Beal, M. Mitochondrial dysfunction in progressive supranuclear palsy. Neurochem. Int. 2002, 40, 559–564. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative Damage Is the Earliest Event in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.A.; Galvis-Escobar, S.; Abisambra, J.F. Tau-mediated dysregulation of RNA: Evidence for a common molecular mechanism of toxicity in frontotemporal dementia and other tauopathies. Neurobiol. Dis. 2020, 141, 104939. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.L.; Wilhelmsen, K.; Sima, A.A.F.; Jones, M.Z.; D’Amato, C.J.; Gilman, S.; Participants, C. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference participants. Ann. Neurol. 1997, 41, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; de Lago, E.; Martínez, A.; Fernández-Ruiz, J. New Statement about NRF2 in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Biomolecules 2022, 12, 1200. [Google Scholar] [CrossRef]
- Dumanchin, C.; Camuzat, A.; Campion, D.; Verpillat, P.; Hannequin, D.; Dubois, B.; Saugier-Veber, P.; Martin, C.; Penet, C.; Charbonnier, F.; et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum. Mol. Genet. 1998, 7, 1825–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Bugiani, O.; Murrell, J.R.; Giaccone, G.; Hasegawa, M.; Ghigo, G.; Tabaton, M.; Morbin, M.; Primavera, A.; Carella, F.; Solaro, C.; et al. Frontotemporal Dementia and Corticobasal Degeneration in a Family with a P301S Mutation in Tau. J. Neuropathol. Exp. Neurol. 1999, 58, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Zhang, Y.; Li, S.; Wei, H.; Yu, H.; Zhou, Q.; Wei, L.; Ke, D.; Wang, Q.; Yang, Y.; et al. P301S-hTau acetylates KEAP1 to trigger synaptic toxicity via inhibiting NRF2/ARE pathway: A novel mechanism underlying hTau-induced synaptic toxicities. Clin. Transl. Med. 2022, 12, e1003. [Google Scholar] [CrossRef]
- Castro-Sánchez, S.; García-Yagüe, Á.J.; Kügler, S.; Lastres-Becker, I. CX3CR1-deficient microglia shows impaired signalling of the transcription factor NRF2: Implications in tauopathies. Redox Biol. 2019, 22, 101118. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef]
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive astrocytes acquire neuroprotective as well as deleterious signatures in response to Tau and Aß pathology. Nat. Commun. 2022, 13, 135. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, T.; Lechat, B.; Demedts, D.; Gielis, L.; Devijver, H.; Borghgraef, P.; Duimel, H.; Verheyen, F.; Kügler, S.; van Leuven, F. Dendritic Degeneration, Neurovascular Defects, and Inflammation Precede Neuronal Loss in a Mouse Model for Tau-Mediated Neurodegeneration. Am. J. Pathol. 2011, 179, 2001–2015. [Google Scholar] [CrossRef]
- Riordan, R.; Rong, W.; Yu, Z.; Ross, G.; Valerio, J.; Dimas-Muñoz, J.; Heredia, V.; Magnusson, K.; Galvan, V.; Perez, V.I. Effect of Nrf2 loss on senescence and cognition of tau-based P301S mice. GeroScience 2023. [Google Scholar] [CrossRef] [PubMed]
- Tamvaka, N.; Manne, S.; Kondru, N.; Ross, O.A. Pick’s Disease, Seeding an Answer to the Clinical Diagnosis Conundrum. Biomedicines 2023, 11, 1646. [Google Scholar] [CrossRef] [PubMed]
- Tacik, P.; DeTure, M.; Hinkle, K.M.; Lin, W.-L.; Sanchez-Contreras, M.; Carlomagno, Y.; Pedraza, O.; Rademakers, R.; Ross, O.A.; Wszolek, Z.K.; et al. A Novel Tau Mutation in Exon 12, p.Q336H, Causes Hereditary Pick Disease. J. Neuropathol. Exp. Neurol. 2015, 74, 1042–1052. [Google Scholar] [CrossRef]
- Siano, G.; Micaelli, M.; Scarlatti, A.; Quercioli, V.; di Primio, C.; Cattaneo, A. The Q336H MAPT Mutation Linked to Pick’s Disease Leads to Increased Binding of Tau to the Microtubule Network via Altered Conformational and Phosphorylation Effects. Front. Mol. Neurosci. 2020, 13, 569395. [Google Scholar] [CrossRef]
- Castellani, R.; Smith, M.; Richey, P.; Kalaria, R.; Gambetti, P.; Perry, G. Evidence for oxidative stress in Pick disease and corticobasal degeneration. Brain Res. 1995, 696, 268–271. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Naudí, A.; Kichev, A.; Ferrer, I.; Pamplona, R.; Portero-Otín, M. Depletion of oxidative and endoplasmic reticulum stress regulators in Pick disease. Free Radic. Biol. Med. 2010, 48, 1302–1310. [Google Scholar] [CrossRef]
- Koziorowski, D.; Figura, M.; Milanowski, M.; Szlufik, S.; Alster, P.; Madetko, N.; Friedman, A. Mechanisms of Neurodegeneration in Various Forms of Parkinsonism—Similarities and Differences. Cells 2021, 10, 656. [Google Scholar] [CrossRef]
- Ling, H.; Macerollo, A. Is it Useful to Classify PSP and CBD as Different Disorders? Yes. Mov. Disord. Clin. Pract. 2018, 5, 145–148. [Google Scholar] [CrossRef]
- Whiteside, D.J.; Street, D.; Murley, A.G.; Jones, P.S.; Malpetti, M.; Ghosh, B.C.P.; Coyle-Gilchrist, I.; Gerhard, A.; Hu, M.T.; Klein, J.C.; et al. Network connectivity and structural correlates of survival in progressive supranuclear palsy and corticobasal syndrome. Hum. Brain Mapp. 2023, 44, 4239–4255. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J. Neurol. 1999, 246 (Suppl. S2), II6–II15. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, S.L.; Kril, J.J.; Halliday, G.M. Cellular and regional vulnerability in frontotemporal tauopathies. Acta Neuropathol. 2019, 138, 705–727. [Google Scholar] [CrossRef]
- Bruch, J.; Xu, H.; Rösler, T.W.; de Andrade, A.; Kuhn, P.; Lichtenthaler, S.F.; Arzberger, T.; Winklhofer, K.F.; Müller, U.; Höglinger, G.U. PERK activation mitigates tau pathology in vitro and in vivo. EMBO Mol. Med. 2017, 9, 371–384. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, I.; Keller-McGandy, C.E.; Albers, D.S.; Beal, M.; Vonsattel, J.-P.; Standaert, D.G.; Augood, S.J. Expression and activity of antioxidants in the brain in progressive supranuclear palsy. Brain Res. 2002, 930, 170–181. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillón, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s Disease Prevention: From Risk Factors to Early Intervention. Alzheimer’s Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2023, 19, 658–670. [Google Scholar] [CrossRef]
- Halliday, G. Pathology and hippocampal atrophy in Alzheimer’s disease. Lancet Neurol. 2017, 16, 862–864. [Google Scholar] [CrossRef]
- Pini, L.; Pievani, M.; Bocchetta, M.; Altomare, D.; Bosco, P.; Cavedo, E.; Galluzzi, S.; Marizzoni, M.; Frisoni, G.B. Brain atrophy in Alzheimer’s Disease and aging. Ageing Res. Rev. 2016, 30, 25–48. [Google Scholar] [CrossRef]
- Jahn, H. Memory loss in Alzheimer’s disease. Dialog.-Clin. Neurosci. 2013, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Von Otter, M.; Landgren, S.; Nilsson, S.; Zetterberg, M.; Celojevic, D.; Bergström, P.; Minthon, L.; Bogdanovic, N.; Andreasen, N.; Gustafson, D.R.; et al. Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech. Ageing Dev. 2010, 131, 105–110. [Google Scholar] [CrossRef]
- Milanesi, E.; Dobre, M.; Cucos, C.A.; Rojo, A.I.; Jiménez-Villegas, J.; Capetillo-Zarate, E.; Matute, C.; Piñol-Ripoll, G.; Manda, G.; Cuadrado, A. Whole Blood Expression Pattern of Inflammation and Redox Genes in Mild Alzheimer’s Disease. J. Inflamm. Res. 2021, 14, 6085–6102. [Google Scholar] [CrossRef]
- Vogrinc, D.; Kramberger, M.G.; Emeršič, A.; Čučnik, S.; Goričar, K.; Dolžan, V. Genetic Polymorphisms in Oxidative Stress and Inflammatory Pathways as Potential Biomarkers in Alzheimer’s Disease and Dementia. Antioxidants 2023, 12, 316. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.-X.; Dai, S.-X.; Guo, Y.; Han, F.-F.; Zheng, J.-J.; Li, G.-H.; Huang, J.-F. Meta-Analysis of Parkinson’s Disease and Alzheimer’s Disease Revealed Commonly Impaired Pathways and Dysregulation of NRF2-Dependent Genes. J. Alzheimer’s Dis. 2017, 56, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.; Leon, J.; Mazzei, G.; Abolhassani, N.; Haruyama, N.; Saito, T.; Saido, T.; Hokama, M.; Iwaki, T.; Ohara, T.; et al. Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 2017, 7, 17762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nuñez, A.; Nevado-Holgado, A.J.; Killik, R.; van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; García-Yagüe, A.J.; Buendia, I.; van Leuven, F.; Yamamoto, M.; López, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Bahn, G.; Park, J.-S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Copple, I.M. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol. Sci. 2023, 44, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer’s disease associated with Down syndrome: A genetic form of dementia. Lancet Neurol. 2021, 20, 930–942. [Google Scholar] [CrossRef]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in Down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Gensous, N.; Bacalini, M.G.; Conte, M.; Salvioli, S. Accelerated bio-cognitive aging in Down syndrome: State of the art and possible deceleration strategies. Aging Cell 2019, 18, e12903. [Google Scholar] [CrossRef]
- McCarron, M.; McCallion, P.; Reilly, E.; Mulryan, N. A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 2014, 58, 61–70. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.J.; Fisher, E.M.C.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Lanzillotta, C.; Zuliani, I.; Tramutola, A.; Barone, E.; Blarzino, C.; Folgiero, V.; Caforio, M.; Valentini, D.; Villani, A.; Locatelli, F.; et al. Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention. Prog. Neurobiol. 2021, 196, 101892. [Google Scholar] [CrossRef]
- Pagnotta, S.; Tramutola, A.; Barone, E.; di Domenico, F.; Pittalà, V.; Salerno, L.; Folgiero, V.; Caforio, M.; Locatelli, F.; Petrini, S.; et al. CAPE and its synthetic derivative VP961 restore BACH1/NRF2 axis in Down Syndrome. Free. Radic. Biol. Med. 2022, 183, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Oyake, T.; Itoh, K.; Motohashi, H.; Hayashi, N.; Hoshino, H.; Nishizawa, M.; Yamamoto, M.; Igarashi, K. Bach Proteins Belong to a Novel Family of BTB-Basic Leucine Zipper Transcription Factors That Interact with MafK and Regulate Transcription through the NF-E2 Site. Mol. Cell Biol. 1996, 16, 6083–6095. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Hoshino, H.; Takaku, K.; Nakajima, O.; Muto, A.; Suzuki, H.; Tashiro, S.; Takahashi, S.; Shibahara, S.; Alam, J.; et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002, 21, 5216–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakoda, E.; Igarashi, K.; Sun, J.; Kurisu, K.; Tashiro, S. Regulation of heme oxygenase-1 by transcription factor Bach1 in the mouse brain. Neurosci. Lett. 2008, 440, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 Competes with Nrf2 Leading to Negative Regulation of the Antioxidant Response Element (ARE)-mediated NAD(P)H: Quinone Oxidoreductase 1 Gene Expression and Induction in Response to Antioxidants. J. Biol. Chem. 2005, 280, 16891–16900. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Butterfield, D.A. The BACH1/Nrf2 Axis in Brain in Down Syndrome and Transition to Alzheimer Disease-Like Neuropathology and Dementia. Antioxidants 2020, 9, 779. [Google Scholar] [CrossRef]
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 Overexpression in Down Syndrome Correlates with the Alteration of the HO-1/BVR-A System: Insights for Transition to Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 1107–1120. [Google Scholar] [CrossRef]
- Breen, P.W.; Krishnan, V. Recent Preclinical Insights into the Treatment of Chronic Traumatic Encephalopathy. Front. Neurosci. 2020, 14, 616. [Google Scholar] [CrossRef]
- Pierre, K.; Dyson, K.; Dagra, A.; Williams, E.; Porche, K.; Lucke-Wold, B. Chronic Traumatic Encephalopathy: Update on Current Clinical Diagnosis and Management. Biomedicines 2021, 9, 415. [Google Scholar] [CrossRef]
- Katsumoto, A.; Takeuchi, H.; Tanaka, F. Tau Pathology in Chronic Traumatic Encephalopathy and Alzheimer’s Disease: Similarities and Differences. Front. Neurol. 2019, 10, 980. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brackhan, M.; Arribas-Blazquez, M.; Lastres-Becker, I. Aging, NRF2, and TAU: A Perfect Match for Neurodegeneration? Antioxidants 2023, 12, 1564. https://doi.org/10.3390/antiox12081564
Brackhan M, Arribas-Blazquez M, Lastres-Becker I. Aging, NRF2, and TAU: A Perfect Match for Neurodegeneration? Antioxidants. 2023; 12(8):1564. https://doi.org/10.3390/antiox12081564
Chicago/Turabian StyleBrackhan, Mirjam, Marina Arribas-Blazquez, and Isabel Lastres-Becker. 2023. "Aging, NRF2, and TAU: A Perfect Match for Neurodegeneration?" Antioxidants 12, no. 8: 1564. https://doi.org/10.3390/antiox12081564
APA StyleBrackhan, M., Arribas-Blazquez, M., & Lastres-Becker, I. (2023). Aging, NRF2, and TAU: A Perfect Match for Neurodegeneration? Antioxidants, 12(8), 1564. https://doi.org/10.3390/antiox12081564