
Carvedilol Phenocopies PGC-1α Overexpression to Alleviate Oxidative Stress, Mitochondrial Dysfunction and Prevent Doxorubicin-Induced Toxicity in Human iPSC-Derived Cardiomyocytes

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Human iPSCs
2.3.1. Reprogramming Human Fibroblasts to iPSCs
2.3.2. iPSC Culture
2.3.3. iPSC-CM Differentiation
2.4. Determination of Cell Viability
2.5. Caspase 3/7 Assay
2.6. Detection of Mitochondrial Oxidants
2.7. Immunocytochemistry
2.8. Western Blot Analysis
2.9. Quantitative Polymerase Chain Reaction (qPCR)
2.10. Measurement of Mitochondrial Oxygen Consumption Rate
2.11. Measurement of Intracellular Reduced Thiols
2.12. Statistical Analysis
3. Results
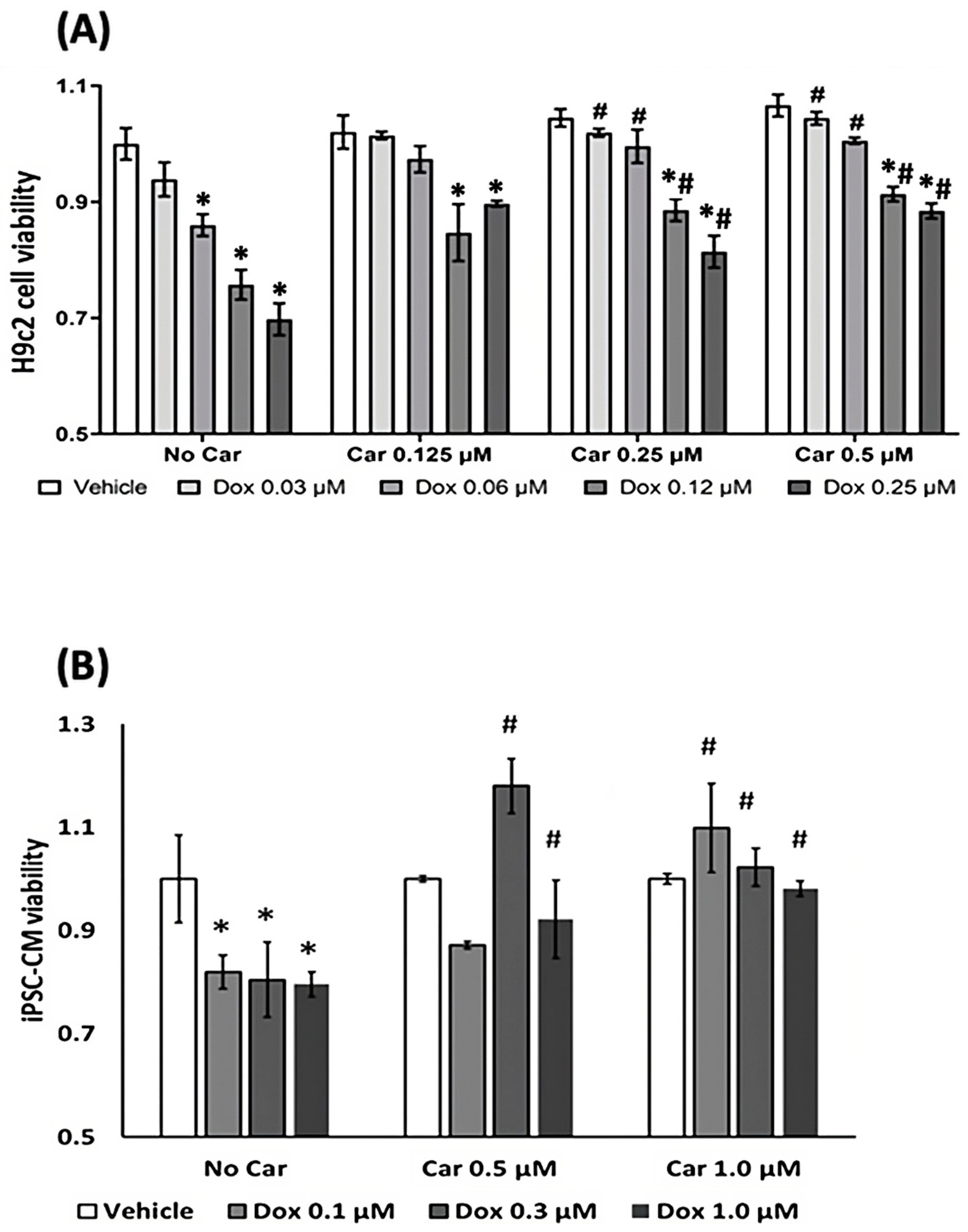
3.1. Carvedilol Exerts Cardioprotective Effects from CTRTOX in Rat Cardiomyoblasts and Human iPSC-CMs
3.1.1. Cell Viability Assay
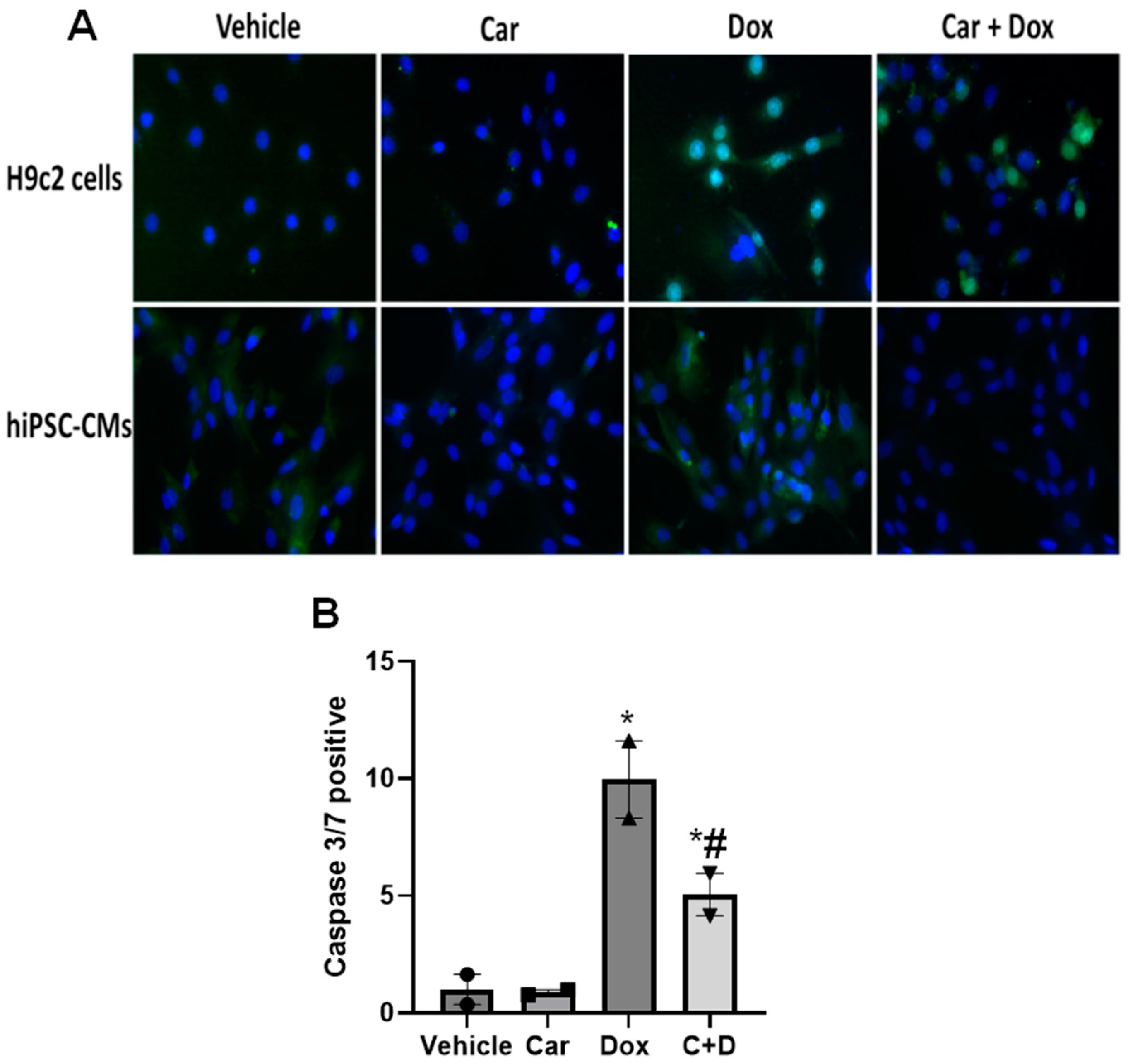
3.1.2. Apoptosis Assay
3.2. Carvedilol Pretreatment Decreases Levels of Mitochondrial Oxidants following DOX Exposure in H9c2 and Human iPSC-CMs
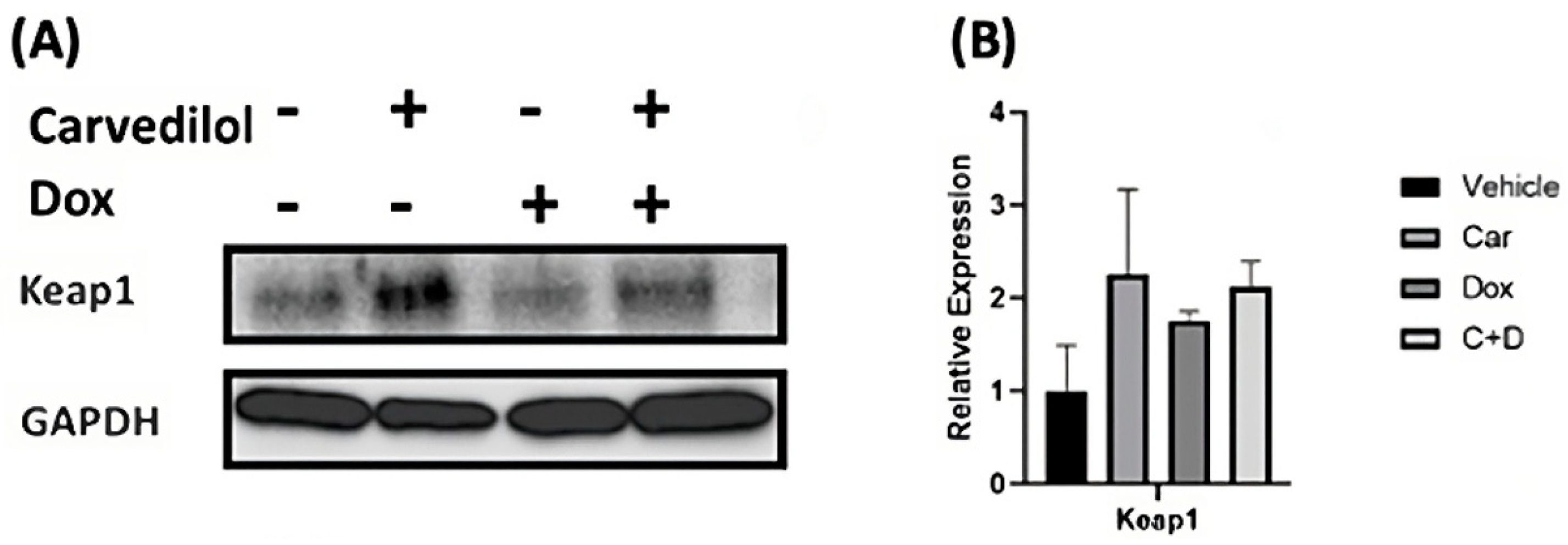
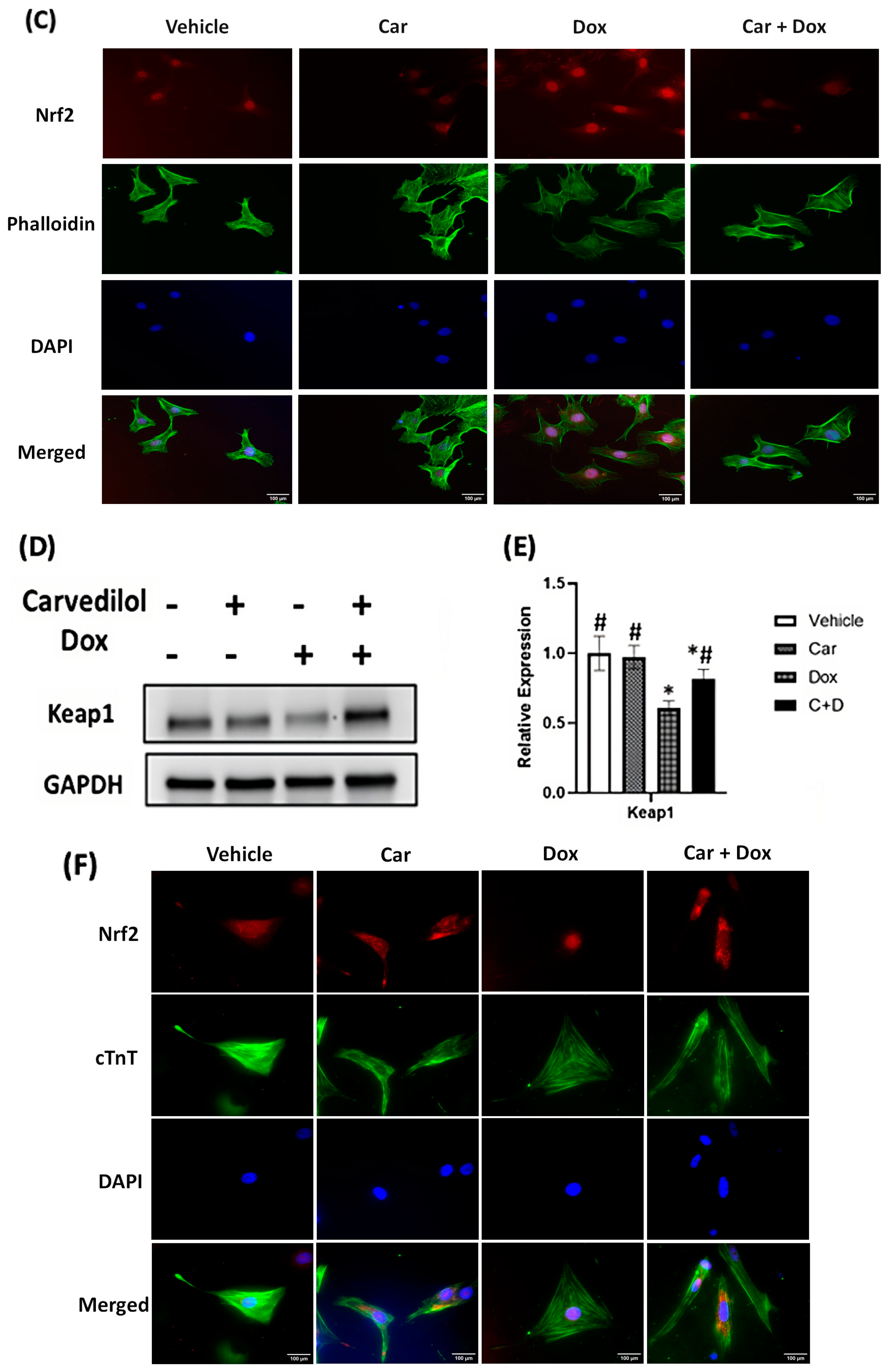
3.3. Carvedilol Pretreatment Sustains Levels of Cytosolic Keap1 and Decreased Nuclear Translocation of Nrf2 in DOX-Treated Rat Cardiomyoblasts and Human iPSC-CMs
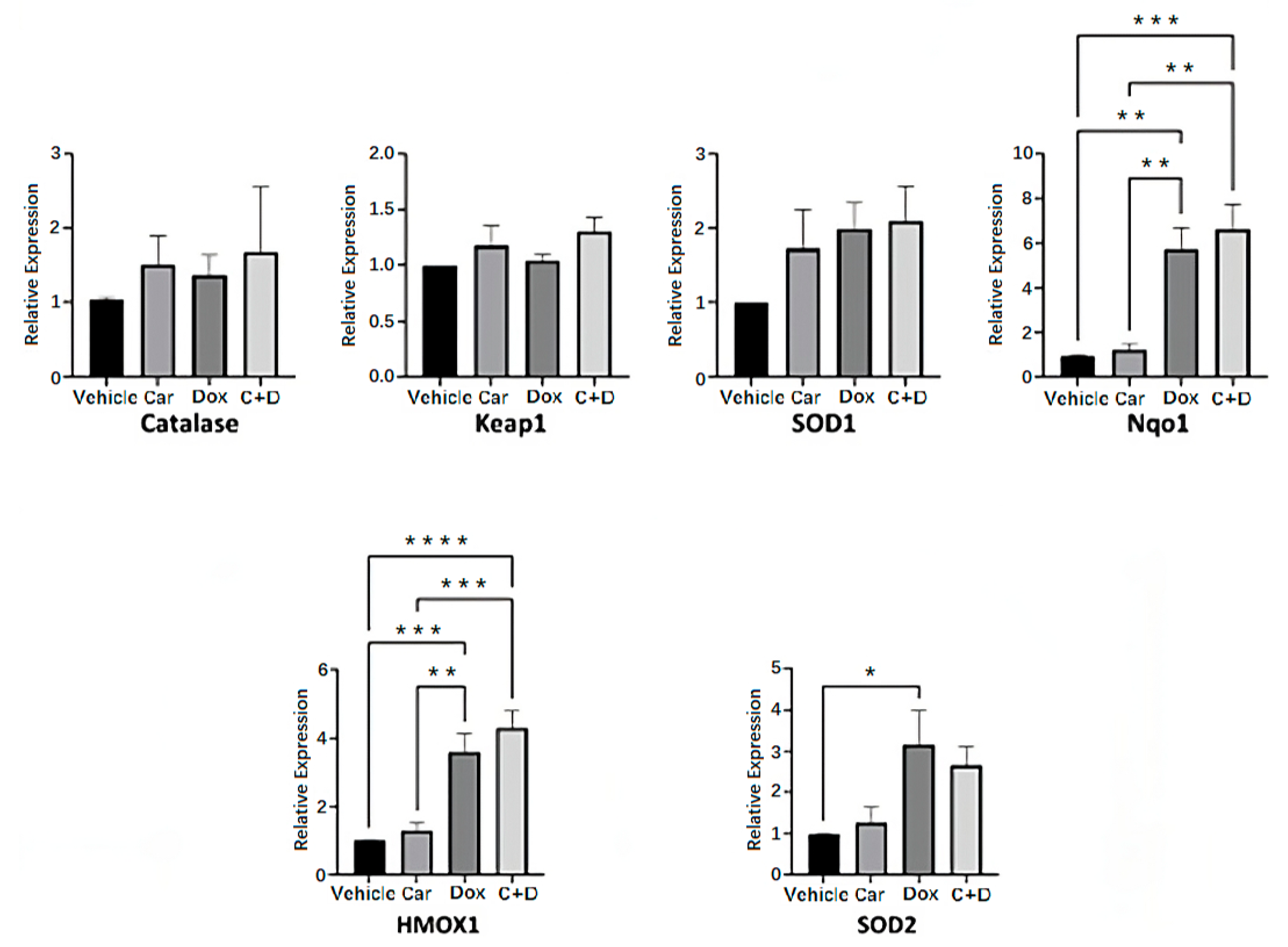
3.4. DOX Treatment Induces Expression of Nrf2-Regulated Genes in Human iPSC-CMs
3.5. Carvedilol Pretreatment Prevents DOX-Induced Decreased Levels of Total Reduced Thiols in H9c2 Cells
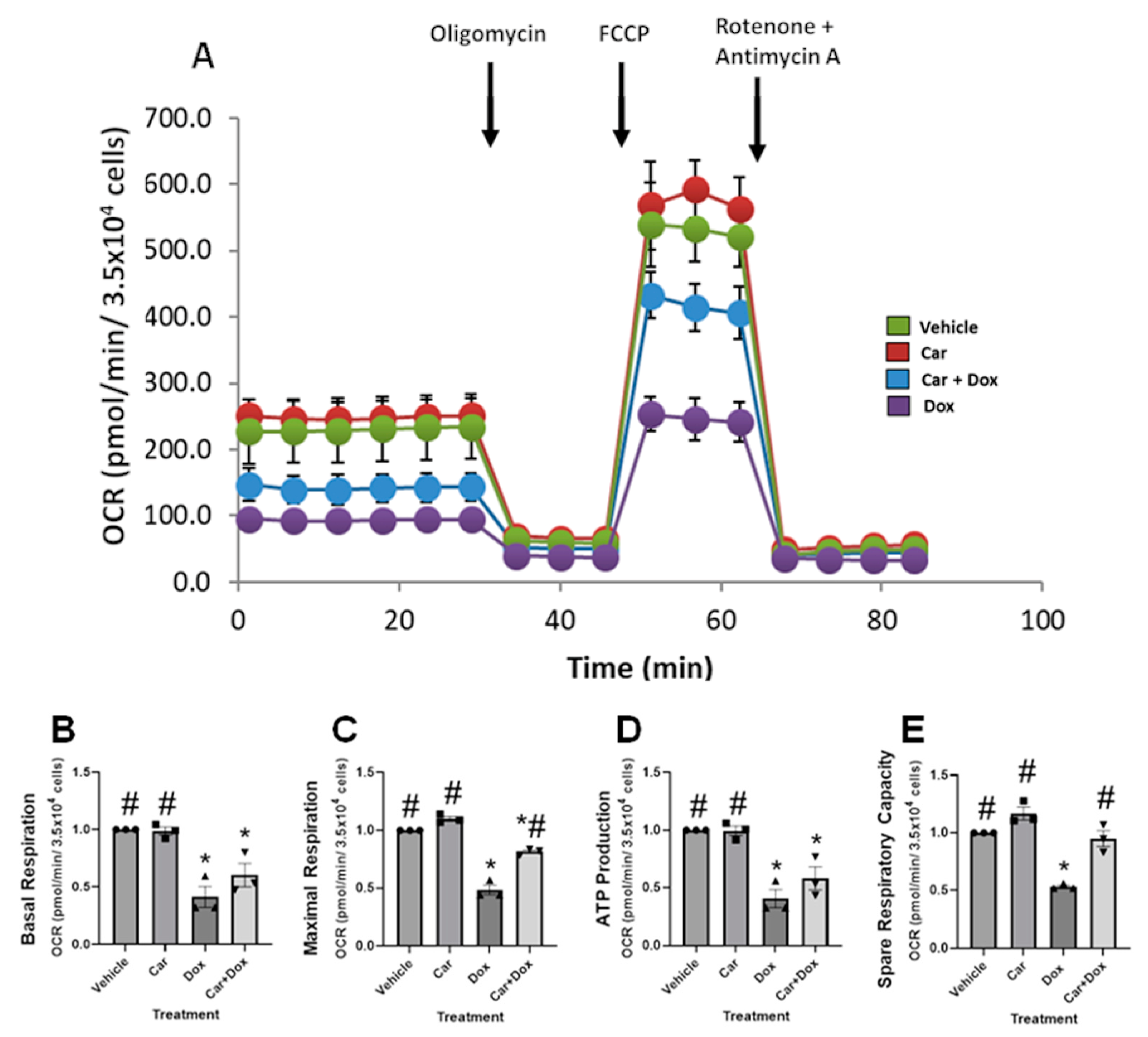
3.6. Carvedilol Prevents DOX-Induced Suppression of Mitochondrial Respiratory Function in Human iPSC-CMs
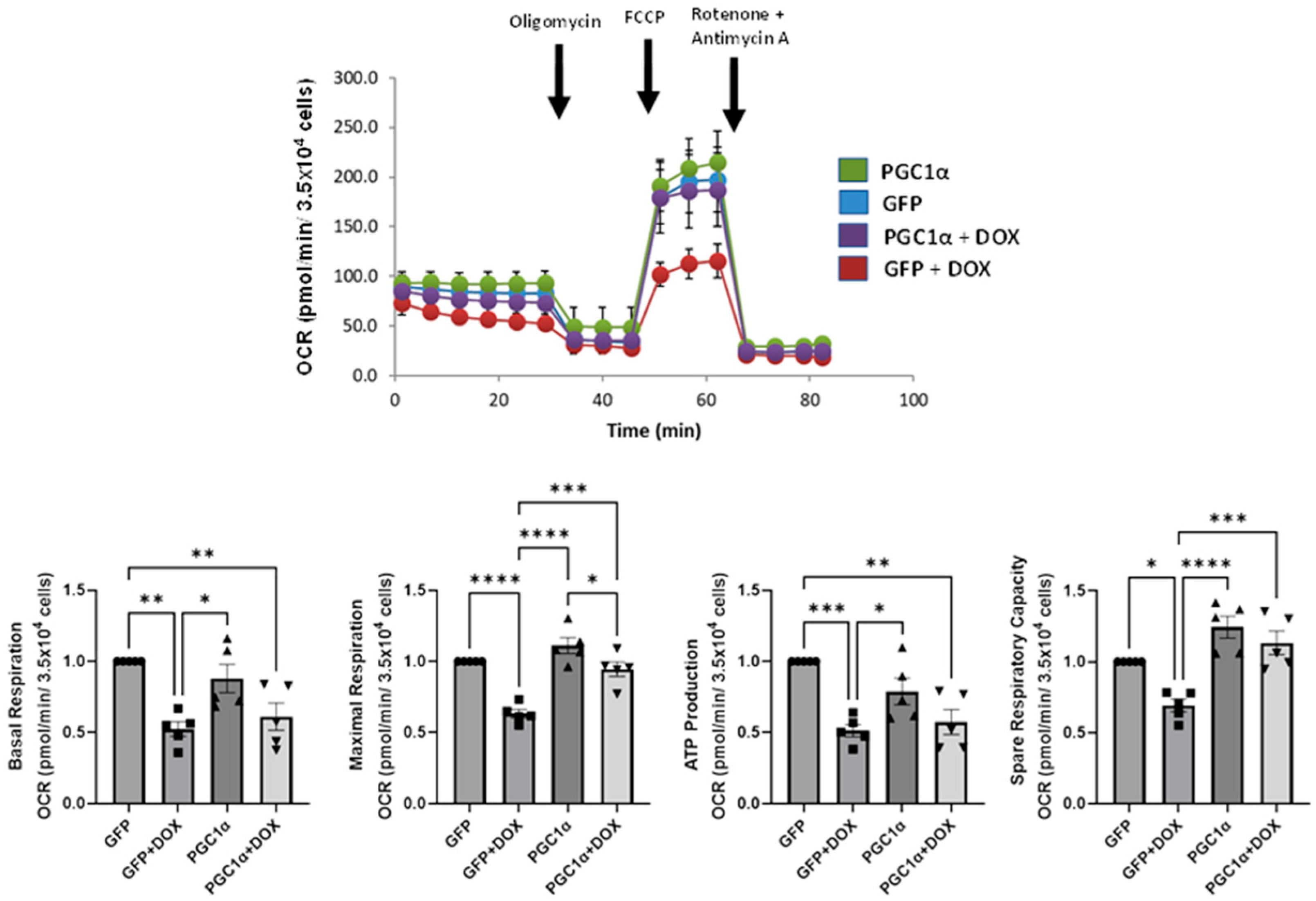
3.7. PGC-1α Overexpression Recapitulates the Pharmacological Prevention by Carvedilol of DOX-Induced Mitochondrial Dysfunction in Human iPSC-CMs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Johnson-Arbor, K.; Dubey, R. StatPearls; StatPearls Publishing LLC: Tampa, FL, USA, 2022. [Google Scholar]
- Bansal, N.; Adams, M.J.; Ganatra, S.; Colan, S.D.; Aggarwal, S.; Steiner, R.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Strategies to prevent anthracycline-induced cardiotoxicity in cancer survivors. Cardiooncology 2019, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Cao, J.; Wang, X.; Zhang, Y.; Sun, Q.; Jiang, Y.; Yao, J.; Li, C.; Wang, Y.; Wang, W. Ferruginol Restores SIRT1-PGC-1α-Mediated Mitochondrial Biogenesis and Fatty Acid Oxidation for the Treatment of DOX-Induced Cardiotoxicity. Front. Pharmacol. 2021, 12, 773834. [Google Scholar] [CrossRef] [PubMed]
- Shakir, D.K.; Rasul, K.I. Chemotherapy induced cardiomyopathy: Pathogenesis, monitoring and management. J. Clin. Med. Res. 2009, 1, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Kalyanaraman, B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- L’Ecuyer, T.; Sanjeev, S.; Thomas, R.; Novak, R.; Das, L.; Campbell, W.; Heide, R.V. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1273–H1280. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Bayır, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirone, L.; D’Ambrosio, L.; Forte, M.; Genovese, R.; Schiavon, S.; Spinosa, G.; Iacovone, G.; Valenti, V.; Frati, G.; Sciarretta, S. Mitochondria and Doxorubicin-Induced Cardiomyopathy: A Complex Interplay. Cells 2022, 11, 2000. [Google Scholar] [CrossRef] [PubMed]
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a specific complex between adriamycin and negatively-charged phospholipids. Biochim. Biophys. Acta 1980, 597, 1–14. [Google Scholar] [CrossRef]
- Govender, J.; Loos, B.; Marais, E.; Engelbrecht, A.M. Mitochondrial catastrophe during doxorubicin-induced cardiotoxicity: A review of the protective role of melatonin. J. Pineal Res. 2014, 57, 367–380. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Lebrecht, D.; Setzer, B.; Ketelsen, U.P.; Haberstroh, J.; Walker, U.A. Time-dependent and tissue-specific accumulation of mtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation 2003, 108, 2423–2429. [Google Scholar] [CrossRef] [Green Version]
- Volkova, M.; Russell, R., 3rd. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Pituskin, E.; Mackey, J.R.; Koshman, S.; Jassal, D.; Pitz, M.; Haykowsky, M.J.; Pagano, J.J.; Chow, K.; Thompson, R.B.; Vos, L.J.; et al. Multidisciplinary Approach to Novel Therapies in Cardio-Oncology Research (MANTICORE 101-Breast): A Randomized Trial for the Prevention of Trastuzumab-Associated Cardiotoxicity. J. Clin. Oncol. 2017, 35, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Gulati, G.; Heck, S.L.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; von Knobelsdorff-Brenkenhoff, F.; Bratland, Å.; Storås, T.H.; et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): A 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur. Heart J. 2016, 37, 1671–1680. [Google Scholar] [CrossRef] [Green Version]
- Bosch, X.; Rovira, M.; Sitges, M.; Domènech, A.; Ortiz-Pérez, J.T.; de Caralt, T.M.; Morales-Ruiz, M.; Perea, R.J.; Monzó, M.; Esteve, J. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: The OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J. Am. Coll. Cardiol. 2013, 61, 2355–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebbi, C.K.; London, W.B.; Friedman, D.; Villaluna, D.; De Alarcon, P.A.; Constine, L.S.; Mendenhall, N.P.; Sposto, R.; Chauvenet, A.; Schwartz, C.L. Dexrazoxane-associated risk for acute myeloid leukemia/myelodysplastic syndrome and other secondary malignancies in pediatric Hodgkin’s disease. J. Clin. Oncol. 2007, 25, 493–500. [Google Scholar] [CrossRef]
- Macedo, A.V.S.; Hajjar, L.A.; Lyon, A.R.; Nascimento, B.R.; Putzu, A.; Rossi, L.; Costa, R.B.; Landoni, G.; Nogueira-Rodrigues, A.; Ribeiro, A.L. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC CardioOncology 2019, 1, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P.; Tabone, M.D.; Mora, J.; Morland, B.; Jones, R.L. Risk-benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: Re-evaluating the European labeling. FutureOncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef]
- Ganatra, S.; Nohria, A.; Shah, S.; Groarke, J.D.; Sharma, A.; Venesy, D.; Patten, R.; Gunturu, K.; Zarwan, C.; Neilan, T.G.; et al. Upfront dexrazoxane for the reduction of anthracycline-induced cardiotoxicity in adults with preexisting cardiomyopathy and cancer: A consecutive case series. Cardiooncology 2019, 5, 1. [Google Scholar] [CrossRef]
- De Oliveira, B.L.; Niederer, S. A Biophysical Systems Approach to Identifying the Pathways of Acute and Chronic Doxorubicin Mitochondrial Cardiotoxicity. PLoS Comput. Biol. 2016, 12, e1005214. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef] [Green Version]
- Chandran, K.; Aggarwal, D.; Migrino, R.Q.; Joseph, J.; McAllister, D.; Konorev, E.A.; Antholine, W.E.; Zielonka, J.; Srinivasan, S.; Avadhani, N.G.; et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: Cardioprotection by Mito-Q. Biophys. J. 2009, 96, 1388–1398. [Google Scholar] [CrossRef] [Green Version]
- Renu, K.; Vinayagam, S.; Madhyastha, H.; Madhyastha, R.; Maruyama, M.; Suman, S.; Arunachalam, S.; Vellingiri, B.; Valsala Gopalakrishnan, A. Exploring the Pattern of Metabolic Alterations Causing Energy Imbalance via PPARα Dysregulation in Cardiac Muscle During Doxorubicin Treatment. Cardiovasc. Toxicol. 2022, 22, 436–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Fan, S.; Shu, L.; Huang, W.; Xie, L.; Bi, C.; Yu, H.; Wang, Y.; Li, Y. Exploration the Mechanism of Doxorubicin-Induced Heart Failure in Rats by Integration of Proteomics and Metabolomics Data. Front. Pharmacol. 2020, 11, 600561. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxid. Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, S.J.; Howden, E.J.; Haykowsky, M.J.; Antill, Y.; Salim, A.; Nightingale, S.S.; Loi, S.; Claus, P.; Janssens, K.; Mitchell, A.M.; et al. Exercise for the Prevention of Anthracycline-Induced Functional Disability and Cardiac Dysfunction: The BREXIT Study. Circulation 2023, 147, 532–545. [Google Scholar] [CrossRef]
- Keating, G.M.; Jarvis, B. Carvedilol: A review of its use in chronic heart failure. Drugs 2003, 63, 1697–1741. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, G.; Egan, C.G. Use of carvedilol in hypertension: An update. Vasc. Health Risk Manag. 2012, 8, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Abreu, R.M.; Santos, D.J.; Moreno, A.J. Effects of carvedilol and its analog BM-910228 on mitochondrial function and oxidative stress. J. Pharmacol. Exp. Ther. 2000, 295, 1022–1030. [Google Scholar]
- Kheiri, B.; Abdalla, A.; Osman, M.; Haykal, T.; Chahine, A.; Ahmed, S.; Osman, K.; Hassan, M.; Bachuwa, G.; Bhatt, D.L. Meta-Analysis of Carvedilol for the Prevention of Anthracycline-Induced Cardiotoxicity. Am. J. Cardiol. 2018, 122, 1959–1964. [Google Scholar] [CrossRef]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R.; das Dores Cruz, F.; Goncalves Brandao, S.M.; Rigaud, V.O.C.; Higuchi-dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M.; et al. Carvedilol for Prevention of Chemotherapy-Related Cardiotoxicity: The CECCY Trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef]
- Szajerski, P.; Zielonka, J.; Sikora, A.; Adamus, J.; Marcinek, A.; Gębicki, J.; Kozlovski, V.I.; Drelicharz, Ł.; Chłopicki, S. Radical scavenging and NO-releasing properties of selected beta-adrenoreceptor antagonists. Free Radic. Res. 2006, 40, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef]
- Riedel, M.; Jou, C.J.; Lai, S.; Lux, R.L.; Moreno, A.P.; Spitzer, K.W.; Christians, E.; Tristani-Firouzi, M.; Benjamin, I.J. Functional and pharmacological analysis of cardiomyocytes differentiated from human peripheral blood mononuclear-derived plu-ripotent stem cells. Stem Cell Rep. 2014, 3, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Bhutani, K.; Nazor, K.L.; Williams, R.; Tran, H.; Dai, H.; Džakula, Ž.; Cho, E.H.; Pang, A.W.; Rao, M.; Cao, H.; et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 2016, 7, 10536. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, S.; Liu, N.; Ma, L.; Wang, T.; Veedu, R.N.; Li, T.; Zhang, F.; Zhou, H.; Cheng, X.; et al. A systematic investigation of key factors of nucleic acid precipitation toward optimized DNA/RNA isolation. Biotechniques 2020, 68, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, I.J.; McMillan, D.R. Stress, (heat shock) proteins: Molecular chaperones in cardiovascular biology and disease. Circ. Res. 1998, 83, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Brewer, A.C.; Mustafi, S.B.; Murray, T.V.; Rajasekaran, N.S.; Benjamin, I.J. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid. Redox Signal. 2013, 18, 1114–1127. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numazawa, S.; Yoshida, T. Nrf2-dependent gene expressions: A molecular toxicological aspect. J. Toxicol. Sci. 2004, 29, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, B.Y.; Ahmad, M.I.; Abdallah, Q.M.; Qatouseh, L.A.; Qinna, N.A. Modulation of NRF2/ARE pathway- and cell death-related genes during drug-induced liver injury. Hum. Exp. Toxicol. 2021, 40, 2223–2236. [Google Scholar] [CrossRef]
- Lenihan, D.J.; Cardinale, D.M. Late cardiac effects of cancer treatment. J. Clin. Oncol. 2012, 30, 3657–3664. [Google Scholar] [CrossRef] [PubMed]
- Aruoma, O.I. Peroxyl radical scavenging activity of the antihypertensive drug carvedilol. Toxicol. Vitr. 1996, 10, 625–629. [Google Scholar] [CrossRef]
- Araujo-Gutierrez, R.; Chitturi, K.R.; Xu, J.; Wang, Y.; Kinder, E.; Senapati, A.; Chebrolu, L.B.; Kassi, M.; Trachtenberg, B.H. Baseline global longitudinal strain predictive of anthracycline-induced cardiotoxicity. Cardiooncology 2021, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Hochster, H.; Wasserheit, C.; Speyer, J. Cardiotoxicity and cardioprotection during chemotherapy. Curr. Opin. Oncol. 1995, 7, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst. Rev. 2005, 6, CD003917. [Google Scholar] [CrossRef]
- Avila, M.S.; Ayub Ferreira, S.M.; Bocchi, E.A. Reply: Can Carvedilol Prevent Chemotherapy-Related Cardiotoxicity?: A Dream to Be Balanced With Tolerability. J. Am. Coll. Cardiol. 2018, 72, 1182–1183. [Google Scholar] [CrossRef]
- Spallarossa, P.; Garibaldi, S.; Altieri, P.; Fabbi, P.; Manca, V.; Nasti, S.; Rossettin, P.; Ghigliotti, G.; Ballestrero, A.; Patrone, F.; et al. Carvedilol prevents doxorubicin-induced free radical release and apoptosis in cardiomyocytes in vitro. J. Mol. Cell. Cardiol. 2004, 37, 837–846. [Google Scholar] [CrossRef]
- Alanazi, A.M.; Fadda, L.; Alhusaini, A.; Ahmad, R.; Hasan, I.H.; Mahmoud, A.M. Liposomal Resveratrol and/or Carvedilol Attenuate Doxorubicin-Induced Cardiotoxicity by Modulating Inflammation, Oxidative Stress and S100A1 in Rats. Antioxid. 2020, 9, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System as a Molecular Target of Cancer Treatment. Cancers 2020, 13, 46. [Google Scholar] [CrossRef]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Samadi, N. Contradictory roles of Nrf2/Keap1 signaling pathway in cancer prevention/promotion and chemoresistance. DNA Repair 2017, 54, 13–21. [Google Scholar] [CrossRef]
- Kourek, C.; Touloupaki, M.; Rempakos, A.; Loritis, K.; Tsougkos, E.; Paraskevaidis, I.; Briasoulis, A. Cardioprotective Strategies from Cardiotoxicity in Cancer Patients: A Comprehensive Review. J. Cardiovasc. Dev. Dis. 2022, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef]
- Yu, W.; Chen, C.; Xu, C.; Xie, D.; Wang, Q.; Liu, W.; Zhao, H.; He, F.; Chen, B.; Xi, Y.; et al. Activation of p62-NRF2 Axis Protects against Doxorubicin-Induced Ferroptosis in Cardiomyocytes: A Novel Role and Molecular Mechanism of Resveratrol. Am. J. Chin. Med. 2022, 50, 2103–2123. [Google Scholar] [CrossRef]
- Lu, G.; Liu, Q.; Gao, T.; Li, J.; Zhang, J.; Chen, O.; Cao, C.; Mao, M.; Xiao, M.; Zhang, X.; et al. Resveratrol and FGF1 Synergistically Ameliorates Doxorubicin-Induced Cardiotoxicity via Activation of SIRT1-NRF2 Pathway. Nutrients 2022, 14, 4017. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, R.; McElhanon, K.; Allen, C.D.; Megyesi, J.K.; Beneš, H.; Singh, S.P. Sulforaphane protects the heart from doxorubicin-induced toxicity. Free Radic. Biol. Med. 2015, 86, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Chen, Q.; Sun, Y.P.; Wang, X.; Lv, L.; Zhang, L.P.; Liu, J.S.; Zhao, S.; Wang, X.L. Sulforaphane protection against the development of doxorubicin-induced chronic heart failure is associated with Nrf2 Upregulation. Cardiovasc. Ther. 2017, 35, e12277. [Google Scholar] [CrossRef]
- Li, B.; Kim, D.S.; Yadav, R.K.; Kim, H.R.; Chae, H.J. Sulforaphane prevents doxorubicin-induced oxidative stress and cell death in rat H9c2 cells. Int. J. Mol. Med. 2015, 36, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Gao, L.; Zhang, A.; Hackfort, B.T.; Zucker, I.H. Therapeutic Effects of Nrf2 Activation by Bardoxolone Methyl in Chronic Heart Failure. J. Pharmacol. Exp. Ther. 2019, 371, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, L.; Lu, Z.Q.; Bentley, R.A.; Colley, H.E.; Murdoch, C.; Webb, S.D.; Cross, M.J.; Copple, I.M.; Sharma, P. Attenuation of doxorubicin-induced cardiotoxicity in a human in vitro cardiac model by the induction of the NRF-2 pathway. Biomed. Pharmacother. 2019, 112, 108637. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Smolina, N.; Bruton, J.; Kostareva, A.; Sejersen, T. Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. Methods Mol. Biol. 2017, 1601, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zhang, L.; Liu, H.; Wang, J.; Li, J.; Yang, Y.; Wang, Y.; Cai, H.; Li, R.; Zhao, Y. Salsolinol Attenuates Doxorubicin-Induced Chronic Heart Failure in Rats and Improves Mitochondrial Function in H9c2 Cardiomyocytes. Front. Pharmacol. 2019, 10, 1135. [Google Scholar] [CrossRef]
- Liu, D.; Ma, Z.; Xu, L.; Zhang, X.; Qiao, S.; Yuan, J. PGC1α activation by pterostilbene ameliorates acute doxorubicin cardiotoxicity by reducing oxidative stress via enhancing AMPK and SIRT1 cascades. Aging 2019, 11, 10061–10073. [Google Scholar] [CrossRef]
- Wen, J.; Wang, J.; Li, P.; Wang, R.; Wang, J.; Zhou, X.; Zhang, L.; Li, H.; Wei, S.; Cai, H.; et al. Protective effects of higenamine combined with [6]-gingerol against doxorubicin-induced mitochondrial dysfunction and toxicity in H9c2 cells and potential mechanisms. Biomed. Pharmacother. 2019, 115, 108881. [Google Scholar] [CrossRef]
- Saleh, M.F.; Elsayad, M.E.; Goda, A.E. Mitigation of doxorubicin-induced cardiotoxicity by dichloroacetate: Potential roles of restoration of PGC-1α/SIRT3 signaling and suppression of oxidative stress and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 6573–6584. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Hang, W.W.; Yao, L.; Yang, S.; Nie, W.; Huang, F. Carvedilol promotes mitochondrial biogenesis by regulating the PGC-1/TFAM pathway in human umbilical vein endothelial cells (HUVECs). Biochem. Biophys. Res. Commun. 2016, 470, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Human Nqo1 | CCTGCCATTCTGAAAGGCTGGT | GTGGTGATGGAAAGCACTGCCT |
| Human HMOX1 | CCAGGCAGAGAATGCTGAGTTC | AAGACTGGGCTCTCCTTGTTGC |
| Human Catalase | GTGCGGAGATTCAACACTGCCA | CGGCAATGTTCTCACACAGACG |
| Human SOD2 | CTGGACAAACCTCAGCCCTAAC | AACCTGAGCCTTGGACACCAAC |
| Human SOD1 | CTCACTCTCAGGAGACCATTGC | CCACAAGCCAAACGACTTCCAG |
| Human Keap1 | CAACTTCGCTGAGCAGATTGGC | TGATGAGGGTCACCAGTTGGCA |
| Human GAPDH | TCCAAAATCAAGTGGGGCGA | TGATGACCCTTTTGGCTCCC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uche, N.; Dai, Q.; Lai, S.; Kolander, K.; Thao, M.; Schibly, E.; Sendaydiego, X.; Zielonka, J.; Benjamin, I.J. Carvedilol Phenocopies PGC-1α Overexpression to Alleviate Oxidative Stress, Mitochondrial Dysfunction and Prevent Doxorubicin-Induced Toxicity in Human iPSC-Derived Cardiomyocytes. Antioxidants 2023, 12, 1585. https://doi.org/10.3390/antiox12081585
Uche N, Dai Q, Lai S, Kolander K, Thao M, Schibly E, Sendaydiego X, Zielonka J, Benjamin IJ. Carvedilol Phenocopies PGC-1α Overexpression to Alleviate Oxidative Stress, Mitochondrial Dysfunction and Prevent Doxorubicin-Induced Toxicity in Human iPSC-Derived Cardiomyocytes. Antioxidants. 2023; 12(8):1585. https://doi.org/10.3390/antiox12081585
Chicago/Turabian StyleUche, Nnamdi, Qiang Dai, Shuping Lai, Kurt Kolander, Mai Thao, Elizabeth Schibly, Xavier Sendaydiego, Jacek Zielonka, and Ivor J. Benjamin. 2023. "Carvedilol Phenocopies PGC-1α Overexpression to Alleviate Oxidative Stress, Mitochondrial Dysfunction and Prevent Doxorubicin-Induced Toxicity in Human iPSC-Derived Cardiomyocytes" Antioxidants 12, no. 8: 1585. https://doi.org/10.3390/antiox12081585
APA StyleUche, N., Dai, Q., Lai, S., Kolander, K., Thao, M., Schibly, E., Sendaydiego, X., Zielonka, J., & Benjamin, I. J. (2023). Carvedilol Phenocopies PGC-1α Overexpression to Alleviate Oxidative Stress, Mitochondrial Dysfunction and Prevent Doxorubicin-Induced Toxicity in Human iPSC-Derived Cardiomyocytes. Antioxidants, 12(8), 1585. https://doi.org/10.3390/antiox12081585