

Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. The Implications of Oxidative Stress in Chronic Diseases in the Era of Precision Medicine
2. Cellular Redox Status in the Pathophysiological Complex of Cardiovascular Diseases
3. Generation of Reactive Oxygen Species and Antioxidants
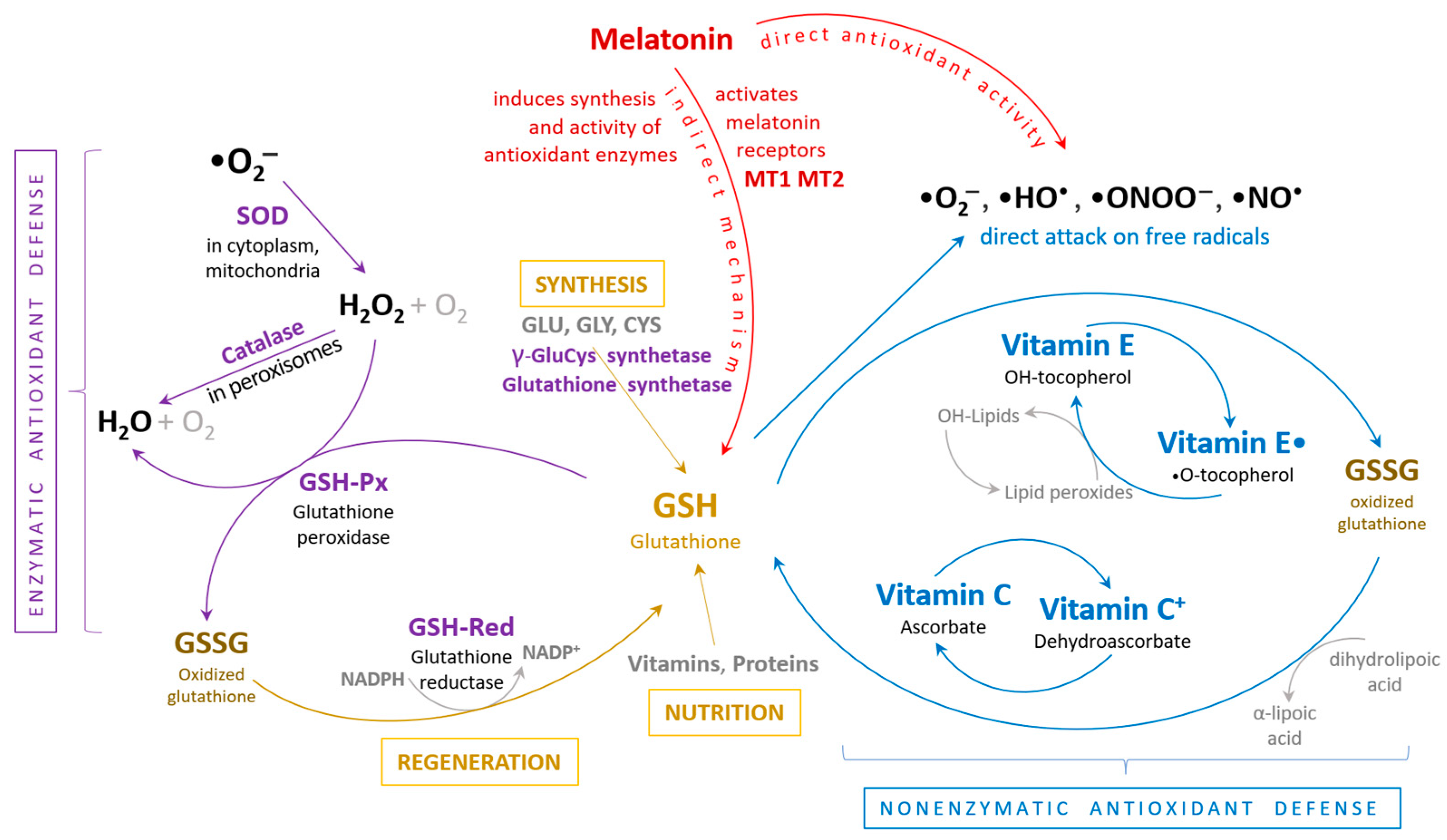
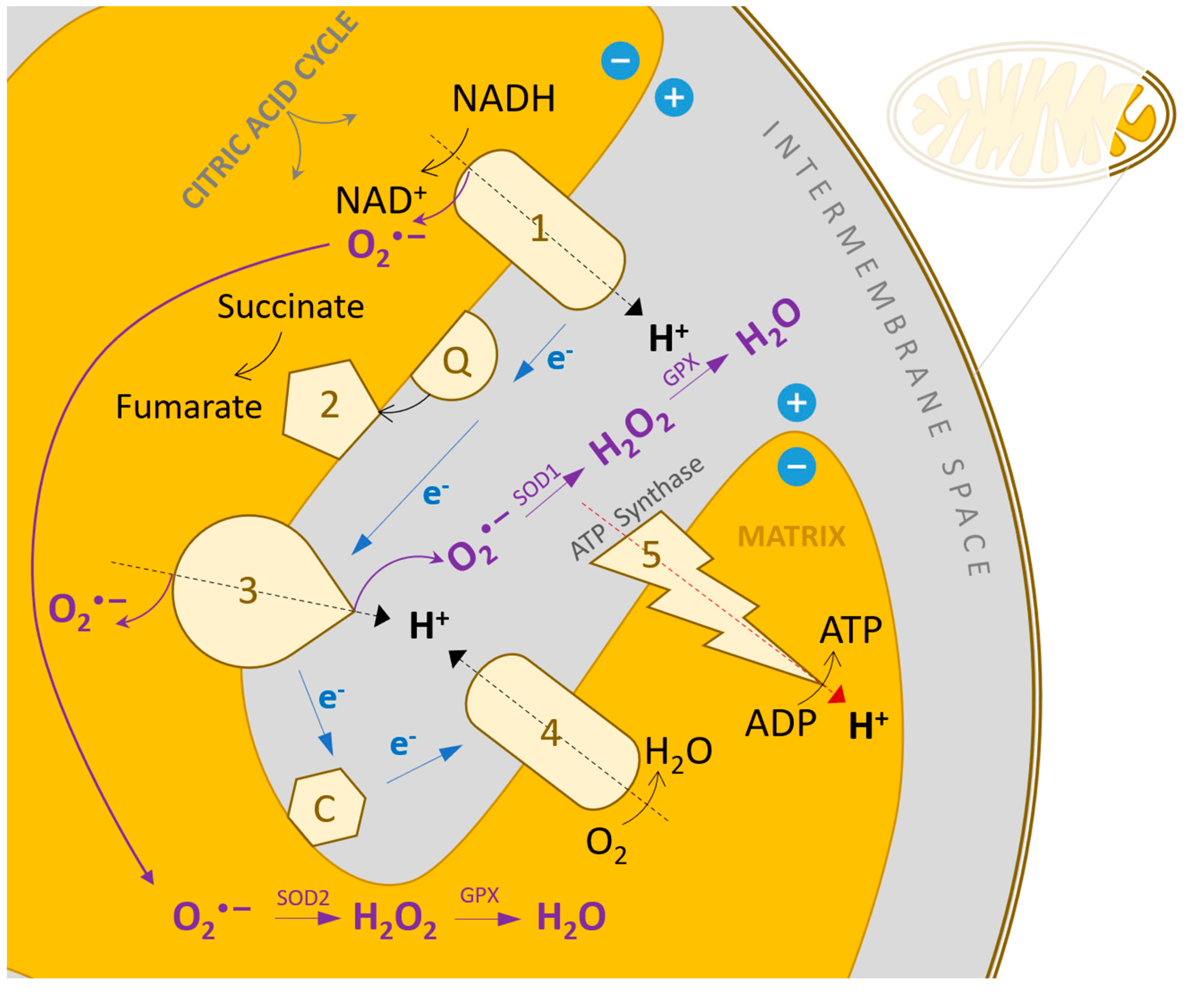
4. Elevated Reactive Oxygen Species Concentration in Heart Failure
5. Sources of Reactive Oxygen Species in the Cardiovascular System
6. Oxidative Stress and Mitochondrial DNA Degradation
7. Oxidative Stress in the Process of Myocardial Remodeling
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Yang, J.; Luo, J.; Tian, X.; Zhao, Y.; Li, Y.; Wu, X. Progress in Understanding Oxidative Stress, Aging, and Aging-Related Diseases. Antioxidants 2024, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Denny, J.C.; Collins, F.S. Precision Medicine in 2030—Seven Ways to Transform Healthcare. Cell 2021, 184, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, G.R.; Sinkunas, V.; Liguori, G.R.; Auler-Júnior, J.O.C. Precision Medicine: Changing the Way We Think about Healthcare. Clinics 2018, 73, e723. [Google Scholar] [CrossRef] [PubMed]
- Tretter, V.; Hochreiter, B.; Zach, M.L.; Krenn, K.; Klein, K.U. Understanding Cellular Redox Homeostasis: A Challenge for Precision Medicine. Int. J. Mol. Sci. 2022, 23, 106. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Tolouee, M.; Hendriks, K.; Lie, F.F.; Gartzke, L.P.; Goris, M.; HoogstraBerends, F.; Henning, R.H. Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion. Cell Transplant. 2022, 31, 9636897221108705. [Google Scholar] [CrossRef]
- Dragoi, C.M.; Nicolae, A.C.; Grigore, C.; Dinu-Pirvu, C.E.; Arsene, A.L. Characteristics of glucose homeostasis and lipidic profile in a hamster metabolic syndrome model, after the co-administration of melatonin and irbesartan in a multiparticulate pharmaceutical formation. In Proceedings of the 2nd International Conference on Interdisciplinary Management of Diabetes Mellitus and its Complications, INTERDIAB 2016, Bucharest, Romania, 3–5 March 2016; pp. 221–229. [Google Scholar]
- Azarova, I.; Polonikov, A.; Klyosova, E. Molecular Genetics of Abnormal Redox Homeostasis in Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2023, 24, 4738. [Google Scholar] [CrossRef]
- Niculae, D.; Dusman, R.; Leonte, R.A.; Chilug, L.E.; Dragoi, C.M.; Nicolae, A.; Serban, R.M.; Niculae, D.A.; Dumitrescu, I.B.; Draganescu, D. Biological Pathways as Substantiation of the Use of Copper Radioisotopes in Cancer Theranostics. Front. Phys. 2021, 8, 568296. [Google Scholar] [CrossRef]
- Choi, E.H.; Kim, M.H.; Park, S.J. Targeting Mitochondrial Dysfunction and Reactive Oxygen Species for Neurodegenerative Disease Treatment. Int. J. Mol. Sci. 2024, 25, 7952. [Google Scholar] [CrossRef]
- Tarta-Arsene, O.; Leanca, M.; Dică, A.; Bran, E.; Rad, F.; Timnea, O.; Păcurar, D.; Velescu, B.S.; Nicolae, A.C.; Drăgoi, C.M. Dietary omega-3 fatty acids supplimentation for attention deficit with hyperactivity disorder in epileptic children. Farmácia 2017, 65, 550–556. [Google Scholar]
- Sirbu, C.A.; Georgescu, R.; Pleşa, F.C.; Paunescu, A.; Marilena Ţânţu, M.; Nicolae, A.C.; Caloianu, I.; Mitrica, M. Cannabis and Cannabinoids in Multiple Sclerosis: From Experimental Models to Clinical Practice-A Review. Am. J. Ther. 2023, 30, e220–e231. [Google Scholar] [CrossRef] [PubMed]
- Axente, M.; Mirea, A.; Sporea, C.; Pădure, L.; Drăgoi, C.M.; Nicolae, A.C.; Ion, D.A. Clinical and Electrophysiological Changes in Pediatric Spinal Muscular Atrophy after 2 Years of Nusinersen Treatment. Pharmaceutics 2022, 14, 2074. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.; Lee, T. The Role of Reactive Oxygen Species in Reproductive and Skeletal Health. Antioxidants 2024, 13, 245–260. [Google Scholar] [CrossRef]
- DeGroat, W.; Abdelhalim, H.; Patel, K.; Mendhe, D.; Zeeshan, S.; Ahmed, Z. Discovering Biomarkers Associated and Predicting Cardiovascular Disease with High Accuracy Using a Novel Nexus of Machine Learning Techniques for Precision Medicine. Sci. Rep. 2024, 14, 1. [Google Scholar] [CrossRef]
- Diaconu, C.C.; Cozma, M.-A.; Dobrică, E.-C.; Gheorghe, G.; Jichitu, A.; Ionescu, V.A.; Nicolae, A.C.; Drăgoi, C.M.; Găman, M.-A. Polypharmacy in the Management of Arterial Hypertension—Friend or Foe? Medicina 2021, 57, 1288. [Google Scholar] [CrossRef]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Rodríguez Guzmán, R.; Centofanti, F.; Doldo, E.; Céspedes Miranda, E.M.; Orlandi, A. Oxidative Stress and New Pathogenetic Mechanisms in Endothelial Dysfunction: Potential Diagnostic Biomarkers and Therapeutic Targets. J. Clin. Med. 2020, 9, 1995. [Google Scholar] [CrossRef]
- Ungurianu, A.; Zanfirescu, A.; Margina, D. Regulation of Gene Expression through Food-Curcumin as a Sirtuin Activity Modulator. Plants 2022, 11, 1741. [Google Scholar] [CrossRef]
- Di Raimondo, D.; Buscemi, S.; Musiari, G.; Rizzo, G.; Pirera, E.; Corleo, D.; Pinto, A.; Tuttolomondo, A. Ketogenic Diet, Physical Activity, and Hypertension—A Narrative Review. Nutrients 2021, 13, 2567. [Google Scholar] [CrossRef]
- Moroșan, E.; Secăreanu, A.A.; Musuc, A.M.; Mititelu, M.; Ioniță, A.C.; Ozon, E.A.; Dărăban, A.M.; Karampelas, O. Advances on the Antioxidant Activity of a Phytocomplex Product Containing Berry Extracts from Romanian Spontaneous Flora. Processes 2022, 10, 646. [Google Scholar] [CrossRef]
- Valenzano, A.; Polito, R.; Trimigno, V.; Di Palma, A.; Moscatelli, F.; Corso, G.; Sessa, F.; Salerno, M.; Montana, A.; Di Nunno, N.; et al. Effects of Very Low Calorie Ketogenic Diet on the Orexinergic System, Visceral Adipose Tissue, and ROS Production. Antioxidants 2019, 8, 643. [Google Scholar] [CrossRef]
- Daraban, A.M.; Olah, N.K.; Câmpean, R.F.; Furtuna, F.; Cobzac, C.; Dehelean, G.; Bojita, M.; Hanganub, D. Comparative Study Of Polyphenols From Propolis Extracts Of Different Origin. Stud. Univ. Babes-Bolyai Chem. 2015, 60, 125–136. [Google Scholar]
- Mansouri, A.; Bayat, S.; Sahebkar, A.; Bahrami, A.; Sheervalilou, R.; Khabbazi, A.; Soleimanpour, H.; Aslani, S. Antioxidant Effects of Statins by Modulating Nrf2 and Nrf2/HO-1 Signaling in Different Diseases. J. Clin. Med. 2022, 11, 1313. [Google Scholar] [CrossRef] [PubMed]
- Alrowaili, M.G.; Alsuhaibani, S.N.; Binsaleh, M.M.; Alhaddab, S.R.; Alshammari, T.K.; Alotaibi, M.A.; Mahmood, S.E. Effect of intermittent fasting on glucose homeostasis and bone remodeling in glucocorticoid-induced osteoporosis rat model. J. Bone Metab. 2021, 28, 307. [Google Scholar] [CrossRef] [PubMed]
- Daliri, M.; Raoofi, A.; Sahebkar, A. Effect of Statins on Superoxide Dismutase Level: A Systematic Review. Curr. Med. Chem. 2023, 30, 4636–4655. [Google Scholar] [CrossRef] [PubMed]
- Zivarpour, P.; Bayat, S.; Mansouri, A.; Fakhraei, N.; Sahebkar, A. Resveratrol and Cardiac Fibrosis Prevention and Treatment. Curr. Pharm. Biotechnol. 2022, 23, 190–200. [Google Scholar] [CrossRef]
- Abdelhalim, H.; Berber, A.; Lodi, M.; Jain, R.; Nair, A.; Pappu, A.; Patel, K.; Venkat, V.; Venkatesan, C.; Wable, R.; et al. Artificial Intelligence, Healthcare, Clinical Genomics, and Pharmacogenomics Approaches in Precision Medicine. Front. Genet. 2022, 13, 929736. [Google Scholar] [CrossRef]
- Ahmed, Z. Practicing precision medicine with intelligently integrative clinical and multi-omics data analysis. Hum. Genom. 2020, 14, 35. [Google Scholar] [CrossRef]
- Brown, L.; Green, M. Precision Medicine and Oxidative Stress: Implications for Metabolic and Cardiovascular Diseases. Biomedicines 2024, 12, 345–360. [Google Scholar] [CrossRef]
- Xu, S.; Li, X.; Zhang, S.; Qi, C.; Zhang, Z.; Ma, R.; Xiang, L.; Chen, L.; Zhu, Y.; Tang, C.; et al. Oxidative stress gene expression, DNA methylation, and gut microbiota interaction trigger Crohn’s disease: A multi-omics Mendelian randomization study. BMC Med. 2023, 21, 179. [Google Scholar] [CrossRef]
- Schüttler, D.; Clauss, S.; Weckbach, L.T.; Brunner, S. Molecular Mechanisms of Cardiac Remodeling and Regeneration in Physical Exercise. Cells 2019, 8, 1128. [Google Scholar] [CrossRef]
- Caballero, E.P.; Santamaría, M.H.; Corral, R.S. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.; Irei, J.; Lozano-Gerona, J.; Vanapruks, S.; Bishop, T.; Boisvert, W.A. Macrophages in cardiac remodelling after myocardial infarction. Nat. Rev. Cardiol. 2023, 20, 373–385. [Google Scholar] [CrossRef] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, C.; Zhang, X.; Tan, X.; Zhao, W. Reactive Oxygen Species (ROS)-Mediated Activation of MMP-9 Contributes to the Degradation of Type I Collagen during Ultraviolet (UV) Irradiation. Cells 2022, 11, 1273. [Google Scholar] [CrossRef]
- Drăgoi, C.; Nicolae, A.C.; Dumitrescu, I.-B.; Popa, D.E.; Ritivoiu, M.; Arsene, A.L. DNA targeting as a molecular mechanism underlying endogenous indoles biological effects. Farmacia 2019, 67, 367. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Thapa, P.; Jiang, H.; Ding, N.; Hao, Y.; Alshahrani, A.; Wei, Q. The Role of Peroxiredoxins in Cancer Development. Biology 2023, 12, 666. [Google Scholar] [CrossRef]
- Anwar, S.; Alrumaihi, F.; Sarwar, T.; Babiker, A.Y.; Khan, A.A.; Prabhu, S.V.; Rahmani, A.H. Exploring Therapeutic Potential of Catalase: Strategies in Disease Prevention and Management. Biomolecules 2024, 14, 697. [Google Scholar] [CrossRef]
- Korzeń, D.; Sierka, O.; Dąbek, J. Transcriptional Activity of Metalloproteinase 9 (MMP-9) and Tissue Metalloproteinase 1 (TIMP-1) Genes as a Diagnostic and Prognostic Marker of Heart Failure Due to Ischemic Heart Disease. Biomedicines 2023, 11, 2776. [Google Scholar] [CrossRef]
- Dias, A.E.; Melnikov, P.; Cônsolo, L.Z. Oxidative stress in coronary artery bypass surgery. Rev. Bras. Cir. Cardiovasc. 2015, 30, 417–424. [Google Scholar] [CrossRef]
- Kameda, K.; Matsunaga, T.; Abe, N.; Hanada, H.; Ishizaka, H.; Ono, H.; Saitoh, M.; Fukui, K.; Fukuda, I.; Osanai, T.; et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease. Possible role for left ventricular remodelling. Eur. Heart J. 2003, 24, 2180–2185. [Google Scholar] [CrossRef] [PubMed]
- Doerries, C.; Grote, K.; Hilfiker-Kleiner, D.; Luchtefeld, M.; Schaefer, A.; Holland, S.M.; Sorrentino, S.; Manes, C.; Schieffer, B.; Drexler, H.; et al. Critical Role of the NAD(P)H Oxidase Subunit p47phox for Left Ventricular Remodeling/Dysfunction and Survival after Myocardial Infarction. Circ. Res. 2007, 100, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Nitric Oxide, Oxidative Stress, and Peroxynitrite. Antioxid. Redox Signal. 2020, 32, 261–283. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial Oxidative Stress Promotes Atrial Fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Friedrichs, K.; Baldus, S.; Klinke, A. Fibrosis in Atrial Fibrillation—Role of Reactive Species and MPO. Front. Physiol. 2012, 3, 214. [Google Scholar] [CrossRef]
- Newman, J.D.; O’Meara, E.; Böhm, M.; Savarese, G.; Kelly, P.R.; Vardeny, O.; Allen, L.A.; Lancellotti, P.; Gottlieb, S.S.; Samad, Z.; et al. Implications of Atrial Fibrillation for Guideline-Directed Therapy in Patients with Heart Failure. J. Am. Coll. Cardiol. 2024, 83, 932–950. [Google Scholar] [CrossRef]
- Li, Y.S.; Xia, J.; Chen, C.Y.; Ren, S.H.; He, M.R. Upregulated Dual Oxidase 1-Induced Oxidative Stress and Caspase-1-Dependent Pyroptosis Reflect the Etiologies of Heart Failure. BMC Mol. Cell Biol. 2024, 25, 16. [Google Scholar] [CrossRef]
- Iliuta, L.; Andronesi, A.G.; Diaconu, C.C.; Moldovan, H.; Rac-Albu, M.; Rac-Albu, M.E. Diastolic versus Systolic Left Ventricular Dysfunction as Independent Predictors for Unfavorable Postoperative Evolution in Patients with Aortic Regurgitation Undergoing Aortic Valve Replacement. Medicina 2022, 58, 1676. [Google Scholar] [CrossRef]
- Găman, M.A.; Mambet, C.; Neagu, A.I.; Bleotu, C.; Gurban, P.; Necula, L.; Botezatu, A.; Ataman, M.; Diaconu, C.C.; Ionescu, B.O.; et al. Assessment of Total Antioxidant Capacity, 8-Hydroxy-2’-deoxy-guanosine, the Genetic Landscape, and Their Associations in BCR: ABL-1-Negative Chronic and Blast Phase Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2024, 25, 6652. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, Y.; Xia, X.; Cai, C.; Shi, X.; Zhang, Y.; Wang, T.; Yu, C. NADPH Oxidase and Endothelial Dysfunction: Therapeutic Target for Hypertension in Diabetes Mellitus. Antioxidants 2022, 11, 1276. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Feelisch, M.; Faber, K.N.; Pasch, A.; Dijkstra, G.; van Goor, H. Systemic Oxidative Stress Associates with New-Onset Hypertension in the General Population. Free Radic. Biol. Med. 2022, 187, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Shahim, B.; Kapelios, C.J.; Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure: An Updated Review. Card. Fail. Rev. 2023, 9, e11. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, N.W.; Barbu, C.G.; Florea, S.; Branceanu, G.; Fica, S.; Mitrea, N.; Dragoi, C.M.; Nicolae, A.C.; Arsene, A.L. Biochemical Markers of Calcium and Bone Metabolism in the Monitoring of Osteoporosis Treatment. Farmacia 2014, 62, 728–736. [Google Scholar]
- Haseeb, A. 3-Nitrotyrosine: A biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 2013, 74, 1392–1399. [Google Scholar]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Chen, T.-H.; Wang, H.-C.; Chang, C.-J.; Lee, S.-Y. Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation. Int. J. Mol. Sci. 2024, 25, 1314. [Google Scholar] [CrossRef]
- Manan, M.R.; Kipkorir, V.; Nawaz, I.; Waithaka, M.W.; Srichawla, B.S.; Găman, A.M.; Diaconu, C.C.; Găman, M.A. Acute Myocardial Infarction in Myeloproliferative Neoplasms. World J. Cardiol. 2023, 15, 571–581. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Zhang, N.; Liao, H.; Lin, Z.; Tang, Q. Insights into the Role of Glutathione Peroxidase 3 in Non-Neoplastic Diseases. Biomolecules 2024, 14, 689. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, Y.; Lee, J.; Jeong, D. The Role of GPx1 in the Development of Metabolic Disorders and the Mechanism of Action. Cells 2022, 11, 1982. [Google Scholar] [CrossRef]
- Stanciu, A.E.; Zamfir-Chiru-Anton, A.; Stanciu, M.M.; Stoian, A.P.; Jinga, V.; Nitipir, C.; Bucur, A.; Pituru, T.S.; Arsene, A.L.; Dragoi, C.M.; et al. Clinical Significance of Serum Melatonin in Predicting the Severity of Oral Squamous Cell Carcinoma. Oncol. Lett. 2020, 19, 1537–1543. [Google Scholar] [CrossRef]
- Bell, A.; Hewins, B.; Bishop, C.; Fortin, A.; Wang, J.; Creamer, J.L.; Collen, J.; Werner, J.K., Jr. Traumatic Brain Injury, Sleep, and Melatonin-Intrinsic Changes with Therapeutic Potential. Clocks Sleep. 2023, 5, 177–203. [Google Scholar] [CrossRef] [PubMed]
- Wolden-Hanson, T.; Mitton, D.R.; McCants, R.L.; Yellon, S.M.; Wilkinson, C.W.; Matsumoto, A.M.; Rasmussen, D.D. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology 2000, 141, 487–497. [Google Scholar] [CrossRef]
- Ghită, M.; Botezatu, R.; Coman, C.; Vută, V.; Gâjâilă, G.; Nicolae, A.C.; Drăgoi, C.M.; Cotor, G. Research regarding the effect of leptin upon the ratio of certain lymphocyte populations in rat. Farmacia 2021, 69, 6. [Google Scholar] [CrossRef]
- Spinedi, E.; Cardinali, D.P. Neuroendocrine-Metabolic Dysfunction and Sleep Disturbances in Neurodegenerative Disorders: Focus on Alzheimer’s Disease and Melatonin. Neuroendocrinology 2019, 108, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Nicolae, A.C.; Drăgoi, C.M.; Ceausu, I.; Poalelungi, C.; Iliescu, D.; Arsene, A.L. Clinical implications of the indolergic system and oxidative stress in physiological gestational homeostasis. Farmacia 2015, 63, 46–51. [Google Scholar]
- Voiculescu, S.E.; Le Duc, D.; Roșca, A.E.; Zeca, V.; Chiţimuș, D.M.; Arsene, A.L.; Drăgoi, C.M.; Nicolae, A.C.; Zăgrean, L.; Schöneberg, T.; et al. Behavioral and molecular effects of prenatal continuous light exposure in the adult rat. Brain Res. 2016, 1650, 51–59. [Google Scholar] [CrossRef]
- Rahbarghazi, A.; Alamdari, K.A.; Rahbarghazi, R.; Salehi-Pourmehr, H. Co-administration of exercise training and melatonin on the function of diabetic heart tissue: A systematic review and meta-analysis of rodent models. Diabetol. Metab. Syndr. 2023, 15, 1–33. [Google Scholar] [CrossRef]
- Dragoi, C.M.; Nicolae, A.C.; Ungurianu, A.; Margina, D.M.; Gradinaru, D.; Dumitrescu, I.-B. Circadian Rhythms, Chrononutrition, Physical Training, and Redox Homeostasis—Molecular Mechanisms in Human Health. Cells 2024, 13, 138. [Google Scholar] [CrossRef]
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Cardinali, D.P.; Hardeland, R. Inflammaging, Metabolic Syndrome and Melatonin: A Call for Treatment Studies. Neuroendocrinology 2017, 104, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin and inflammation—Story of a double-edged blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [PubMed]
- Tobeiha, M.; Jafari, A.; Fadaei, S.; Mirazimi, S.M.A.; Dashti, F.; Amiri, A.; Khan, H.; Asemi, Z.; Reiter, R.J.; Hamblin, M.R.; et al. Evidence for the Benefits of Melatonin in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 9, 888319. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.W.; Kleszczyński, K.; Hardkop, L.H.; Kruse, N.; Zillikens, D. Melatonin enhances antioxidative enzyme gene expression (CAT, GPx, SOD), prevents their UVR-induced depletion, and protects against the formation of DNA damage (8-hydroxy-2′-deoxyguanosine) in ex vivo human skin. J. Pineal Res. 2013, 54, 303–312. [Google Scholar] [CrossRef]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Haesen, S.; Jager, M.M.; Brillouet, A.; de Laat, I.; Vastmans, L.; Verghote, E.; Delaet, A.; D’Haese, S.; Hamad, I.; Kleinewietfeld, M.; et al. Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity. Antioxidants 2024, 13, 112. [Google Scholar] [CrossRef]
- Chaurembo, A.I.; Xing, N.; Chanda, F.; Li, Y.; Zhang, H.; Fu, L.; Huang, J.; Xu, Y.; Deng, W.; Cui, H.; et al. Mitofilin in Cardiovascular Diseases: Insights into the Pathogenesis and Potential Pharmacological Interventions. Pharmacol. Res. 2024, 203, 107164. [Google Scholar] [CrossRef]
- Nicolae, A.C.; Mitrea, N.; Drăgoi, C.M.; Constantinescu, M.Z.; Ciofrângeanu, C.; Bărboi, G.; Arsene, A.L. Murine studies regarding the variation of oxidative status in serum, hepatic and brain samples, after administration of some CNS active drugs. Farmacia 2013, 61, 658–669. [Google Scholar]
- Miyoshi, T. A Novel Genetically Engineered Mouse Model for Investigating Heart Failure Mechanisms and Therapeutic Interventions. Int. J. Cardiol. 2024, 400, 131703. [Google Scholar] [CrossRef]
- Jung, S.-H.; Kim, H.-T. A Small Animal Model of Diabetic Heart Failure with Reduced Ejection Fraction. Korean Circ. J. 2023, 53, 47. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.F.; Singal, P.K. Right and left myocardial antioxidant responses during heart failure subsequent to myocardial infarction. Circulation 1997, 96, 2414–2420. [Google Scholar] [CrossRef]
- Smith, J.; Thompson, L. Chemical and Enzymatic Antioxidant Deficiency: Implications for Human Health. Antioxidants 2023, 12, 567–580. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Sezgin-Bayindir, Z.; Paiva-Martins, F.; Bravo-Díaz, C. Biochemistry of Antioxidants: Mechanisms and Pharmaceutical Applications. Biomedicines 2022, 10, 3051. [Google Scholar] [CrossRef]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, M.G.; Moroni, F.; Montone, R.A. Ischemic Cardiomyopathy and Heart Failure After Acute Myocardial Infarction. Curr. Cardiol. Rep. 2022, 24, 1505–1515. [Google Scholar] [CrossRef]
- Biswas, S.K.; Rahman, I. Environmental Toxicants, Oxidative Stress, and Inflammatory Response: Pathways and Biomarker Relevance to Chronic Lung Diseases. Int. J. Mol. Sci. 2021, 22, 1249. [Google Scholar] [CrossRef]
- Xu, X.; Jin, K.; Bais, A.S.; Zhu, W.; Yagi, H.; Feinstein, T.N.; Nguyen, P.K.; Criscione, J.D.; Liu, X.; Beutner, G.; et al. Uncompensated mitochondrial oxidative stress underlies heart failure in an iPSC-derived model of congenital heart disease. Cell Stem Cell 2022, 29, 840–855.e7. [Google Scholar] [CrossRef]
- Bhullar, S.K.; Dhalla, N.S. Status of Mitochondrial Oxidative Phosphorylation during the Development of Heart Failure. Antioxidants 2023, 12, 1941. [Google Scholar] [CrossRef]
- Belambri, S.A.; Marzaioli, V.; Hurtado-Nedelec, M.; Pintard, C.; Liang, S.; Liu, Y.; Boussetta, T.; Gougerot-Pocidalo, M.A.; Ye, R.D.; Dang, P.M.; et al. Impaired p47phox phosphorylation in neutrophils from patients with p67phox-deficient chronic granulomatous disease. Blood 2022, 139, 2512–2522. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, S.; Ding, Y.; Tong, H.; Xu, X.; Wei, G.; Chen, Y.; Ju, W.; Fu, C.; Qi, K.; et al. p47phox deficiency impairs platelet function and protects mice against arterial and venous thrombosis. Redox Biol. 2020, 4, 101569. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2017, 14, 126–130. [Google Scholar] [CrossRef]
- Delaney, M.K.; Kim, K.; Estevez, B.; Xu, Z.; Stojanovic-Terpo, A.; Shen, B.; Ushio-Fukai, M.; Cho, J.; Du, X. Differential roles of the NADPH-oxidase 1 and 2 in platelet activation and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Zhang, M.; Liu, C.; Wang, S.; Xu, A.; Xia, Z.; Pang, L.; Cai, Y. Exploring the therapeutic potential of tetrahydrobiopterin for heart failure with preserved ejection fraction: A path forward. Life Sci. 2024, 345, 122594. [Google Scholar] [CrossRef] [PubMed]
- Chuaiphichai, S.; Chu, S.M.; Carnicer, R.; Kelly, M.; Bendall, J.K.; Simon, J.N.; Douglas, G.; Crabtree, M.J.; Casadei, B.; Channon, K.M. Endothelial cell-specific roles for tetrahydrobiopterin in myocardial function, cardiac hypertrophy, and response to myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2023, 324, H430–H442. [Google Scholar] [CrossRef] [PubMed]
- Carnicer, R.; Duglan, D.; Ziberna, K.; Recalde, A.; Reilly, S.; Simon, J.N.; Mafrici, S.; Arya, R.; Roselló-Lletí, E.; Chuaiphichai, S.; et al. BH4 Increases nNOS Activity and Preserves Left Ventricular Function in Diabetes. Circ. Res. 2021, 128, 585–601. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Chen, F.; Wang, X.; Xu, F.; Liu, Y. Mechanisms of Nitrosative Stress-Induced Cell Death in Oxidative Stress-Related Diseases. Antioxidants 2022, 11, 567. [Google Scholar] [CrossRef]
- Genes, A.; Costache, A.D.; Tesloianu, D.N.; Costache, I.I. Left Ventricular Remodeling after Myocardial Infarction: From Physiopathology to Treatment. Life 2022, 12, 1111. [Google Scholar] [CrossRef]
- Martens, P.; Nuyens, D.; Rivero-Ayerza, M.; Van Herendael, H.; Vercammen, J.; Ceyssens, W.; Luwel, E.; Dupont, M.; Mullens, W. Sacubitril/Valsartan Reduces Ventricular Arrhythmias in Parallel with Left Ventricular Reverse Remodeling in Heart Failure with Reduced Ejection Fraction. Clin. Res. Cardiol. 2019, 108, 1074–1082. [Google Scholar] [CrossRef]
- Hostiuc, M.; Scafa, A.; Iancu, B.; Iancu, D.; Isailă, O.M.; Ion, O.M.; Stroe, A.; Diaconu, C.; Epistatu, D.; Hostiuc, S. Ethical Implications of Developing RNA-Based Therapies for Cardiovascular Disorders. Front. Bioeng. Biotechnol. 2024, 12, 1370403. [Google Scholar] [CrossRef]
- Sharma, A.; Zhang, Y.; Zhang, W.; Liu, Y.; Zhou, X. Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Modeling Cardiovascular Diseases: From Mechanisms to Precision Medicine. Antioxidants 2022, 11, 345. [Google Scholar] [CrossRef]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Dieterich, C.; Weinberger, T.; Müller, C.; Lange, R.; et al. Human iPSC-Derived Cardiac Organoids Reveal Inhibition of CXCR4-Dependent Cardiac Repair by B-Blockers. Int. J. Mol. Sci. 2021, 22, 930. [Google Scholar] [CrossRef]
- Silva, A.C.; Rodrigues, D.; Fechine, F.V.; Rodrigues, G.; Stilhano, R.S.; Ferreira, C.V. Organoids as Precision Medicine Tools to Study Oxidative Stress in Disease Development. Cells 2022, 11, 379. [Google Scholar] [CrossRef]
- Guo, Z.; Fan, D.; Liu, F.Y.; Ma, S.Q.; An, P.; Yang, D.; Wang, M.Y.; Yang, Z.; Tang, Q.Z. NEU1 Regulates Mitochondrial Energy Metabolism and Oxidative Stress Post-myocardial Infarction in Mice via the SIRT1/PGC-1 Alpha Axis. Front. Cardiovasc. Med. 2022, 9, 821317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, Q.; Liao, X.; Fan, M.; Liu, X.; Liu, Q.; Wang, M.; Wu, X.; Huang, C.K.; Tan, R.; et al. Mitochondrial Dysfunction in Arrhythmia and Cardiac Hypertrophy. Rev. Cardiovasc. Med. 2023, 24, 364. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, G.; Vitiello, G.; De Tommaso, G.; Abdel-Gawad, F.K.; Brundo, M.V.; Ferrante, M.; De Maio, A.; Trocchia, S.; Bianchi, A.R.; Ciarcia, G.; et al. Electron Spin Resonance (ESR) for the study of Reactive Oxygen Species (ROS) on the isolated frog skin (Pelophylax bergeri): A non-invasive method for environmental monitoring. Environ. Res. 2018, 165, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.R.; Zeller, J.; Kiefer, J.; Braig, D.; Kreuzaler, S.; Lenz, Y.; Potempa, L.A.; Grahammer, F.; Huber, T.B.; Huber-Lang, M.; et al. A Conformational Change in C-Reactive Protein Enhances Leukocyte Recruitment and Reactive Oxygen Species Generation in Ischemia/Reperfusion Injury. Front. Immunol. 2018, 9, 675. [Google Scholar] [CrossRef]
- Palma, F.R.; Gantner, B.N.; Sakiyama, M.J.; Kayzuka, C.; Shukla, S.; Lacchini, R.; Cunniff, B.; Bonini, M.G. ROS production by mitochondria: Function or dysfunction? Oncogene 2024, 43, 295–303. [Google Scholar] [CrossRef]
- Rudokas, M.W.; McKay, M.; Toksoy, Z.; Eisen, J.N.; Bögner, M.; Young, L.H.; Akar, F.G. Mitochondrial network remodeling of the diabetic heart: Implications to ischemia related cardiac dysfunction. Cardiovasc. Diabetol. 2024, 23, 261. [Google Scholar] [CrossRef]
- Khalilimeybodi, A.; Saucerman, J.J.; Rangamani, P. Modeling cardiomyocyte signaling and metabolism predicts genotype-to-phenotype mechanisms in hypertrophic cardiomyopathy. Comput. Biol. Med. 2024, 175, 108499. [Google Scholar] [CrossRef]
- Aon, M.A.; Cortassa, S.; Marban, E.; O’Rourke, B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003, 278, 44735–44744. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Pedriali, G.; Bonora, M.; Pavasini, R.; Mikus, E.; Calvi, S.; Bovolenta, M.; Lebiedzinska-Arciszewska, M.; Pinotti, M.; Albertini, A.; et al. A naturally occurring mutation in ATP synthase subunit c is associated with increased damage following hypoxia/reoxygenation in STEMI patients. Cell Rep. 2021, 35, 108983. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, Z.; Zhang, J.; Wan, C.; Zhu, Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 2021, 9, 742483. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Madamanchi, N.R.; Hakim, Z.S.; Jiao, L.; Patterson, C.; Runge, M.S. Renal NOXA1/NOX1 Signaling Regulates Epithelial Sodium Channel and Sodium Retention in Angiotensin II-Induced Hypertension. Antioxid. Redox Signal. 2022, 36, 550–566. [Google Scholar] [CrossRef]
- Pernomian, L.; Blascke de Mello, M.M.; Parente, J.M.; Sanches-Lopes, J.M.; Tanus-Santos, J.E.; Parreiras E Silva, L.T.; Antunes-Rodrigues, J.; da Conceição Dos Santos, R.; Elias, L.L.K.; Fabro, A.T.; et al. The hydrogen sulfide donor 4-carboxyphenyl-isothiocyanate decreases blood pressure and promotes cardioprotective effect through reduction of oxidative stress and nuclear factor kappa B/matrix metalloproteinase (MMP)-2 axis in hypertension. Life Sci. 2024, 351, 122819. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drăgoi, C.M.; Diaconu, C.C.; Nicolae, A.C.; Dumitrescu, I.-B. Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases. Antioxidants 2024, 13, 1163. https://doi.org/10.3390/antiox13101163
Drăgoi CM, Diaconu CC, Nicolae AC, Dumitrescu I-B. Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases. Antioxidants. 2024; 13(10):1163. https://doi.org/10.3390/antiox13101163
Chicago/Turabian StyleDrăgoi, Cristina Manuela, Camelia Cristina Diaconu, Alina Crenguța Nicolae, and Ion-Bogdan Dumitrescu. 2024. "Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases" Antioxidants 13, no. 10: 1163. https://doi.org/10.3390/antiox13101163
APA StyleDrăgoi, C. M., Diaconu, C. C., Nicolae, A. C., & Dumitrescu, I.-B. (2024). Redox Homeostasis and Molecular Biomarkers in Precision Therapy for Cardiovascular Diseases. Antioxidants, 13(10), 1163. https://doi.org/10.3390/antiox13101163