
Mitochondrial Quality Control in Alzheimer’s Disease: Insights from Caenorhabditis elegans Models



Abstract
:1. Introduction
2. C. elegans as a Model Organism to Study Neurodegeneration and Mitochondrial Biology in Alzheimer’s Disease (AD)
2.1. APL-1 Models
2.2. Amyloid-β Models
2.3. PTL-1 Models
2.4. Tau Models
2.5. APOE Models
2.6. Presenilin Models
2.7. The Limitations of the C. elegans Model for Studying AD Pathology
3. Mitophagy in Alzheimer’s Disease
4. Mitophagy in Worms: Mechanisms and Regulation
4.1. PINK-1-PDR-1-Dependent Mitophagy Pathway
4.2. PINK-1/PDR-1-Independent Mitophagy Pathway
5. Contribution of the C. elegans Model Organism to Better Understand Mitophagy Dysregulation in Alzheimer’s Disease
6. Potential Causes of Dysfunctional MQC in AD
6.1. Failure to Recruit Mitophagy Receptors and Autophagy Receptors Required for Mitophagy
6.2. Compromised Mitochondrial Dynamics
6.3. Impaired Mitochondrial Biogenesis
6.4. Impaired Organellar Communication and Trafficking
6.5. Mito-UPR as an Intersecting Stress Signaling Pathway
6.6. Impaired Lysosomal Activity
6.7. Signaling Pathways That Control Both Mitophagy and Autophagy—The mTOR-AMPK Axis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer’s Association. 2024 Alzheimer’s disease facts and figures. Alzheimers Dement 2024, 20, 3708–3821. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Lee, V.M.; Balin, B.J.; Otvos, L., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Piaceri, I. Genetics of familial and sporadic Alzheimer s disease. Front. Biosci. 2013, E5, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, B.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sa, J.; da Cruz, E.S.O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Latina, V.; Valenti, D. Therapeutic Potential of Targeting Mitochondria for Alzheimer’s Disease Treatment. J. Clin. Med. 2022, 11, 6742. [Google Scholar] [CrossRef]
- Ng MY, W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Dev. Cell 2021, 56, 881–905. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Kim, W.; Hendricks, G.L.; Lee, K.; Mylonakis, E. An update on the use of C. elegans for preclinical drug discovery: Screening and identifying anti-infective drugs. Expert Opin. Drug Discov. 2017, 12, 625–633. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, Y.; Chen, Y.; Cheng, B.; Peng, A.; Huang, K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur. J. Pharmacol. 2018, 819, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Markaki, M.; Tavernarakis, N. Modeling human diseases in Caenorhabditis elegans. Biotechnol. J. 2010, 5, 1261–1276. [Google Scholar] [CrossRef] [PubMed]
- Roussos, A.; Kitopoulou, K.; Borbolis, F.; Palikaras, K. Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders. Biomolecules 2023, 13, 478. [Google Scholar] [CrossRef] [PubMed]
- Onraet, T.; Zuryn, S. C. elegans as a model to study mitochondrial biology and disease. Semin. Cell Dev. Biol. 2024, 154 Pt A, 48–58. [Google Scholar] [CrossRef]
- Campbell, D.; Zuryn, S. The mechanisms and roles of mitochondrial dynamics in C. elegans. Semin. Cell Dev. Biol. 2024, 156, 266–275. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Kaymak, E.; Farley, B.M.; Hay, S.A.; Li, C.; Ho, S.; Hartman, D.J.; Ryder, S.P. Efficient generation of transgenic reporter strains and analysis of expression patterns in Caenorhabditis elegans using library MosSCI. Dev. Dyn. 2016, 245, 925–936. [Google Scholar] [CrossRef]
- Apostolakou, A.E.; Sula, X.K.; Nastou, K.C.; Nasi, G.I.; Iconomidou, V.A. Exploring the conservation of Alzheimer-related pathways between H. sapiens and C. elegans: A network alignment approach. Sci. Rep. 2021, 11, 4572. [Google Scholar] [CrossRef]
- Alvarez, J.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines 2022, 10, 288. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Cheng, R.; Tolen, L.; Shah, V.; Gillani, A.; Nasrin, A.; Li, C. Pan-neuronal expression of APL-1, an APP-related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans. J. Neurosci. 2012, 32, 10156–10169. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D.; Taft, A.; Kapulkin, V.; Duke, K.; Kim, S.; Fei, Q.; Wood, D.E.; Sahagan, B.G. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol. Aging 2023, 24, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Gallrein, C.; Iburg, M.; Michelberger, T.; Kocak, A.; Puchkov, D.; Liu, F.; Mariscal, S.M.A.; Nayak, T.; Schierle, G.S.K.; Korstein, J. Novel amyloid-beta pathology C. elegans model reveals distinct neurons as seeds of pathogenicity. Prog. Neurobiol. 2021, 198, 101907. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef]
- Guha, S.; Fischer, S.; Johnson GV, W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Russell, J.C.; Lei, H.; Chaliparambil, R.K.; Fish, S.; Markiewicz, S.M.; Lee, T.I.; Noori, A.; Kaeberlein, M. Generation and characterization of a tractable C. elegans model of tauopathy. Geroscience 2021, 43, 2621–2631. [Google Scholar] [CrossRef]
- Levitan, D.; Greenwald, I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995, 377, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef]
- Sae-Lee, W.; Scott, L.L.; Brose, L.; Encarnacion, A.J.; Shi, T.; Kore, P.; Oyibo, L.O.; Ye, C.; Rozmiarek, S.; Pierce, J.T. APP-Induced Patterned Neurodegeneration Is Exacerbated by APOE4 in Caenorhabditis elegans. G3 2020, 10, 2851–2861. [Google Scholar] [CrossRef]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Li, C. Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct. Funct. 2010, 214, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Hornsten, A.; Lieberthal, J.; Fadia, S.; Malins, R.; Ha, L.; Xu, X.; Daigle, I.; Markowitz, M.; O’ Connor, G.; Plasterk, R.; et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc. Natl. Acad. Sci. USA 2007, 104, 1971–1976. [Google Scholar] [CrossRef]
- Kim, B.; Elzinga, S.E.; Henn, R.E.; McGinley, L.M.; Feldman, E.L. The effects of insulin and insulin-like growth factor I on amyloid precursor protein phosphorylation in in vitro and in vivo models of Alzheimer’s disease. Neurobiol. Dis. 2019, 132, 104541. [Google Scholar] [CrossRef] [PubMed]
- Ezkurdia, A.; Ramirez, M.J.; Solas, M. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: A Focus on Insulin Resistance. Int. J. Mol. Sci. 2023, 24, 4354. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Goncalves, R.A.; Ferreira, S.T. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat. Rev. Neurosci. 2022, 23, 215–230. [Google Scholar] [CrossRef]
- Athanasaki, A.; Melanis, K.; Tsantzali, I.; Stefanou, M.I.; Ntymenou, S.; Paraskevas, S.G.; Kalamatianos, T.; Boutati, E.; Lambadiari, V.; Voumvoukaris, K.I.; et al. Type 2 Diabetes Mellitus as a Risk Factor for Alzheimer’s Disease: Review and Meta-Analysis. Biomedicines 2022, 10, 778. [Google Scholar] [CrossRef]
- Fay, D.S.; Fluet, A.; Johnson, C.J.; Link, C.D. In vivo aggregation of beta-amyloid peptide variants. J. Neurochem. 1998, 71, 1616–1625. [Google Scholar] [CrossRef]
- Fonte, V.; Kapulkin, W.J.; Taft, A.; Fluet, A.; Friedman, D.; Link, C.D. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 9439–9444. [Google Scholar] [CrossRef]
- Fonte, V.; Kipp, D.R.; Yerg, J., 3rd; Merin, D.; Forrestal, M.; Wagner, E.; Roberts, C.M.; Link, C.D. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J. Biol. Chem. 2008, 283, 784–791. [Google Scholar] [CrossRef]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Vowels, J.J.; Thomas, J.H. Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics 1992, 130, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Honda, S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J. 1999, 13, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 13091–13096. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Johnson, T.E. A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics 1996, 143, 1207–1218. [Google Scholar] [CrossRef]
- Johnson, T.E.; Cypser, J.; de Castro, E.; de Castro, S.; Henderson, S.; Murakami, S.; Rikke, B.; Tedesco, P.; Link, C. Gerontogenes mediate health and longevity in nematodes through increasing resistance to environmental toxins and stressors. Exp. Gerontol. 2000, 35, 687–694. [Google Scholar] [CrossRef]
- Ahmad, W.; Ebert, P.R. Metformin Attenuates Abeta Pathology Mediated Through Levamisole Sensitive Nicotinic Acetylcholine Receptors in a C. elegans Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 5427–5439. [Google Scholar] [CrossRef]
- Steinkraus, K.A.; Smith, E.D.; Davis, C.; Carr, D.; Pendergrass, W.R.; Sutphin, G.L.; Kennedy, B.K.; Kaeberlein, M. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell 2008, 7, 394–404. [Google Scholar] [CrossRef]
- Mouton, P.R.; Chachich, M.E.; Quigley, C.; Spangler, E.; Ingram, D.K. Caloric restriction attenuates amyloid deposition in middle-aged dtg APP/PS1 mice. Neurosci. Lett. 2009, 464, 184–187. [Google Scholar] [CrossRef]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef]
- Schafer, M.J.; Alldred, M.J.; Lee, S.H.; Calhoun, M.E.; Petkova, E.; Mathews, P.M.; Ginsberg, S.D. Reduction of beta-amyloid and gamma-secretase by calorie restriction in female Tg2576 mice. Neurobiol. Aging 2015, 36, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ho, L.; Qin, W.; Rocher, A.B.; Seror, I.; Humala, N.; Maniar, K.; Dolios, G.; Wang, R.; Hof, P.R.; et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2005, 19, 659–661. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Power Guerra, N.; Stenzel, J.; Ruhlmann, C.; Lindner, T.; Krause, B.J.; Vollmar, B.; Teipel, S.; Kuhla, A. Long-Term Caloric Restriction Attenuates beta-Amyloid Neuropathology and Is Accompanied by Autophagy in APPswe/PS1delta9 Mice. Nutrients 2021, 13, 985. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef]
- Gomez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef]
- Morsch, R.; Simon, W.; Coleman, P.D. Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 1999, 58, 188–197. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef]
- Goedert, M.; Baur, C.P.; Ahringer, J.; Jakes, R.; Hasegawa, M.; Spillantini, M.G.; Smith, M.J.; Hill, F. PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J. Cell Sci. 1996, 109 Pt 11, 2661–2672. [Google Scholar] [CrossRef]
- Gordon, P.; Hingula, L.; Krasny, M.L.; Swienckowski, J.L.; Pokrywka, N.J.; Raley-Susman, K.M. The invertebrate microtubule-associated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev. Genes Evol. 2008, 218, 541–551. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E. Caenorhabditis elegans models of tauopathy. FASEB J. 2017, 31, 5137–5148. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.W.; Wu, G.H.; Hsu, C.C.; Chang, C.Y.; Wagner, O.I. Tau/PTL-1 associates with kinesin-3 KIF1A/UNC-104 and affects the motor’s motility characteristics in C. elegans neurons. Neurobiol. Dis. 2011, 43, 495–506. [Google Scholar] [CrossRef]
- Chew, Y.L.; Fan, X.; Gotz, J.; Nicholas, H.R. PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 2013, 126 Pt 9, 2079–2091. [Google Scholar] [CrossRef]
- Chew, Y.L.; Gotz, J.; Nicholas, H.R. Neuronal protein with tau-like repeats (PTL-1) regulates intestinal SKN-1 nuclear accumulation in response to oxidative stress. Aging Cell 2015, 14, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Krieg, M.; Stuhmer, J.; Cueva, J.G.; Fetter, R.; Spilker, K.; Cremers, D.; Shen, K.; Dunn, A.R.; Goodman, M.B. Genetic defects in beta-spectrin and tau sensitize C. elegans axons to movement-induced damage via torque-tension coupling. eLife 2017, 6, e20172. [Google Scholar] [CrossRef] [PubMed]
- Lins, J.; Hopkins, C.E.; Brock, T.; Hart, A.C. The use of CRISPR to generate a whole-gene humanized MAPT and the examination of P301L and G272V clinical variants, along with the creation of deletion null alleles of ptl-1, pgrn-1 and alfa-1 loci. MicroPubl. Biol. 2022, 2022. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 33. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Butler, V.J.; Salazar, D.A.; Soriano-Castell, D.; Alves-Ferreira, M.; Dennissen, F.J.A.; Vohra, M.; Oses-Prieto, J.A.; Li, K.H.; Wang, A.L.; Jing, B.; et al. Tau/MAPT disease-associated variant A152T alters tau function and toxicity via impaired retrograde axonal transport. Hum. Mol. Genet. 2019, 28, 1498–1514. [Google Scholar] [CrossRef]
- Han, M.; Saxton, A.; Currey, H.; Waldherr, S.M.; Liachko, N.F.; Kraemer, B.C. Transgenic Dendra2::tau expression allows in vivo monitoring of tau proteostasis in Caenorhabditis elegans. Dis. Model. Mech. 2024, 17, dmm050473. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.A.; Liachko, N.F.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443. [Google Scholar] [CrossRef]
- Liu, S.Y.; Wang, W.; Cai, Z.Y.; Yao, L.F.; Chen, Z.W.; Wang, C.Y.; Zhao, B.; Li, K.S. Polymorphism -116C/G of human X-box-binding protein 1 promoter is associated with risk of Alzheimer’s disease. CNS Neurosci. Ther. 2013, 19, 229–234. [Google Scholar] [CrossRef]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8, 180024. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER stress and UPR in Alzheimer’s disease: Mechanisms, pathogenesis, treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar] [CrossRef]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Cheng, A.; Carroll, T.; King, D.; Koren, S.A.; Swords, S.; Nehrke, K.; Johnson, G.V.W. Selective disruption of Drp1-independent mitophagy and mitolysosome trafficking by an Alzheimer’s disease relevant tau modification in a novel Caenorhabditis elegans model. Genetics 2022, 222, iyac104. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Achanta, K.; Choi, S.; Akbari, M.; Bohr, V.A. Alteration of mitochondrial homeostasis is an early event in a C. elegans model of human tauopathy. Aging 2021, 13, 23876–23894. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Islam, S.; Sun, Y.; Gao, Y.; Nakamura, T.; Noorani, A.A.; Li, T. Presenilin Is Essential for ApoE Secretion, a Novel Role of Presenilin Involved in Alzheimer’s Disease Pathogenesis. J. Neurosci. 2022, 42, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, C.C.; Chen, X.F.; Zhang, Y.W.; Xu, H.; Bu, G. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and Abeta metabolism in apoE4-targeted replacement mice. Mol. Neurodegener. 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Griffin, E.F.; Scopel, S.E.; Stephen, C.A.; Holzhauer, A.C.; Vaji, M.A.; Tuckey, R.A.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. ApoE-associated modulation of neuroprotection from Abeta-mediated neurodegeneration in transgenic Caenorhabditis elegans. Dis. Model. Mech. 2019, 12, dmm037218. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.I.; Cao, Y.; Xue, Y.; Ji, Y.; Winer, B.Y.; Zhang, M.; Singhal, N.S.; Pierce, J.T.; Chen, S.; Ma, D.K. Suppressing APOE4-induced mortality and cellular damage by targeting VHL. bioRxiv 2024. [Google Scholar] [CrossRef]
- Koks, S.; Pfaff, A.L.; Bubb, V.J.; Quinn, J.P. Transcript Variants of Genes Involved in Neurodegeneration Are Differentially Regulated by the APOE and MAPT Haplotypes. Genes 2021, 12, 423. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, Y.; Xiao, Z.; Zhao, Q.; Wu, W.; Liang, X.; Luo, J.; Cao, Y.; Shao, M.; Guo, Q.; et al. TOMM40 and APOE variants synergistically increase the risk of Alzheimer’s disease in a Chinese population. Aging Clin. Exp. Res. 2021, 33, 1667–1675. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wijsman, E.M.; Nemens, E.; Anderson, L.; Goddard, K.A.; Weber, J.L.; Bird, T.D.; Schellenberg, G.D. A familial Alzheimer’s disease locus on chromosome 1. Science 1995, 269, 970–973. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Petit, D.; Fernandez, S.G.; Zoltowska, K.M.; Enzlein, T.; Ryan, N.S.; O’Connor, A.; Szaruga, M.; Hill, E.; Vandenberghe, R.; Fox, N.C.; et al. Abeta profiles generated by Alzheimer’s disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol. Psychiatry 2022, 27, 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Duff, K.; Eckman, C.; Zehr, C.; Yu, X.; Prada, C.M.; Perez-tur, J.; Hutton, M.; Buee, L.; Harigaya, Y.; Yager, D.; et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin. Nature 1996, 383, 710–713. [Google Scholar] [CrossRef]
- Sun, L.; Zhou, R.; Yang, G.; Shi, Y. Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Abeta42 and Abeta40 peptides by gamma-secretase. Proc. Natl. Acad. Sci. USA 2017, 114, E476–E485. [Google Scholar] [CrossRef]
- Saura, C.A.; Choi, S.Y.; Beglopoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chattarji, S.; Kelleher, R.J., 3rd; Kandel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Wines-Samuelson, M.; Schulte, E.C.; Smith, M.J.; Aoki, C.; Liu, X.; Kelleher, R.J., 3rd; Shen, J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS ONE 2010, 5, e10195. [Google Scholar] [CrossRef]
- L’Hernault, S.W.; Arduengo, P.M. Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J. Cell Biol. 1992, 119, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Cinar, H.N.; Sweet, K.L.; Hosemann, K.E.; Earley, K.; Newman, A.P. The SEL-12 presenilin mediates induction of the Caenorhabditis elegans uterine pi cell fate. Dev. Biol. 2001, 237, 173–182. [Google Scholar] [CrossRef]
- Levitan, D.; Doyle, T.G.; Brousseau, D.; Lee, M.K.; Thinakaran, G.; Slunt, H.H.; Sisodia, S.S.; Greenwald, I. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1996, 93, 14940–14944. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Lukinova, N.; Fortini, M.E. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature 1999, 398, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 1999, 398, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Wittenburg, N.; Eimer, S.; Lakowski, B.; Rohrig, S.; Rudolph, C.; Baumeister, R. Presenilin is required for proper morphology and function of neurons in C. elegans. Nature 2000, 406, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; Greenwald, I. Suppressors of the egg-laying defective phenotype of sel-12 presenilin mutants implicate the CoREST corepressor complex in LIN-12/Notch signaling in C. elegans. Genes Dev. 2002, 16, 2713–2728. [Google Scholar] [CrossRef]
- Kitagawa, N.; Shimohama, S.; Oeda, T.; Uemura, K.; Kohno, R.; Kuzuya, A.; Shibasaki, H.; Ishii, N. The role of the presenilin-1 homologue gene sel-12 of Caenorhabditis elegans in apoptotic activities. J. Biol. Chem. 2003, 278, 12130–12134. [Google Scholar] [CrossRef]
- Sarasija, S.; Norman, K.R. A gamma-Secretase Independent Role for Presenilin in Calcium Homeostasis Impacts Mitochondrial Function and Morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. eLife 2018, 7, e33052. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Sarasija, S.; Laboy, J.T.; Samarakoon, R.; Norman, K.R. Increased mitochondrial calcium uptake and concomitant mitochondrial activity by presenilin loss promotes mTORC1 signaling to drive neurodegeneration. Aging Cell 2021, 20, e13472. [Google Scholar] [CrossRef]
- Ryan, K.C.; Laboy, J.T.; Norman, K.R. Deregulation of Mitochondrial Calcium Handling Due to Presenilin Loss Disrupts Redox Homeostasis and Promotes Neuronal Dysfunction. Antioxidants 2022, 11, 1642. [Google Scholar] [CrossRef]
- Bargmann, C.I. Neurobiology of the Caenorhabditis elegans genome. Science 1998, 282, 2028–2033. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.B.; Hall, D.H.; Avery, L.; Lockery, S.R. Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron 1998, 20, 763–772. [Google Scholar] [CrossRef]
- Vargas JN, S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2023, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X.; Song, Y.Q.; Tu, J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell. 2014, 53, 167–178. [Google Scholar] [CrossRef]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif-crucial for selective autophagy. J. Cell Sci. 2013, 126 Pt 15, 3237–3247. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Rambold, A.S.; Lippincott-Schwartz, J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 2011, 10, 4032–4038. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.J.; Misrani, A.; Tabassum, S.; Yang, L. Mitophagy pathways and Alzheimer’s disease: From pathogenesis to treatment. Mitochondrion 2021, 59, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Yu, X.; Mahaman, Y.A.R.; Wang, J.Z.; Liu, R.; Li, Y.; Wang, X. Defective mitophagy and the etiopathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 32. [Google Scholar] [CrossRef]
- Mary, A.; Eysert, F.; Checler, F.; Chami, M. Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol. Psychiatry 2023, 28, 202–216. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, L.; Lu, L.; Sun, H.; Shao, M.; Beck, S.J.; Li, L.; Ramachandran, J.; Du, Y.; Du, H. Synaptosomal Mitochondrial Dysfunction in 5xFAD Mouse Model of Alzheimer’s Disease. PLoS ONE 2016, 11, e0150441. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.H.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef]
- Martin-Maestro, P.; Gargini, R.; Garcia, E.; Perry, G.; Avila, J.; Garcia-Escudero, V. Slower Dynamics and Aged Mitochondria in Sporadic Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 9302761. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Lefterov, I.; Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Letronne, F.; Biedrzycki, R.J.; Kofler, J.; Han, X.; Wang, J.; Schug, J.; et al. APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res. Ther. 2019, 11, 113. [Google Scholar] [CrossRef]
- Dawson, G.R.; Seabrook, G.R.; Zheng, H.; Smith, D.W.; Graham, S.; O’Dowd, G.; Bowery, B.J.; Boyce, S.; Trumbauer, M.E.; Chen, H.Y.; et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 1999, 90, 1–13. [Google Scholar] [CrossRef]
- Phinney, A.L.; Calhoun, M.E.; Wolfer, D.P.; Lipp, H.P.; Zheng, H.; Jucker, M. No hippocampal neuron or synaptic bouton loss in learning-impaired aged beta-amyloid precursor protein-null mice. Neuroscience 1999, 90, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Senechal, Y.; Prut, L.; Kelly, P.H.; Staufenbiel, M.; Natt, F.; Hoyer, D.; Wiessner, C.; Dev, K.K. Increased exploratory activity of APP23 mice in a novel environment is reversed by siRNA. Brain Res. 2008, 1243, 124–133. [Google Scholar] [CrossRef]
- Ikegami, S.; Harada, A.; Hirokawa, N. Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci. Lett. 2000, 279, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar] [CrossRef]
- Muller, U.; Cristina, N.; Li, Z.W.; Wolfer, D.P.; Lipp, H.P.; Rulicke, T.; Brandner, S.; Aguzzi, A.; Weissmann, C. Behavioral and anatomical deficits in mice homozygous for a modified beta-amyloid precursor protein gene. Cell 1994, 79, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.K.; Borchelt, D.R.; Olson, K.; Johannsdottir, R.; Kitt, C.; Yunis, W.; Xu, S.; Eckman, C.; Younkin, S.; Price, D.; et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron 1995, 15, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Gotz, J.; Wiederhold, K.H.; Tolnay, M.; Mistl, C.; Jaton, A.L.; Hong, M.; Ishihara, T.; Lee, V.M.; Trojanowski, J.Q.; et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000, 99, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Yamashita, S.; Fukuda, T.; Park, J.M.; Murayama, M.; Mizoroki, T.; Yoshiike, Y.; Sahara, N.; Takashima, A. Hyperphosphorylated tau in parahippocampal cortex impairs place learning in aged mice expressing wild-type human tau. EMBO J. 2007, 26, 5143–5152. [Google Scholar] [CrossRef]
- Murakami, T.; Paitel, E.; Kawarabayashi, T.; Ikeda, M.; Chishti, M.A.; Janus, C.; Matsubara, E.; Sasaki, A.; Kawarai, T.; Phinney, A.L.; et al. Cortical neuronal and glial pathology in TgTauP301L transgenic mice: Neuronal degeneration, memory disturbance, and phenotypic variation. Am. J. Pathol. 2006, 169, 1365–1375. [Google Scholar] [CrossRef]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; De Ture, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Gamache, J.E.; Kemper, L.; Steuer, E.; Leinonen-Wright, K.; Choquette, J.M.; Hlynialuk, C.; Benzow, K.; Vossel, K.A.; Xia, W.; Koob, M.D.; et al. Developmental Pathogenicity of 4-Repeat Human Tau Is Lost with the P301L Mutation in Genetically Matched Tau-Transgenic Mice. J. Neurosci. 2020, 40, 220–236. [Google Scholar] [CrossRef]
- Wiese, M.; Antebi, A.; Zheng, H. Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans. PLoS ONE 2010, 5, e12790. [Google Scholar] [CrossRef]
- Sconiers, C.; Ko, S.-H.; Chen, L. Isoform-Specific Function of C. elegans Tau, PTL-1. Innov. Aging 2023, 7 (Suppl. S1), 974. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Iorio, R.; Celenza, G.; Petricca, S. Mitophagy: Molecular Mechanisms, New Concepts on Parkin Activation and the Emerging Role of AMPK/ULK1 Axis. Cells 2021, 11, 30. [Google Scholar] [CrossRef]
- Leboutet, R.; Chen, Y.; Legouis, R.; Culetto, E. Mitophagy during development and stress in C. elegans. Mech. Ageing Dev. 2020, 189, 111266. [Google Scholar] [CrossRef] [PubMed]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef] [PubMed]
- Rolland, S.G. How to analyze mitochondrial morphology in healthy cells and apoptotic cells in Caenorhabditis elegans. Methods Enzymol. 2014, 544, 75–98. [Google Scholar] [CrossRef]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Malide, D.; Liu, J.; Rovira, I.I.; Combs, C.A.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef]
- Laker, R.C.; Xu, P.; Ryall, K.A.; Sujkowski, A.; Kenwood, B.M.; Chain, K.H.; Zhang, M.; Royal, M.A.; Hoehn, K.L.; Driscoll, M.; et al. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014, 289, 12005–12015. [Google Scholar] [CrossRef]
- Terskikh, A.; Fradkov, A.; Ermakova, G.; Zaraisky, A.; Tan, P.; Kajava, A.V.; Zhao, X.; Lukyanov, S.; Matz, M.; Kim, S.; et al. “Fluorescent timer”: Protein that changes color with time. Science 2000, 290, 1585–1588. [Google Scholar] [CrossRef]
- Hernandez, G.; Thornton, C.; Stotland, A.; Lui, D.; Sin, J.; Ramil, J.; Magee, N.; Andres, A.; Quarato, G.; Carreira, R.S.; et al. MitoTimer: A novel tool for monitoring mitochondrial turnover. Autophagy 2013, 9, 1852–1861. [Google Scholar] [CrossRef]
- Manil-Segalen, M.; Culetto, E.; Legouis, R.; Lefebvre, C. Interactions between endosomal maturation and autophagy: Analysis of ESCRT machinery during Caenorhabditis elegans development. Methods Enzymol. 2014, 534, 93–118. [Google Scholar] [CrossRef]
- Largeau, C.; Legouis, R. Correlative Light and Electron Microscopy to Analyze LC3 Proteins in Caenorhabditis elegans Embryo. Methods Mol. Biol. 2019, 1880, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Goode, A.E.; Skepper, J.N.; Thorley, A.J.; Seiffert, J.M.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.P.; Ryan, M.P.; Porter, A.E. Avoiding artefacts during electron microscopy of silver nanomaterials exposed to biological environments. J. Microsc. 2016, 261, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Melentijevic, I.; Toth, M.L.; Arnold, M.L.; Guasp, R.J.; Harinath, G.; Nguyen, K.C.; Taub, D.; Parker, J.A.; Neri, C.; Gabel, C.V.; et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Turek, M.; Banasiak, K.; Piechota, M.; Shanmugam, N.; Macias, M.; Sliwinska, M.A.; Niklewicz, M.; Kowalski, K.; Nowak, N.; Chacinska, A.; et al. Muscle-derived exophers promote reproductive fitness. EMBO Rep. 2021, 22, e52071. [Google Scholar] [CrossRef]
- Bonifati, V.; Dekker, M.C.; Vanacore, N.; Fabbrini, G.; Squitieri, F.; Marconi, R.; Antonini, A.; Brustenghi, P.; Libera, A.D.; De Mari, M.; et al. Autosomal recessive early onset parkinsonism is linked to three loci: PARK2, PARK6, and PARK7. Neurol. Sci. 2022, 23 (Suppl. S2), S59–S60. [Google Scholar] [CrossRef]
- Springer, W.; Hoppe, T.; Schmidt, E.; Baumeister, R. A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 2005, 14, 3407–3423. [Google Scholar] [CrossRef]
- Samann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef]
- Castello, P.R.; Drechsel, D.A.; Patel, M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J. Biol. Chem. 2007, 282, 14186–14193. [Google Scholar] [CrossRef]
- Dostal, V.; Wood, S.D.; Thomas, C.T.; Han, Y.; Lau, E.; Lam, M.P.Y. Proteomic signatures of acute oxidative stress response to paraquat in the mouse heart. Sci. Rep. 2020, 10, 18440. [Google Scholar] [CrossRef]
- Choubey, V.; Zeb, A.; Kaasik, A. Molecular Mechanisms and Regulation of Mammalian Mitophagy. Cells 2021, 11, 38. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Rubio-Pena, K.; Sobraske, P.J.; Molina, P.A.; Brookes, P.S.; Galy, V.; Nehrke, K. Fndc-1 contributes to paternal mitochondria elimination in C. elegans. Dev. Biol. 2019, 454, 15–20. [Google Scholar] [CrossRef]
- Lim, Y.; Berry, B.; Viteri, S.; McCall, M.; Park, E.C.; Rongo, C.; Brookes, P.S.; Nehrke, K. FNDC-1-mediated mitophagy and ATFS-1 coordinate to protect against hypoxia-reoxygenation. Autophagy 2021, 17, 3389–3401. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.C.; Sumpter, R.; Jr Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.E10. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef]
- Sakamoto, T.; Inoue, T.; Otomo, Y.; Yokomori, N.; Ohno, M.; Arai, H.; Nakagawa, Y. Deficiency of cardiolipin synthase causes abnormal mitochondrial function and morphology in germ cells of Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 4590–4601. [Google Scholar] [CrossRef]
- Liu, Y.; Samuel, B.S.; Breen, P.C.; Ruvkun, G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014, 508, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Gotz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, M.; Dai, Y.; Sun, Y.; Aman, Y.; Xu, Y.; Yu, P.; Zheng, Y.; Yang, J.; Zhu, X. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging 2020, 12, 16852–16866. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Yu, L.; He, C.L.; Wu, J.M.; Law, B.Y.; Yu, C.L.; Qin, D.L.; Zhou, X.G.; Wu, A.G. Two 18-norspirostane steroidal saponins as novel mitophagy enhancers improve Alzheimer’s disease. Clin. Transl. Med. 2023, 13, e1390. [Google Scholar] [CrossRef]
- Tjahjono, E.; Pei, J.; Revtovich, A.V.; Liu, T.E.; Swadi, A.; Hancu, M.C.; Tolar, J.G.; Kirienko, N.V. Mitochondria-affecting small molecules ameliorate proteostasis defects associated with neurodegenerative diseases. Sci. Rep. 2021, 11, 17733. [Google Scholar] [CrossRef]
- Xie, C.; Zhuang, X.X.; Niu, Z.; Ai, R.; Lautrup, S.; Zheng, S.; Jiang, Y.; Han, R.; Sen Gupta, T.; Cao, S.; et al. Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat. Biomed. Eng. 2022, 6, 76–93. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Barbosa, I.A.; Bandres-Ciga, S.; Quinn, J.P.; Bubb, V.J.; Deshpande, C.; Botia, J.A.; Reynolds, R.H.; Zhang, D.; Simpson, M.A.; et al. Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Park. Dis. 2019, 5, 8. [Google Scholar] [CrossRef]
- Poole, L.P.; Macleod, K.F. Mitophagy in tumorigenesis and metastasis. Cell. Mol. Life Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef]
- Madruga, E.; Maestro, I.; Martinez, A. Mitophagy Modulation, a New Player in the Race against ALS. Int. J. Mol. Sci. 2021, 22, 740. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Chiong, M.; Lavandero, S.; Klionsky, D.J.; Ren, J. Mitophagy in cardiovascular diseases: Molecular mechanisms, pathogenesis, and treatment. Trends Mol. Med. 2022, 28, 836–849. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Kuang, Y.; Ma, K.; Zhou, C.; Ding, P.; Zhu, Y.; Chen, Q.; Xia, B. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 2016, 12, 2363–2373. [Google Scholar] [CrossRef]
- Chamoli, M.; Rane, A.; Foulger, A.; Chinta, S.J.; Shahmirzadi, A.A.; Kumsta, C.; Nambiar, D.K.; Hall, D.; Holcom, A.; Angeli, S.; et al. A drug-like molecule engages nuclear hormone receptor DAF-12/FXR to regulate mitophagy and extend lifespan. Nat. Aging 2023, 3, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Kanazawa, T.; Zappaterra, M.D.; Hasegawa, A.; Wright, A.P.; Newman-Smith, E.D.; Buttle, K.F.; McDonald, K.; Mannella, C.A.; van der Bliek, A.M. The C. elegans Opa1 homologue EAT-3 is essential for resistance to free radicals. PLoS Genet. 2008, 4, e1000022. [Google Scholar] [CrossRef]
- Rolland, S.G.; Lu, Y.; David, C.N.; Conradt, B. The BCL-2-like protein CED-9 of C. elegans promotes FZO-1/Mfn1,2- and EAT-3/Opa1-dependent mitochondrial fusion. J. Cell Biol. 2009, 186, 525–540. [Google Scholar] [CrossRef]
- Machiela, E.; Liontis, T.; Dues, D.J.; Rudich, P.D.; Traa, A.; Wyman, L.; Kaufman, C.; Cooper, J.F.; Lew, L.; Nadarajan, S.; et al. Disruption of mitochondrial dynamics increases stress resistance through activation of multiple stress response pathways. FASEB J. 2020, 34, 8475–8492. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.I.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Delettre, C.; Lenaers, G.; Griffoin, J.M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodrigues, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Knott, A.B.; Bossy-Wetzel, E. Impairing the mitochondrial fission and fusion balance: A new mechanism of neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 283–292. [Google Scholar] [CrossRef]
- Oettinghaus, B.; Schulz, J.M.; Restelli, L.M.; Licci, M.; Savoia, C.; Schmidt, A.; Schmitt, K.; Grimm, A.; More, L.; Hench, J.; et al. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 2016, 23, 18–28. [Google Scholar] [CrossRef]
- Robertson, G.; Patel, M.; Riffle, S.; Marshall, A.; Beasley, H.; Garza-Lopez, E.; Hinton, A.O.; Stoll, M.; Mears, J.; Gama, V. Mitochondrial Fission is Essential to Maintain Cristae Morphology and Bioenergetics. FASEB J. 2022, 36, 4502–4507. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Hu, Z.; Tan, J.; Yang, S.; Zeng, L. Parkin Protects against Oxygen-Glucose Deprivation/Reperfusion Insult by Promoting Drp1 Degradation. Oxid. Med. Cell Longev. 2016, 2016, 8474303. [Google Scholar] [CrossRef]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.; Wang, C.; Cho, J.H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell. 2014, 25, 145–159. [Google Scholar] [CrossRef]
- Haeussler, S.; Kohler, F.; Witting, M.; Premm, M.F.; Rolland, S.G.; Fischer, C.; Chauve, L.; Casanueva, O.; Conradt, B. Autophagy compensates for defects in mitochondrial dynamics. PLoS Genet. 2020, 16, e1008638. [Google Scholar] [CrossRef]
- Kim, S.; Ramalho, T.R.; Haynes, C.M. Regulation of proteostasis and innate immunity via mitochondria-nuclear communication. J. Cell Biol. 2024, 223, e202310005. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: Regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Pradeepkiran, J.A.; Morton, H.; Reddy, P.H. A partial reduction of Drp1 improves cognitive behavior and enhances mitophagy, autophagy and dendritic spines in a transgenic Tau mouse model of Alzheimer disease. Hum. Mol. Genet. 2022, 31, 1788–1805. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Kaur, S.; Mishra, J.; Dibbanti, H.; Singh, A.; Reddy, A.P.; Bhatti, G.K.; Reddy, P.H. Targeting dynamin-related protein-1 as a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166798. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G.; Wang, J.; Wilhelmsson, H.; Oldfors, A.; Rustin, P.; Lewandoski, M.; Barsh, G.S.; Clayton, D.A. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 1998, 18, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Chu, C.T.; Kaufman, B.A. The mitochondrial transcription factor TFAM in neurodegeneration: Emerging evidence and mechanisms. FEBS Lett. 2018, 592, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, M.A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef]
- Kuroda, Y.; Mitsui, T.; Kunishige, M.; Shono, M.; Akaike, M.; Azuma, H.; Matsumoto, T. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum. Mol. Genet. 2006, 15, 883–895. [Google Scholar] [CrossRef]
- Gegg, M.E.; Cooper, J.M.; Schapira, A.H.; Taanman, J.W. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 2009, 4, e4756. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, J.; Zhang, D.; Wu, H.; Li, W.; Wei, H.; Ta, N.; Fan, Y.; Liu, Y.; et al. Mitophagy receptor FUNDC1 is regulated by PGC-1alpha/NRF1 to fine tune mitochondrial homeostasis. EMBO Rep. 2021, 22, e50629. [Google Scholar] [CrossRef]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P., Jr. Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s disease hippocampi. J. Alzheimers Dis. 2014, 40, 319–330. [Google Scholar] [CrossRef]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Campbell, J.M. Supplementation with NAD(+) and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients 2022, 14, 3231. [Google Scholar] [CrossRef]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Elbaz-Alon, Y.; Eisenberg-Bord, M.; Shinder, V.; Stiller, S.B.; Shimoni, E.; Wiedemann, N.; Geiger, T.; Schuldiner, M. Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep. 2015, 12, 7–14. [Google Scholar] [CrossRef]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef]
- Ivankovic, D.; Chau, K.Y.; Schapira, A.H.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Laboy, J.T.; Wang, L.; Barroso, M.; Norman, K.R. C. elegans Presenilin Mediates Inter-Organelle Contacts and Communication that Is Required for Lysosome Activity. Aging Dis. 2024, 16, 2. [Google Scholar] [CrossRef]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef]
- Chakravorty, A.; Jetto, C.T.; Manjithaya, R. Dysfunctional Mitochondria and Mitophagy as Drivers of Alzheimer’s Disease Pathogenesis. Front. Aging Neurosci. 2019, 11, 311. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yang, W.Y. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat. Commun. 2013, 4, 2428. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef]
- Wu, S.; Lei, L.; Song, Y.; Liu, M.; Lu, S.; Lou, D.; Shi, Y.; Wang, Z.; He, D. Mutation of hop-1 and pink-1 attenuates vulnerability of neurotoxicity in C. elegans: The role of mitochondria-associated membrane proteins in Parkinsonism. Exp. Neurol. 2018, 309, 67–78. [Google Scholar] [CrossRef]
- Perrone, M.; Patergnani, S.; Di Mambro, T.; Palumbo, L.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Calcium Homeostasis in the Control of Mitophagy. Antioxid. Redox Signal. 2023, 38, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sanz, S.; Caldero-Escudero, E.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M.; Alvarez, J. SERCA inhibition improves lifespan and healthspan in a chemical model of Parkinson disease in Caenorhabditis elegans. Front. Pharmacol. 2023, 14, 1182428. [Google Scholar] [CrossRef]
- Garcia-Casas, P.; Arias-Del-Val, J.; Alvarez-Illera, P.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Inhibition of Sarco-Endoplasmic Reticulum Ca(2+) ATPase Extends the Lifespan in C. elegans Worms. Front. Pharmacol. 2018, 9, 669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPRmt. Cell Rep. 2019, 28, 1659–1669.e5. [Google Scholar] [CrossRef]
- Haynes, C.M.; Ron, D. The mitochondrial UPR–protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Michaeli, L.; Spector, E.; Haeussler, S.; Carvalho, C.A.; Grobe, H.; Abu-Shach, U.B.; Zinger, H.; Conradt, B.; Broday, L. ULP-2 SUMO protease regulates UPR(mt) and mitochondrial homeostasis in Caenorhabditis elegans. Free Radic. Biol. Med. 2024, 214, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, P.; Anderson, N.S.; Shpilka, T.; Du, Y.; Naresh, N.U.; Li, R.; Zhu, L.J.; Luk, K.; Lavelle, J.; et al. LONP-1 and ATFS-1 sustain deleterious heteroplasmy by promoting mtDNA replication in dysfunctional mitochondria. Nat. Cell Biol. 2022, 24, 181–193. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Cordeiro, A.V.; Bricola, R.S.; Braga, R.R.; Lenhare, L.; Silva, V.R.R.; Anaruma, C.P.; Katashima, C.K.; Crisol, B.M.; Simabuco, F.M.; Silva, A.S.R.; et al. Aerobic Exercise Training Induces the Mitonuclear Imbalance and UPRmt in the Skeletal Muscle of Aged Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2258–2261. [Google Scholar] [CrossRef]
- Li, T.Y.; Sleiman, M.B.; Li, H.; Gao, A.W.; Mottis, A.; Bachmann, A.M.; Alam, G.E.; Li, X.; Goeminne, L.J.E.; Schoonjans, K.; et al. The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 2021, 1, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Durieux, J.; Wolff, S.; Dillin, A. The Cell-Non-Autonomous Nature of Electron Transport Chain-Mediated Longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Baker, B.M.; Nargund, A.M.; Sun, T.; Haynes, C.M. Protective Coupling of Mitochondrial Function and Protein Synthesis via the eIF2α Kinase GCN. PLoS Genet. 2012, 8, e1002760. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chang, S.-Y.; Yin, S.-G.; Liu, Z.-Y.; Cheng, X.; Liu, X.-J.; Jiang, Q.; Gao, G.; Lin, D.Y.; Kang, X.L.; et al. Two conserved epigenetic regulators prevent healthy ageing. Nature 2020, 579, 118–122. [Google Scholar] [CrossRef]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Zeng, J. Defective lysosomal acidification: A new prognostic marker and therapeutic target for neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef]
- Yang, C.; Wang, X. Lysosome biogenesis: Regulation and functions. J. Cell Biol. 2021, 220, e202102001. [Google Scholar] [CrossRef]
- Clokey, G.V.; Jacobson, L.A. The autofluorescent “lipofuscin granules” in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech. Ageing Dev. 1986, 35, 79–94. [Google Scholar] [CrossRef]
- Sigmond, T.; Vellai, T. Lysosomal alteration links food limitation to longevity. Nat. Aging. 2023, 3, 1048–1050. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Zhao, D.; Li, X.; Yang, C.; Wang, X. Lysosome activity is modulated by multiple longevity pathways and is important for lifespan extension in C. elegans. eLife 2020, 9, e55745. [Google Scholar] [CrossRef]
- Gelino, S.; Chang, J.T.; Kumsta, C.; She, X.; Davis, A.; Nguyen, C.; Panowski, S.; Hansen, M. Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLoS Genet. 2016, 12, e1006135. [Google Scholar] [CrossRef]
- Villalobos, T.V.; Ghosh, B.; DeLeo, K.R.; Alam, S.; Ricaurte-Perez, C.; Wang, A.; Mercola, B.M.; Butsch, T.J.; Ramos, C.D.; Das, S.; et al. Tubular lysosome induction couples animal starvation to healthy aging. Nat. Aging 2023, 3, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.; Foster, O.K.; Handa, S.; Peloza, K.; Voss, L.; Somhegyi, H.; Jian, Y.; Vo, M.V.; Harp, M.; Rambo, F.M.; et al. Function and regulation of the Caenorhabditis elegans Rab32 family member GLO-1 in lysosome-related organelle biogenesis. PLoS Genet. 2018, 14, e1007772. [Google Scholar] [CrossRef]
- Hermann, G.J.; Schroeder, L.K.; Hieb, C.A.; Kershner, A.M.; Rabbitts, B.M.; Fonarev, P.; Grant, B.D.; Priess, J.R. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell 2005, 16, 3273–3288. [Google Scholar] [CrossRef] [PubMed]
- de Voer, G.; Peters, D.; Taschner, P.E. Caenorhabditis elegans as a model for lysosomal storage disorders. Biochim. Biophys. Acta 2008, 1782, 433–446. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Zeng, W.; Li, C.; Wu, R.; Yang, X.; Wang, Q.; Lin, B.; Wei, Y.; Li, H.; Shan, G.; Qu, L.; et al. Optogenetic manipulation of lysosomal physiology and autophagy-dependent clearance of amyloid beta. PLoS Biol. 2024, 22, e3002591. [Google Scholar] [CrossRef]
- Wang, B.; Martini-Stoica, H.; Qi, C.; Lu, T.C.; Wang, S.; Xiong, W.; Qi, Y.; Xu, Y.; Sardiello, M.; Li, H.; et al. TFEB-vacuolar ATPase signaling regulates lysosomal function and microglial activation in tauopathy. Nat. Neurosci. 2024, 27, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.A.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8, 998. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Sewell, A.K.; Wu, Z.; Han, M. TOR Signaling in Caenorhabditis elegans Development, Metabolism, and Aging. Genetics 2019, 213, 329–360. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef]
- Garza-Lombo, C.; Schroder, A.; Reyes-Reyes, E.M.; Franco, R. mTOR/AMPK signaling in the brain: Cell metabolism, proteostasis and survival. Curr. Opin. Toxicol. 2018, 8, 102–110. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Yuan, J.; Zhao, X.; Hu, Y.; Sun, H.; Gong, G.; Huang, X.; Chen, X.; Xia, M.; Sun, C.; Huang, Q.; et al. Autophagy regulates the degeneration of the auditory cortex through the AMPK-mTOR-ULK1 signaling pathway. Int. J. Mol. Med. 2018, 41, 2086–2098. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Hajdu, B.; Lorincz, T.; Szarka, A.; Banhegyi, G.; Kapuy, O. A Double Negative Feedback Loop between mTORC1 and AMPK Kinases Guarantees Precise Autophagy Induction upon Cellular Stress. Int. J. Mol. Sci. 2019, 20, 5543. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; Di Domenico, F.; Barone, E.; Butterfield, D.A. mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder. Free Radic. Biol. Med. 2021, 169, 382–396. [Google Scholar] [CrossRef]
- Bartolome, A.; Garcia-Aguilar, A.; Asahara, S.I.; Kido, Y.; Guillen, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lanjuin, A.; Chowdhury, S.R.; Mistry, M.; Silva-Garcia, C.G.; Weir, H.J.; Lee, C.L.; Escoubas, C.C.; Tabakovic, E.; Mair, W.B. Neuronal TORC1 modulates longevity via AMPK and cell nonautonomous regulation of mitochondrial dynamics in C. elegans. eLife 2019, 8, e49158. [Google Scholar] [CrossRef]
- Aspernig, H.; Heimbucher, T.; Qi, W.; Gangurde, D.; Curic, S.; Yan, Y.; von Gromoff, E.D.; Baumeister, R.; Thien, A. Mitochondrial Perturbations Couple mTORC2 to Autophagy in C. elegans. Cell Rep. 2019, 29, 1399–1409.e5. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, H.; Xue, T.; Kong, X.; Tian, D.; Luo, L.; Yang, Y.; Xu, K.; Wei, Y.; Zhuang, Z. Ribavirin extends the lifespan of Caenorhabditis elegans through AMPK-TOR Signaling. Eur. J. Pharmacol. 2023, 946, 175548. [Google Scholar] [CrossRef]
- Shpilka, T.; Du, Y.; Yang, Q.; Melber, A.; Uma Naresh, N.; Lavelle, J.; Kim, S.; Liu, P.; Weidberg, H.; Li, R.; et al. UPRmt scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat. Commun. 2021, 12, 479. [Google Scholar] [CrossRef]
- Gitschlag, B.L.; Tate, A.T.; Patel, M.R. Nutrient status shapes selfish mitochondrial genome dynamics across different levels of selection. eLife 2020, 9, e56686. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivela, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e5. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Licata, J.; Tsukikawa, M.; Marty, E.; Thomas, J.; Felix, C.A.; Xiao, R.; et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015, 24, 4829–4847. [Google Scholar] [CrossRef] [PubMed]
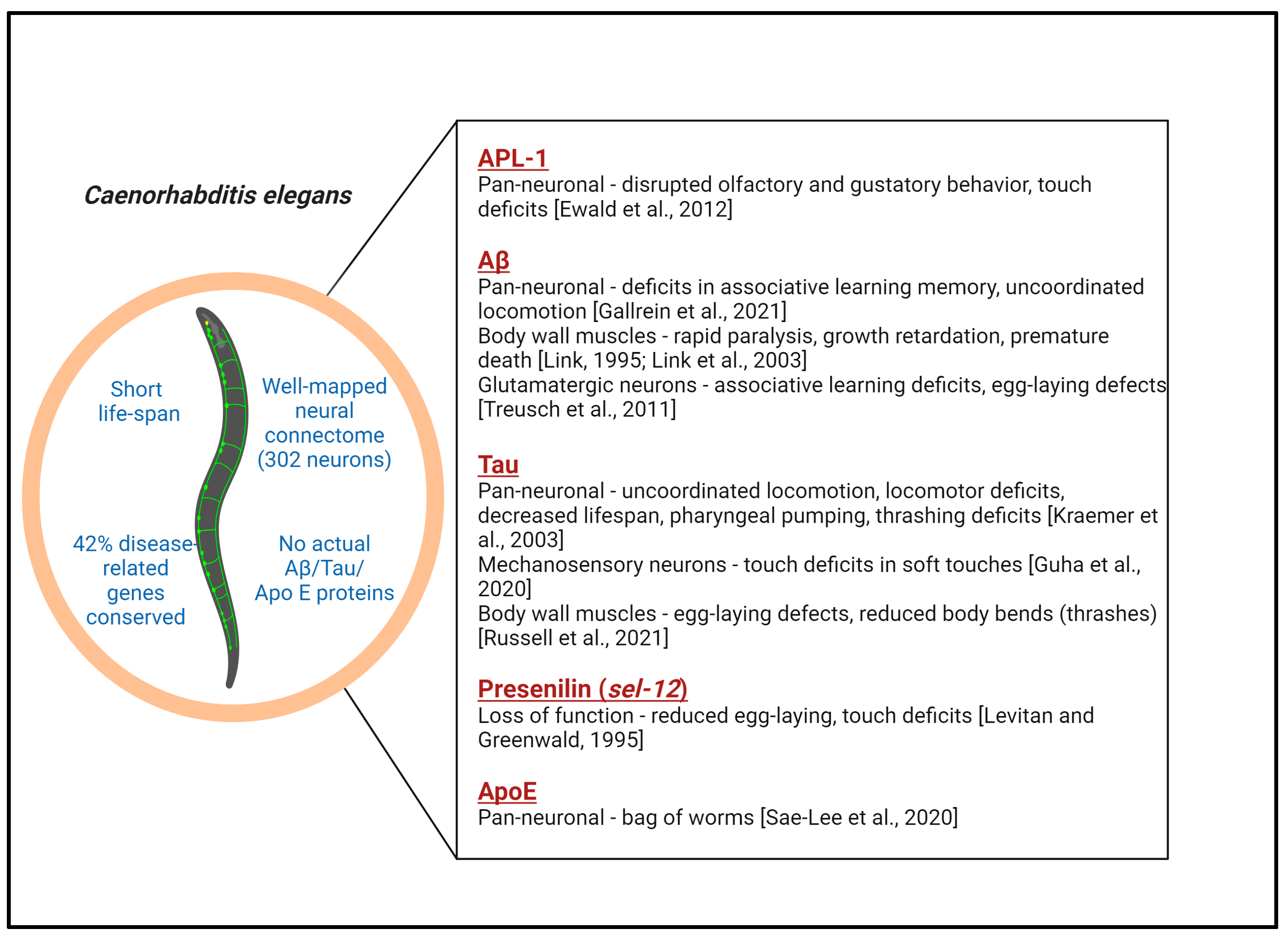
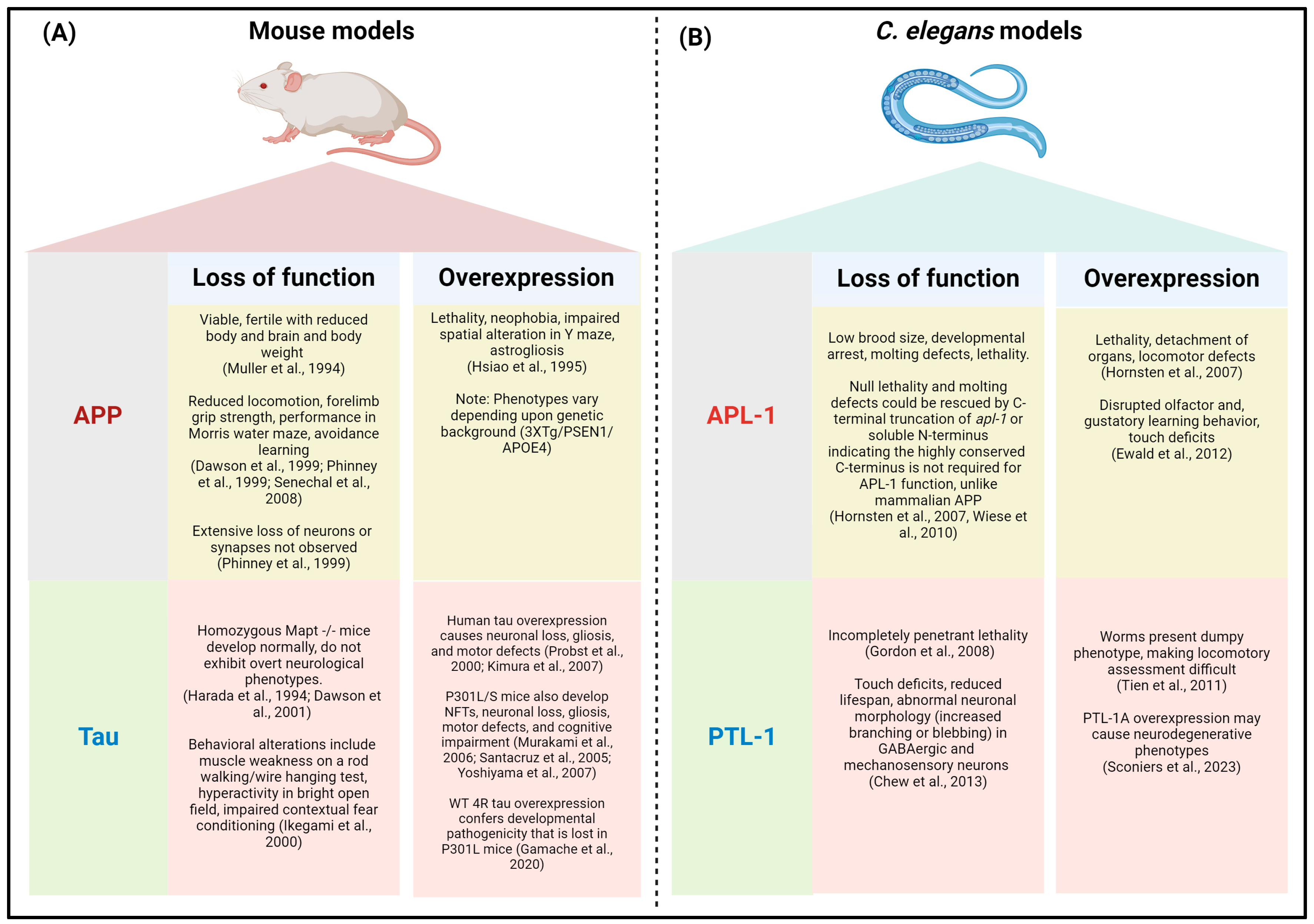


{kind=link}
{kind=link}
{kind=link}
| Tools Utilized to Assess Mitophagy | References |
|---|---|
| 1. Fluorescence co-localization of mitochondria with autophagosomal markers | |
| Mitochondria can either be stained by standard mitochondria-specific dyes like tetramethyl rhodamine ethyl ester (TMRE), mitotracker, or rhodamine 6G, or transgenic lines of worms can be generated that express different mitochondrial proteins (localized in OMM, IMM, or matrix) tagged to fluorophores. Autophagosomes can be marked by generating transgenic lines expressing GFP/mCherry-tagged LGG-1 or LGG-2. The co-localization of mitophagic and autophagic markers is indicative of mitophagy. For example, mitochondria-targeted GFP and DsRed-fused LGG-1 were used in this way to assess mitophagy. The limitation of this method is the high rates of false positive results owing to the ability of LGG-1/LGG-2 to aggregate in an autophagy-independent manner. | [163,165,166] |
| 2. Fluorescence quenching | |
| Mitochondria-targeted Rosella is a fluorescent biosensor consisting of a fusion of pH-insensitive DsRed to pH-sensitive GFP. When mitochondria are engulfed by autophagosomes and fused with lysosomes, the GFP part of the biosensor (sensitive to the acidic interior of lysosomes) is quenched, leaving only the red fluorescence of DsRed. A reduction in the GFP/DsRed fluorescence ratio indicates mitophagy stimulation. | [165] |
| 3. Fluorescent ratiometry: pH | |
| mKeima is a red-emitting fluorescent protein with a pH-dependent Stokes shift that is excited the most strongly at ~440 nm under neutral conditions and at ~550 nm under acidic conditions. Targeting mKeima to the mitochondrial matrix allows it to be used to monitor mitophagy. Under normal physiological conditions, the pH of the mitochondrial matrix is slightly alkaline (pH 8.0), and 440 nm excitation predominates. Upon the delivery of mitochondria to lysosomes, the environment becomes acidic (pH 4.0), and since mito-mKeima is resistant to acid proteases, the excitation maximum shifts to 550 nm. Moreover, there is also a shift in morphology, with round lysosomes clearly distinguishable from the normal tubular–reticular mitochondria, albeit not from fragmented mitochondria. The ratio of emissions and distinct morphologies has been used to estimate mitophagy in transgenic worms. | [35,167,168] |
| 4. Fluorescent ratiometry: redox | |
| Another fluorescent probe used to monitor mitophagy is the Timer probe targeted at mitochondria (MitoTimer). Timer is a mutant of the fluorescent protein DsRed (DsRed 1-E5) that fluoresces green (Ex 483 nm, Em 500 nm) when newly synthesized but shifts irreversibly to red (Ex 558 nm, Em 583 nm) when the protein is oxidized (dehydrogenization of Tyr-67). The red/green fluorescence of MitoTimer can be used to monitor mitochondrial dynamics and has been carefully utilized to monitor mitophagy, particularly under circumstances where mitophagy closely matches mitochondrial biogenesis. | [169,170,171] |
| 5. Electron microscopy | |
| Transmission electron microscopy (TEM) allows us to directly visualize mitochondria surrounded by autophagic (early mitophagy) or lysosomal (late mitophagy) membranes, allowing for an ultrastructural detection of mitophagy. Correlative light and electron microscopy (CLEM) allows us to combine EM and the detection of fluorescent-tagged LC3 proteins (LGG1 and LGG2) in C. elegans. However, a major drawback of TEM is the misinterpretation of data due to methodological artifacts that require an expert eye for correct analysis | [172,173,174] |
| 6. Western blotting | |
| The Western blotting of cell extracts can be used to detect different proteins in the mitochondrial sub-compartments (TOMM20, TIMM23, CYPD, HSP60) and the autophagic proteins required for mitophagy (LC3, p62/SQSTM1). However, the limitation of Western blotting is the inability to monitor mitophagy in different tissues or cell types in worms. | [174] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ganguly, U.; Carroll, T.; Nehrke, K.; Johnson, G.V.W. Mitochondrial Quality Control in Alzheimer’s Disease: Insights from Caenorhabditis elegans Models. Antioxidants 2024, 13, 1343. https://doi.org/10.3390/antiox13111343
Ganguly U, Carroll T, Nehrke K, Johnson GVW. Mitochondrial Quality Control in Alzheimer’s Disease: Insights from Caenorhabditis elegans Models. Antioxidants. 2024; 13(11):1343. https://doi.org/10.3390/antiox13111343
Chicago/Turabian StyleGanguly, Upasana, Trae Carroll, Keith Nehrke, and Gail V. W. Johnson. 2024. "Mitochondrial Quality Control in Alzheimer’s Disease: Insights from Caenorhabditis elegans Models" Antioxidants 13, no. 11: 1343. https://doi.org/10.3390/antiox13111343
APA StyleGanguly, U., Carroll, T., Nehrke, K., & Johnson, G. V. W. (2024). Mitochondrial Quality Control in Alzheimer’s Disease: Insights from Caenorhabditis elegans Models. Antioxidants, 13(11), 1343. https://doi.org/10.3390/antiox13111343