
Thiol-Based Redox Molecules: Potential Antidotes for Acrylamide Toxicity


Abstract
:Highlights
- ACR induces activation of the MAPKs: ERK1/2, p38MAPK and JNK.
- Thioredoxin mimetic (TXM) peptides effectively inhibit ACR-induced activation of MAPKs.
- Inhibitory potency TXM-CB16/TXM-CB13 > AD4/NACA >NAC.
- TXM peptides reverse MAPK signaling and are promising ACR antidotes
Abstract
1. Introduction
2. Experimental Procedures
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Acrylamide Toxicity Treatment Assay
2.4. Acrylamide Toxicity Prophylaxis Assay
2.5. Cell Viability and Methylene Blue Assay
2.6. Western Blot Analysis
2.7. Data Analysis and Statistics
3. Results
3.1. ACR-Induced Activation of ERK1/2 and p38MAPK; Time-Depndence
3.2. ACR-Induced Activation of ERK1/2 and p38MAPK; Concentration-Dependence
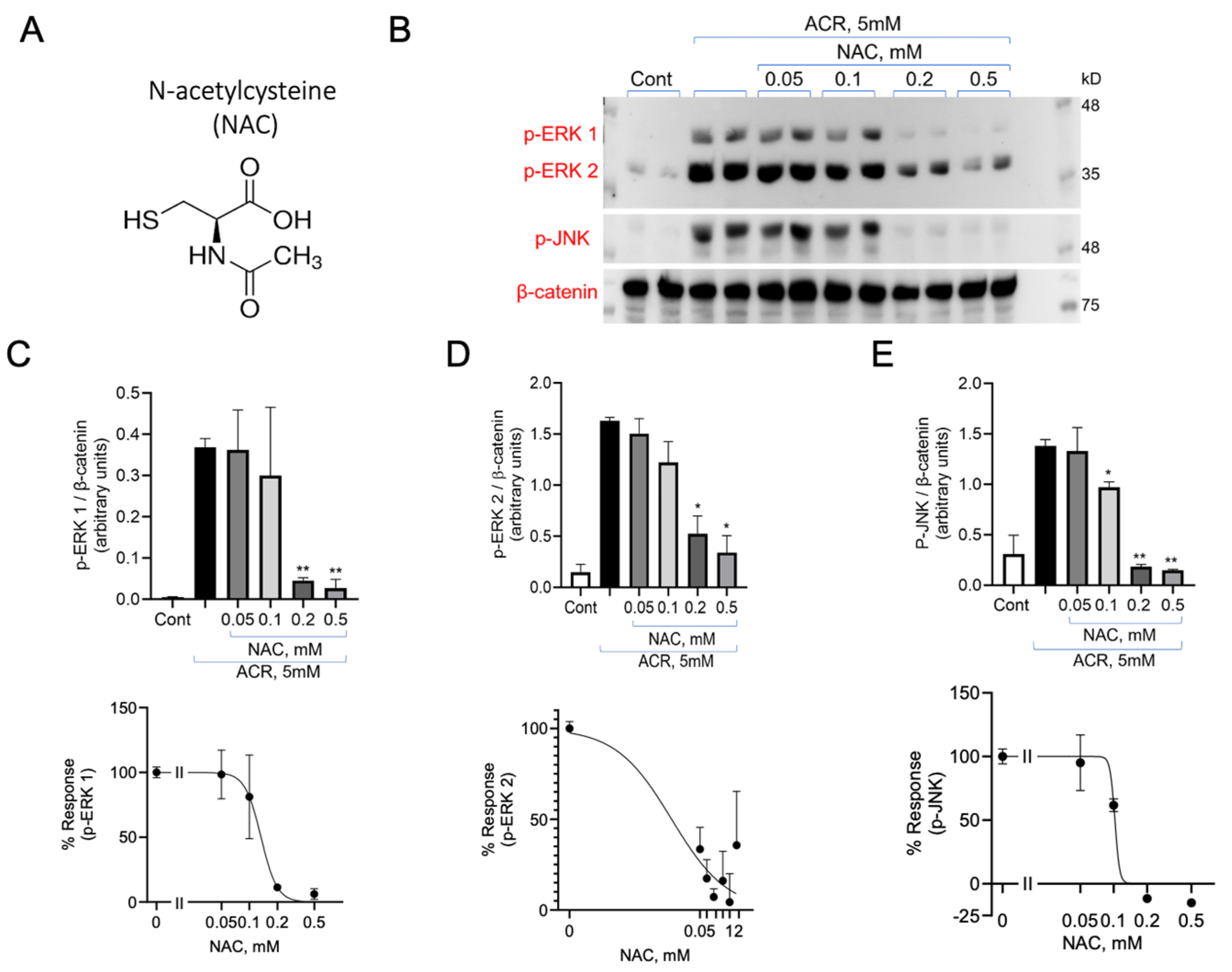
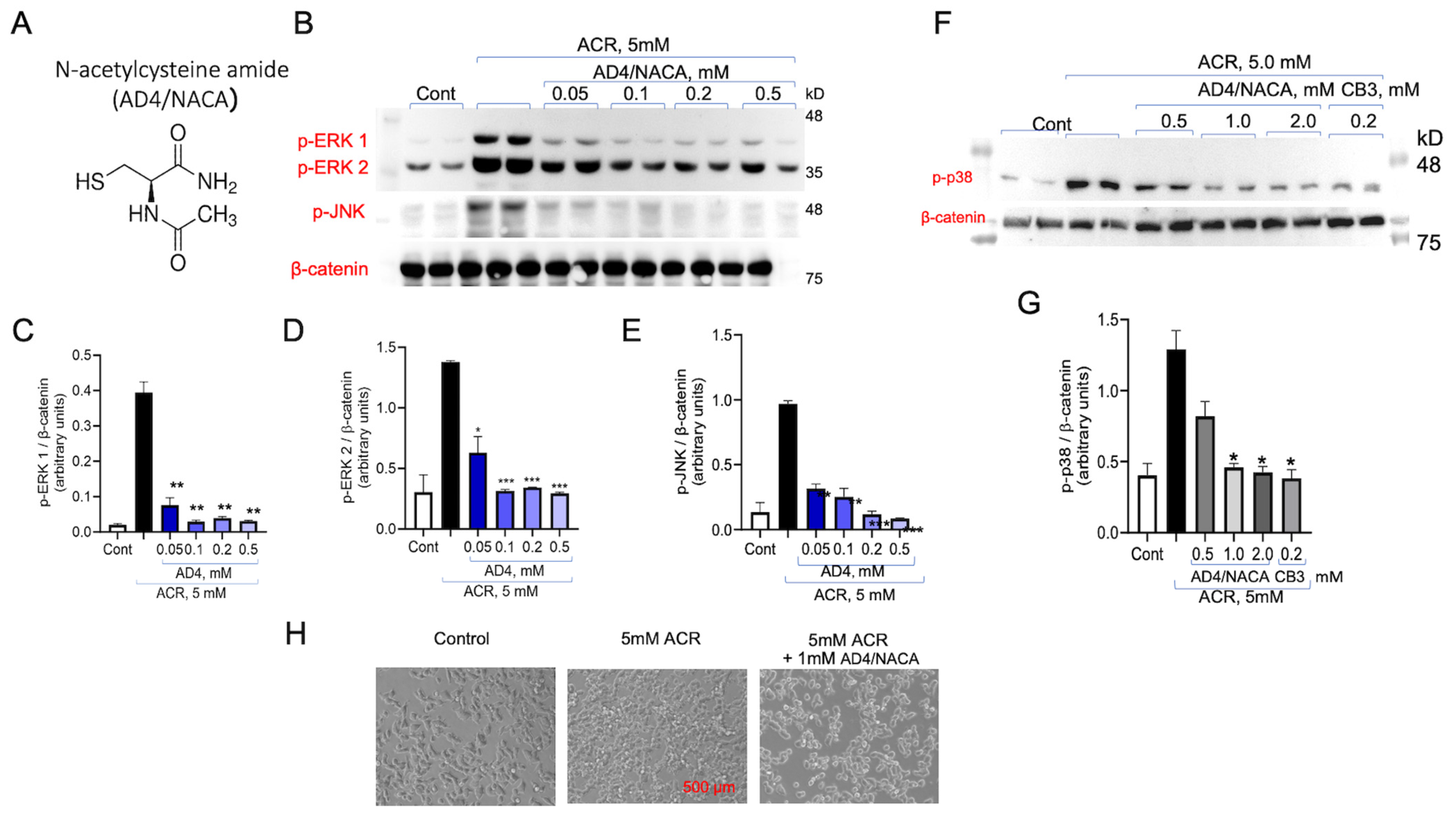
3.3. Inhibition of ACR Induced ERK1/2 and p38MAPK Phosphorylation by N-Acetylcysteine (NAC) and N-Acetylcysteine Amide (AD4/NACA)
3.4. AD4/NACA and AcCys-Pro-CysNH2 (TXM-CB3) Prevent ACR-Induced ERK1/2 Activity
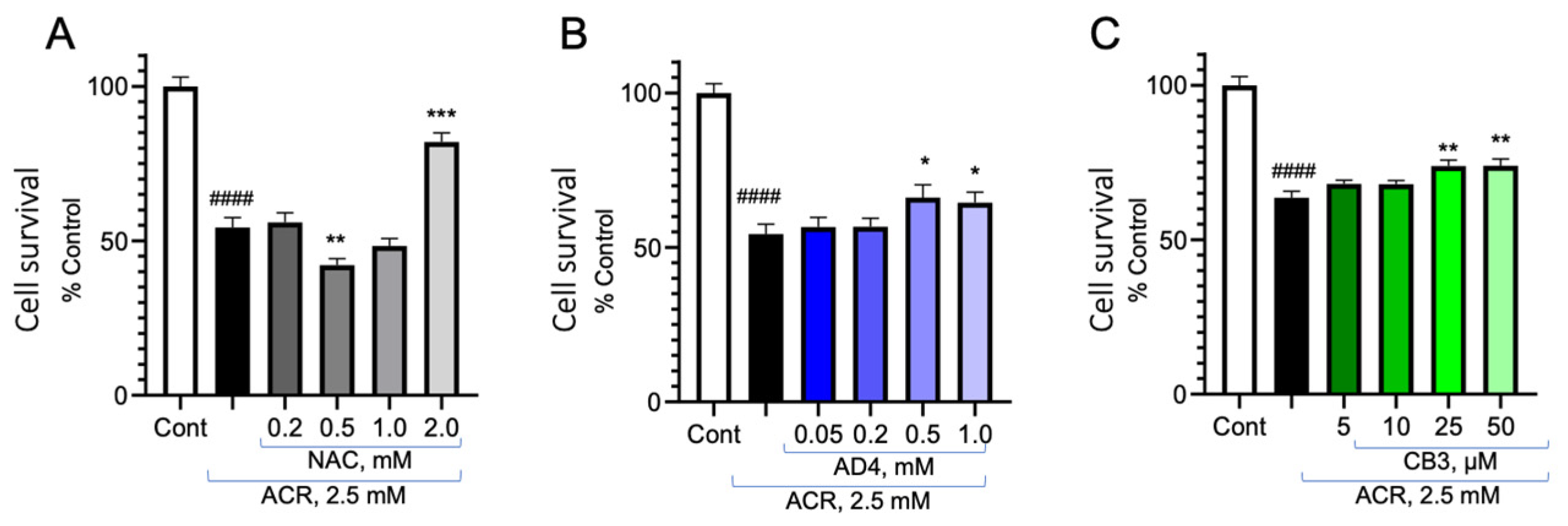
3.5. Reversal of ACR Toxicity on Cell Viability by NAC, AD4/NACA and TXM-CB3
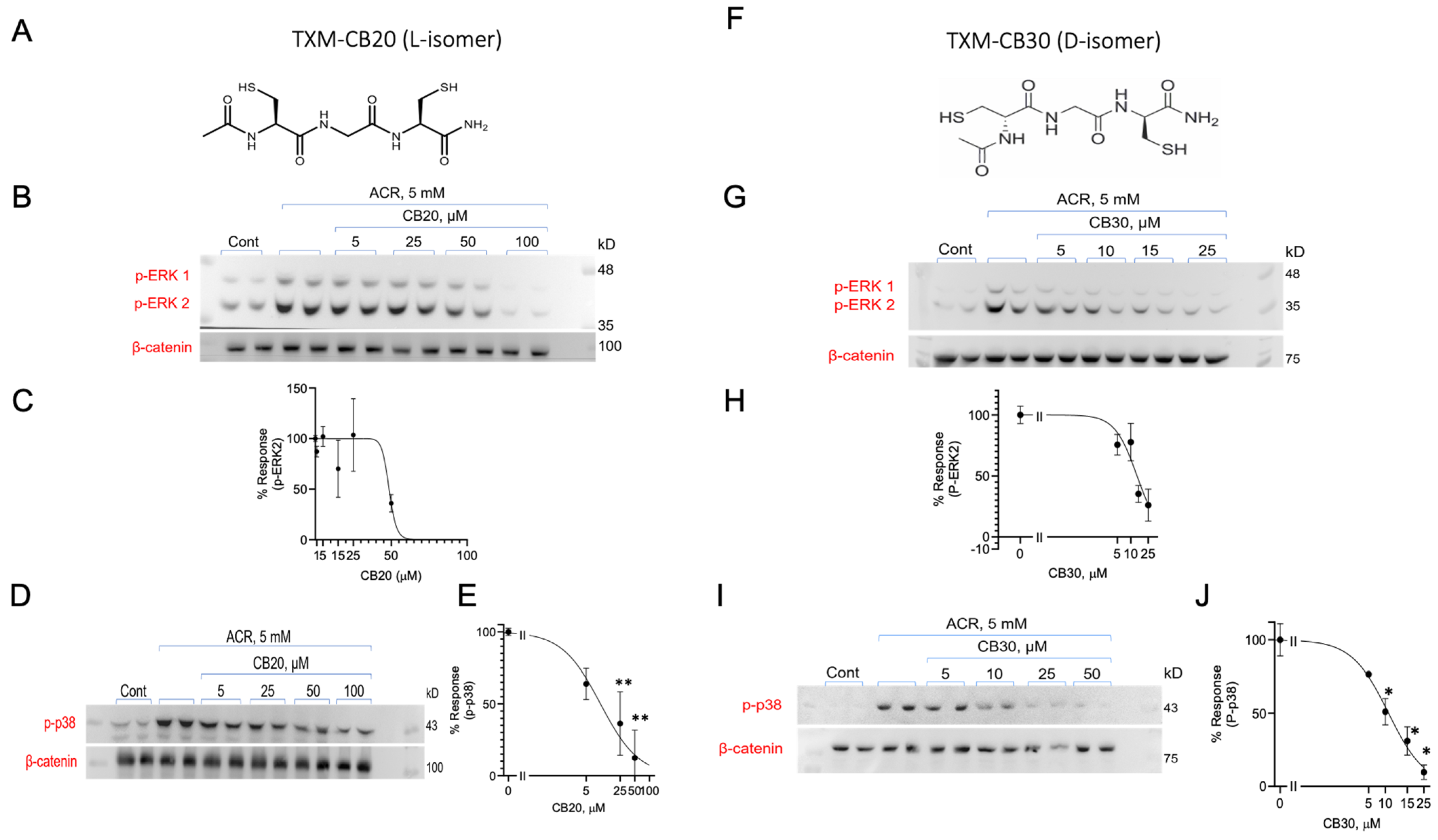
3.6. Inhibition of ACR-Induced ERK1/2 and p38MAPK Phosphorylation by TXM-CB20 (L-Form) and TXM-CB30 (D-Form)
3.7. Inhibition of ACR-Induced ERK1/2 and p38MAPK Phosphorylation by the TXM-CB13
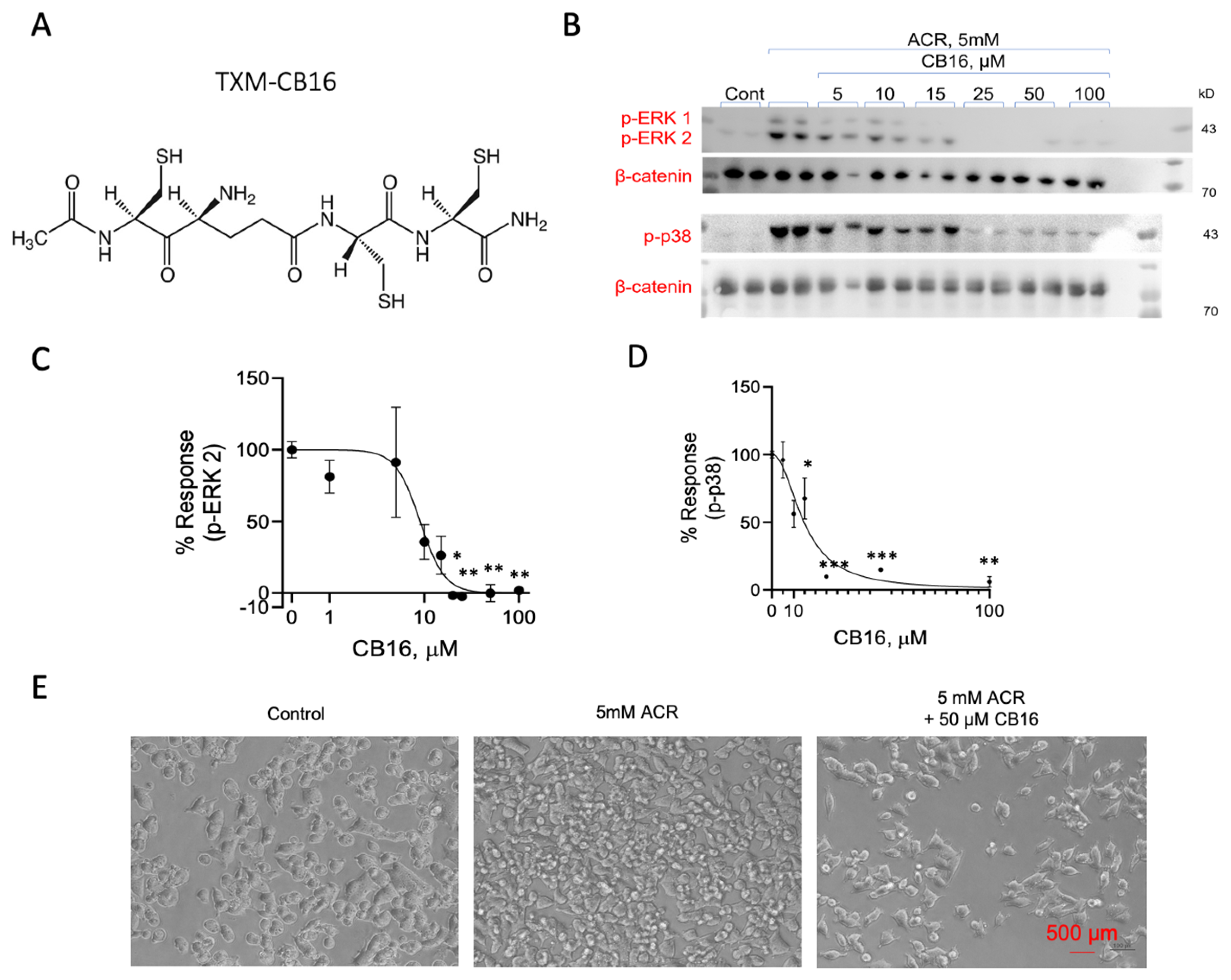
3.8. Inhibition of ACR-Induced ERK1/2 and p38MAPK Phosphorylation by TXM-CB16, a 3-Cys Residues Peptide
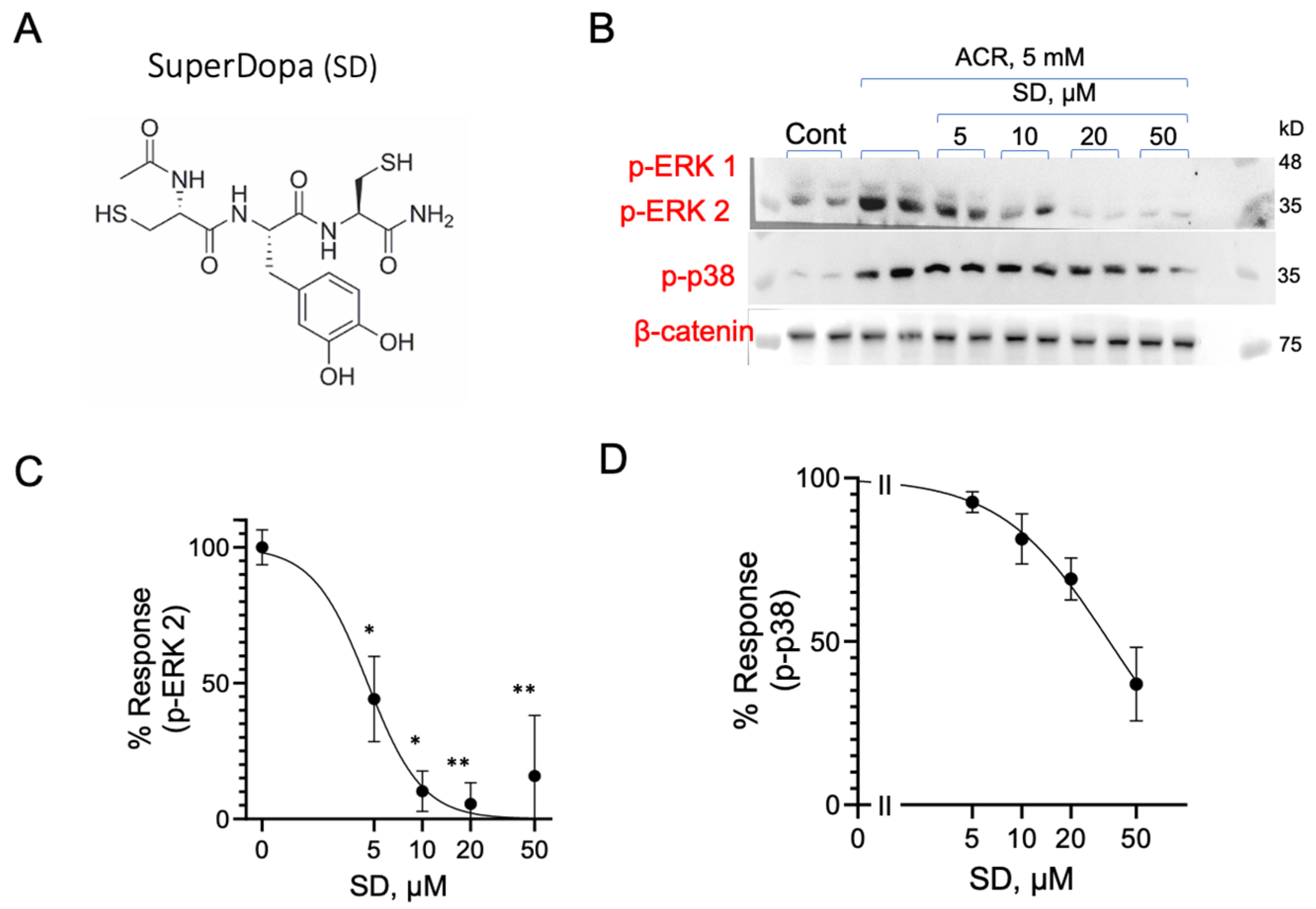
3.9. Inhibition of ACR-Induced ERK1/2 and p38MAPK Phosphorylation by Dopamine Precursor SuperDopa (SD)
4. Discussion
4.1. ACR-Induced Activation of Intracellular Pathways
4.2. Reversal of ACR-Induced MAPK Activation
4.3. Inhibition of ACR-Induced MAPK Activation by TXM-Peptides
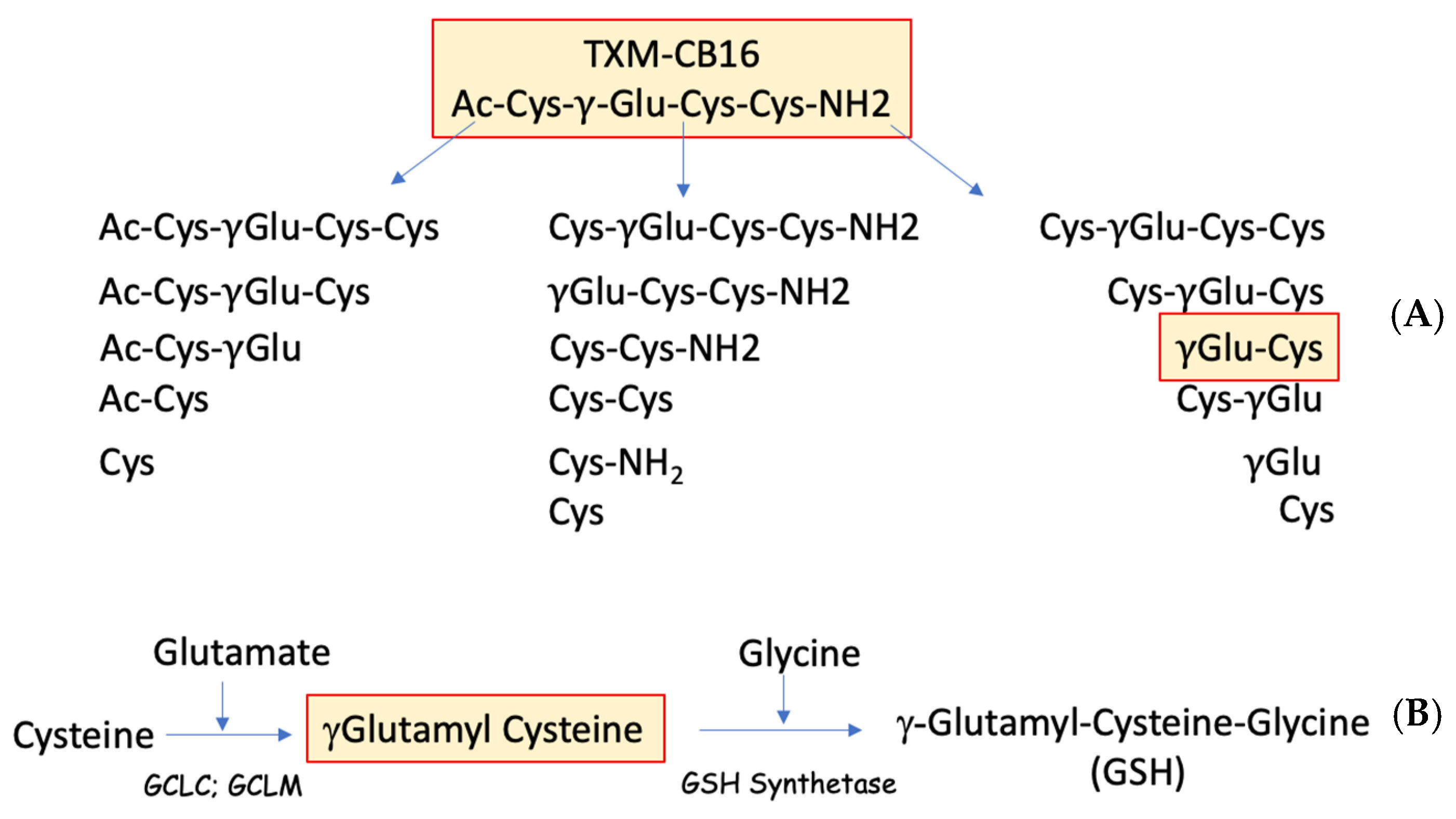
4.4. Inhibition of ACR-Induced MAPK by TXM-CB16, a 3-Cys Tetra-TXM Peptide
4.5. Inhibition of ACR-Induced MAPK Activation by SuperDopa, a Dithiol Levodopa Peptide
Limitation of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- He, F.; Zhang, S.; Wang, H.; Li, G.; Zhang, Z.; Li, F.; Dong, X.; Hu, F. Neurological and electroneuromyographic assessment of the adverse effects of acrylamide on occupationally exposed workers. Scand. J. Work. Environ. Health 1989, 15, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Doerge, D.R.; Gamboa Da Costa, G.; McDaniel, L.P.; Churchwell, M.I.; Twaddle, N.C.; Beland, F.A. DNA adducts derived from administration of acrylamide and glycidamide to mice and rats. Mutat. Res. 2005, 580, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M. Mechanisms of acrylamide induced rodent carcinogenesis. Adv. Exp. Med. Biol. 2005, 561, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Puppel, N.; Tjaden, Z.; Fueller, F.; Marko, D. DNA strand breaking capacity of acrylamide and glycidamide in mammalian cells. Mutat. Res. 2005, 580, 71–80. [Google Scholar] [CrossRef]
- Tareke, E.; Lyn-Cook, B.D.; Duhart, H.; Newport, G.; Ali, S. Acrylamide decreased dopamine levels and increased 3-nitrotyrosine (3-NT) levels in PC 12 cells. Neurosci. Lett. 2009, 458, 89–92. [Google Scholar] [CrossRef]
- Koszucka, A.; Nowak, A.; Nowak, I.; Motyl, I. Acrylamide in human diet, its metabolism, toxicity, inactivation and the associated European Union legal regulations in food industry. Crit. Rev. Food Sci. Nutr. 2020, 60, 1677–1692. [Google Scholar] [CrossRef]
- Calleman, C.J.; Bergmark, E.; Costa, L.G. Acrylamide is metabolized to glycidamide in the rat: Evidence from hemoglobin adduct formation. Chem. Res. Toxicol. 1990, 3, 406–412. [Google Scholar] [CrossRef]
- Fennell, T.R.; Sumner, S.C.J.; Snyder, R.W.; Burgess, J.; Spicer, R.; Bridson, W.E.; Friedman, M.A. Metabolism and hemoglobin adduct formation of acrylamide in humans. Toxicol. Sci. 2005, 85, 447–459. [Google Scholar] [CrossRef]
- Huchthausen, J.; Escher, B.I.; Grasse, N.; König, M.; Beil, S.; Henneberger, L. Reactivity of Acrylamides Causes Cytotoxicity and Activates Oxidative Stress Response. Chem. Res. Toxicol. 2023, 36, 1374–1385. [Google Scholar] [CrossRef]
- Peivasteh-Roudsari, L.; Karami, M.; Barzegar-Bafrouei, R.; Samiee, S.; Karami, H.; Tajdar-Oranj, B.; Mahdavi, V.; Alizadeh, A.M.; Sadighara, P.; Oliveri Conti, G.; et al. Toxicity, metabolism, and mitigation strategies of acrylamide: A comprehensive review. Int. J. Environ. Health Res. 2024, 34, 1–29. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Geohagen, B.C.; Nordstroem, L.U. Mechanisms of soft and hard electrophile toxicities. Toxicology 2019, 418, 62–69. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Ross, J.F.; Lehning, E.J. Nerve terminals as the primary site of acrylamide action: A hypothesis. Neurotoxicology 2002, 23, 43–59. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Zhang, X.; Jiao, Y.; Duan, L.; Dai, L.; Yan, H. Chronic acrylamide exposure resulted in dopaminergic neuron loss, neuroinflammation and motor impairment in rats. Toxicol. Appl. Pharmacol. 2022, 451, 116190. [Google Scholar] [CrossRef]
- Li, S.X.; Cui, N.; Zhang, C.L.; Zhao, X.L.; Yu, S.F.; Xie, K.Q. Effect of subchronic exposure to acrylamide induced on the expression of bcl-2, bax and caspase-3 in the rat nervous system. Toxicology 2006, 217, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wu, X.; Yan, D.; Peng, C.; Rao, C.; Yan, H. Acrylamide-induced oxidative stress and inflammatory response are alleviated by N-acetylcysteine in PC12 cells: Involvement of the crosstalk between Nrf2 and NF-κB pathways regulated by MAPKs. Toxicol. Lett. 2018, 288, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Komoike, Y.; Matsuoka, M. Endoplasmic reticulum stress-mediated neuronal apoptosis by acrylamide exposure. Toxicol. Appl. Pharmacol. 2016, 310, 68–77. [Google Scholar] [CrossRef]
- Katen, A.L.; Roman, S.D. The genetic consequences of paternal acrylamide exposure and potential for amelioration. Mutat. Res. 2015, 777, 91–100. [Google Scholar] [CrossRef]
- Bo, N.; Yilin, H.; Chaoyue, Y.; Lu, L.; Yuan, Y. Acrylamide induces NLRP3 inflammasome activation via oxidative stress- and endoplasmic reticulum stress-mediated MAPK pathway in HepG2 cells. Food Chem. Toxicol. 2020, 145, 111679. [Google Scholar] [CrossRef]
- Cuello, F.; Eaton, P. Cysteine-Based Redox Sensing and Its Role in Signaling by Cyclic Nucleotide-Dependent Kinases in the Cardiovascular System. Annu. Rev. Physiol. 2019, 81, 63–87. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef]
- Humphries, K.M.; Pennypacker, J.K.; Taylor, S.S. Redox regulation of cAMP-dependent protein kinase signaling: Kinase versus phosphatase inactivation. J. Biol. Chem. 2007, 282, 22072–22079. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Oka, S.I.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, H.; Zhuang, S.; Pilz, R.B.; Casteel, D.E. The activity of cGMP-dependent protein kinase Iα is not directly regulated by oxidation-induced disulfide formation at cysteine 43. J. Biol. Chem. 2017, 292, 8262–8268. [Google Scholar] [CrossRef] [PubMed]
- Sheehe, J.L.; Bonev, A.D.; Schmoker, A.M.; Ballif, B.A.; Nelson, M.T.; Moon, T.M.; Dostmann, W.R. Oxidation of cysteine 117 stimulates constitutive activation of the type Iα cGMP-dependent protein kinase. J. Biol. Chem. 2018, 293, 16791–16802. [Google Scholar] [CrossRef]
- Byrne, D.P.; Shrestha, S.; Galler, M.; Cao, M.; Daly, L.A.; Campbell, A.E.; Eyers, C.E.; Veal, E.A.; Kannan, N.; Eyers, P.A. Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 2020, 13, eaax2713. [Google Scholar] [CrossRef]
- Graham, M.E.; Tunik, M.G.; Farmer, B.M.; Bendzans, C.; McCrillis, A.M.; Nelson, L.S.; Portelli, I.; Smith, S.; Goldberg, J.D.; Zhang, M.; et al. Agent of opportunity risk mitigation: People, engineering, and security efficacy. Disaster Med. Public Health Prep. 2010, 4, 291–299. [Google Scholar] [CrossRef]
- Tomassoni, A.J.; French, R.N.E.; Walter, F.G. Toxic industrial chemicals and chemical weapons: Exposure, identification, and management by syndrome. Emerg. Med. Clin. N. Am. 2015, 33, 13–36. [Google Scholar] [CrossRef]
- Kianfar, M.; Nezami, A.; Mehri, S.; Hosseinzadeh, H.; Hayes, A.W.; Karimi, G. The protective effect of fasudil against acrylamide-induced cytotoxicity in PC12 cells. Drug Chem. Toxicol. 2020, 43, 595–601. [Google Scholar] [CrossRef]
- He, Y.; Tan, D.; Mi, Y.; Bai, B.; Jiang, D.; Zhou, X.; Ji, S. Effect of epigallocatechin-3-gallate on acrylamide-induced oxidative stress and apoptosis in PC12 cells. Hum. Exp. Toxicol. 2017, 36, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ramiro, I.; Martín, M.Á.; Ramos, S.; Bravo, L.; Goya, L. Olive oil hydroxytyrosol reduces toxicity evoked by acrylamide in human Caco-2 cells by preventing oxidative stress. Toxicology 2011, 288, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Kacar, S.; Sahinturk, V. The Protective Agents Used against Acrylamide Toxicity: An In Vitro Cell Culture Study-Based Review. Cell J. 2021, 23, 367–381. [Google Scholar] [CrossRef]
- Bachnoff, N.; Trus, M.; Atlas, D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radic. Biol. Med. 2011, 50, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kutner, M.; Khomsky, L.; Trus, M.; Aisner, Y.; Niv, M.Y.Y.; Benhar, M.; Atlas, D. Thioredoxin-mimetic peptides (TXM) reverse auranofin induced apoptosis and restore insulin secretion in insulinoma cells. Biochem. Pharmacol. 2013, 85, 977–990. [Google Scholar] [CrossRef]
- Eligini, S.; Munno, M.; Modafferi, G.; Atlas, D.; Banfi, C. N-Acetylcysteine, N-Acetylcysteine Amide, and Thioredoxin Mimetic Peptides Regenerate Mercaptoalbumin and Exhibit Antioxidant Activity. Antioxidants 2024, 13, 351. [Google Scholar] [CrossRef]
- Jastrzębska, J.; Frankowska, M.; Filip, M.; Atlas, D.; Jastrzebska, J.; Frankowska, M.; Filip, M.; Atlas, D. N-acetylcysteine amide (AD4) reduces cocaine-induced reinstatement. Psychopharmacology 2016, 233, 3437–3448. [Google Scholar] [CrossRef]
- Baratz-Goldstein, R.; Deselms, H.; Heim, L.R.; Khomski, L.; Hoffer, B.J.; Atlas, D.; Pick, C.G. Thioredoxin-Mimetic-Peptides Protect Cognitive Function after Mild Traumatic Brain Injury (mTBI). PLoS ONE 2016, 11, e0157064. [Google Scholar] [CrossRef]
- Atlas, D. Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free Radic. Biol. Med. 2021, 176, 120–141. [Google Scholar] [CrossRef]
- Wiesen, T.; Atlas, D. Novel anti-apoptotic L-DOPA precursors SuperDopa and SuperDopamide as potential neuroprotective agents for halting/delaying progression of Parkinson’s disease. Cell Death Dis. 2022, 13, 227. [Google Scholar] [CrossRef]
- Kim, S.R.R.; Lee, K.S.A.K.A.S.; Park, S.J.J.; Min, K.H.H.; Lee, M.H.H.; Lee, K.S.A.K.A.S.; Bartov, O.; Atlas, D.; Lee, Y.C.C. A novel dithiol amide CB3 attenuates allergic airway disease through negative regulation of p38 mitogen-activated protein kinase. Am. J. Respir. Crit. Care Med. 2011, 183, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Govednik, T.; Lainšček, D.; Kuhar, U.; Lachish, M.; Janežič, S.; Štrbenc, M.; Krapež, U.; Jerala, R.; Atlas, D.; Manček-Keber, M. TXM peptides inhibit SARS-CoV-2 infection, syncytia formation, and lower inflammatory consequences. Antivir. Res. 2024, 222, 105806. [Google Scholar] [CrossRef] [PubMed]
- Medali, T.; Couchie, D.; Mougenot, N.; Mihoc, M.; Bergmann, O.; Derks, W.; Szweda, L.I.; Yacoub, M.; Soliman, S.; Aguib, Y.; et al. Thioredoxin-1 and its mimetic peptide improve systolic cardiac function and remodeling after myocardial infarction. FASEB J. 2024, 38, e23291. [Google Scholar] [CrossRef] [PubMed]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J. Neurochem. 2004, 89, 1241–1251. [Google Scholar] [CrossRef]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic. Biol. Med. 2005, 38, 136–145. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, H.S.; Park, S.J.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Hong, S.H.; Han, H.J.; Lee, Y.R.; et al. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Exp. Mol. Med. 2007, 39, 756–768. [Google Scholar] [CrossRef]
- Lejnev, K.; Khomsky, L.; Bokvist, K.; Mistriel-Zarbib, S.; Naveh, T.; Farb, T.B.B.; Alsina-Fernandez, J.; Atlas, D.; Mistriel-Zerbib, S.; Naveh, T.; et al. Thioredoxin-mimetic peptides (TXM) inhibit inflammatory pathways associated with high-glucose and oxidative stress. Free. Radic. Biol. Med. 2016, 99, 557–571. [Google Scholar] [CrossRef]
- Canesi, F.; Mateo, V.; Couchie, D.; Karabina, S.; Nègre-Salvayre, A.; Rouis, M.; El Hadri, K. A thioredoxin-mimetic peptide exerts potent anti-inflammatory, antioxidant, and atheroprotective effects in ApoE2.Ki mice fed high fat diet. Cardiovasc. Res. 2019, 115, 292–301. [Google Scholar] [CrossRef]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int. J. Cardiol. 2020, 308, 73–81. [Google Scholar] [CrossRef]
- Eligini, S.; Munno, M.; Atlas, D.; Banfi, C. N-acetylcysteine Amide AD4/NACA and Thioredoxin Mimetic Peptides Inhibit Platelet Aggregation and Protect against Oxidative Stress. Antioxidants 2023, 12, 1395. [Google Scholar] [CrossRef]
- Bartov, O.; Sultana, R.; Butterfield, D.A.; Atlas, D. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Abeta(1–42) toxicity. Brain Res. 2006, 1069, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Olchawa, M.M.; Szewczyk, G.; Lachish, M.; Sarna, T.; Atlas, D. SuperDopa (SD), SuperDopa amide (SDA) and Thioredoxin-mimetic peptides protect ARPE-19 cells from photic- and non-photic stress. J. Photochem. Photobiol. 2024, 19, 100225. [Google Scholar] [CrossRef]
- Birkholz, A.; Kopecky, D.J.; Volak, L.P.; Bartberger, M.D.; Chen, Y.; Tegley, C.M.; Arvedson, T.; McCarter, J.D.; Fotsch, C.; Cee, V.J. Systematic Study of the Glutathione Reactivity of N-Phenylacrylamides: 2. Effects of Acrylamide Substitution. J. Med. Chem. 2020, 63, 11602–11614. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Prats, E.; Gómez-Canela, C.; Hsu, C.Y.; Arick, M.A.; Bedrossiantz, J.; Orozco, M.; Garcia-Reyero, N.; Ziv, T.; Ben-Lulu, S.; et al. Therapeutic potential of N-acetylcysteine in acrylamide acute neurotoxicity in adult zebrafish. Sci. Rep. 2019, 9, 16467. [Google Scholar] [CrossRef]
- Attoff, K.; Johansson, Y.; Cediel-Ulloa, A.; Lundqvist, J.; Gupta, R.; Caiment, F.; Gliga, A.; Forsby, A. Acrylamide alters CREB and retinoic acid signalling pathways during differentiation of the human neuroblastoma SH-SY5Y cell line. Sci. Rep. 2020, 10, 16714. [Google Scholar] [CrossRef]
- Bahat-Stroomza, M.; Gilgun-Sherki, Y.; Offen, D.; Panet, H.; Saada, A.; Krool-Galron, N.; Barzilai, A.; Atlas, D.; Melamed, E. A novel thiol antioxidant that crosses the blood brain barrier protects dopaminergic neurons in experimental models of Parkinson’s disease. Eur. J. Neurosci. 2005, 21, 637–646. [Google Scholar] [CrossRef]
- El-Habta, R.; af Bjerkén, S.; Virel, A. N-acetylcysteine increases dopamine release and prevents the deleterious effects of 6-OHDA on the expression of VMAT2, α-synuclein, and tyrosine hydroxylase. Neurol. Res. 2024, 46, 406–415. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| * IC50, µM | |||||
|---|---|---|---|---|---|
| NAME | STRUCTURE | p-ERK 1 | p-ERK 2 | p-p38 | pJNK |
| NAC | Ac Cys | 131 ± 0.4 | 133 ± 0.4 | ND | 102 ± 22 |
| AD4/NACA | Ac Cys-NH2 | 47 ± 0.1 | 63 ± 0.1 | ND | 21 ± 0.7 |
| CB20 | Ac Cys-Gly-Cys NH2 | 97.0 ± 23 | 48.6 ± 11 | 9.8 ± 6.8 | 3.2 ± 1.7 |
| CB30 | Ac DCys-Gly-DCys NH2 | 3.06 ± 1.2 | 17.9 ± 4.8 | 11.5 ± 1.9 | ND |
| CB13 | Ac Cys-Met-Lys-Cys NH2 | 10.4 ± 4.5 | 8.0 ± 3.6 | 12.4 ± 1.3 | ND |
| CB16 | Ac Cys-γGlu-Cys-Cys NH2 | ND | 9.01 ± 2.5 | 14.8 ± 1.7 | ND |
| SD | Ac Cys-Levodopa-Cys NH2 | 4.7 ± 0.7 | 4.55 ± 0.4 | 34.2 ± 5.5 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, V.; Trus, M.; Atlas, D. Thiol-Based Redox Molecules: Potential Antidotes for Acrylamide Toxicity. Antioxidants 2024, 13, 1431. https://doi.org/10.3390/antiox13121431
Martin V, Trus M, Atlas D. Thiol-Based Redox Molecules: Potential Antidotes for Acrylamide Toxicity. Antioxidants. 2024; 13(12):1431. https://doi.org/10.3390/antiox13121431
Chicago/Turabian StyleMartin, Valeria, Michael Trus, and Daphne Atlas. 2024. "Thiol-Based Redox Molecules: Potential Antidotes for Acrylamide Toxicity" Antioxidants 13, no. 12: 1431. https://doi.org/10.3390/antiox13121431
APA StyleMartin, V., Trus, M., & Atlas, D. (2024). Thiol-Based Redox Molecules: Potential Antidotes for Acrylamide Toxicity. Antioxidants, 13(12), 1431. https://doi.org/10.3390/antiox13121431