

Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells from Patients, Cell Lines, and Culture
2.2. Reagents and Antibodies
2.3. Viability Assay
2.4. Apoptosis Assay
2.5. Autophagy Assay
2.6. Intracellular ROS and Superoxide Measurement
2.7. Western Blot Analysis
2.8. NRF2 shRNAi Transfection
2.9. Statistical Analysis
3. Results
3.1. DFX-Induced Programmed Cell Death in ALL Cells
3.2. DFX Treatment Increased ROS Production by Regulating NRF2 Activity in ALL Cells
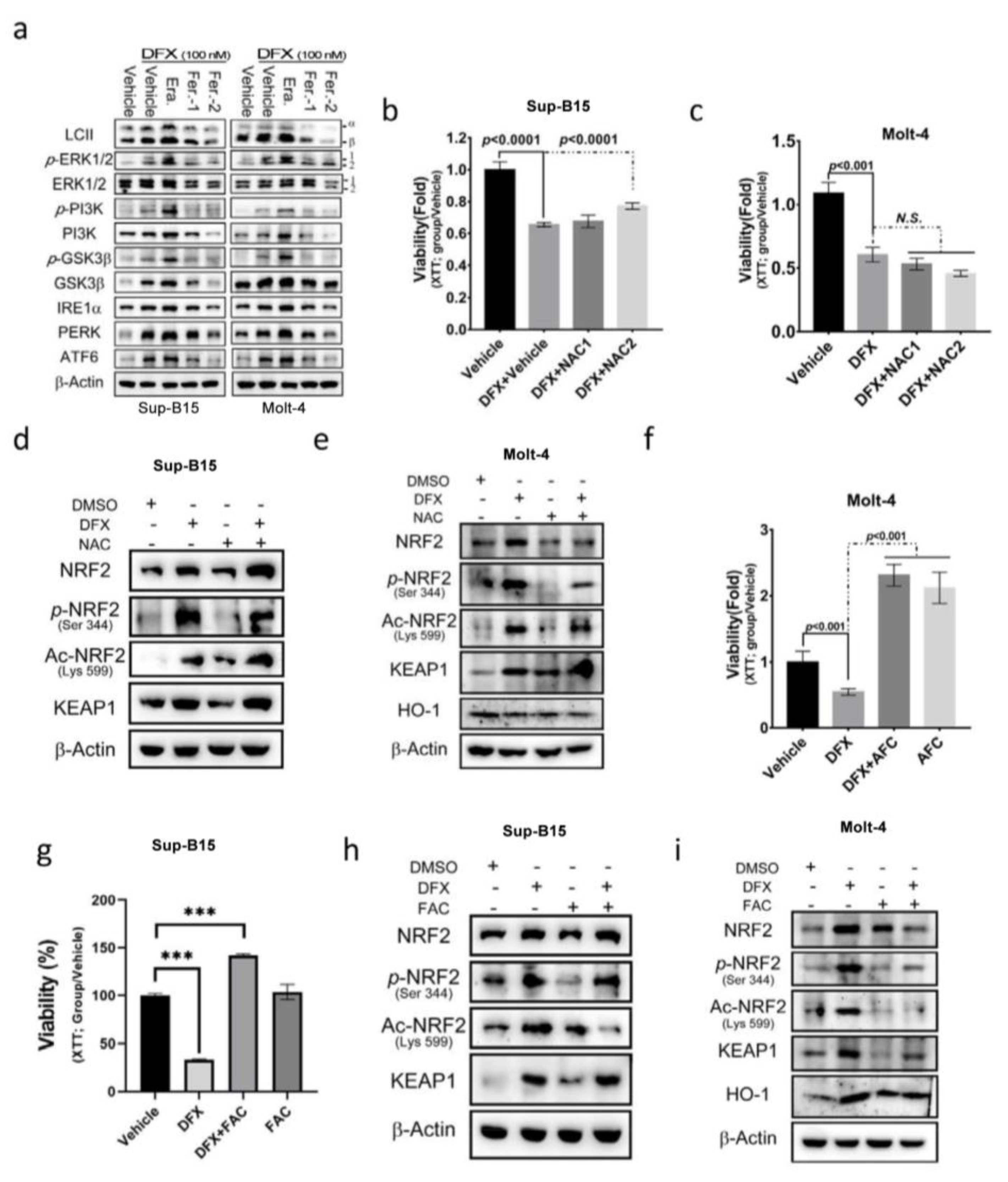
3.3. DFX Induces Ferroptosis via the NRF2 Signalling Pathway in Leukaemia Cell Lines
3.4. N-acetylcysteine (NAC) Cannot Reverse the Cytotoxic Effect of DFX on ALL Cells
3.5. The E1A Binding Protein p300/CREB Binding Protein (p300/CBP) Inhibitor Can Reverse the Effect of DFX on ALL Cells
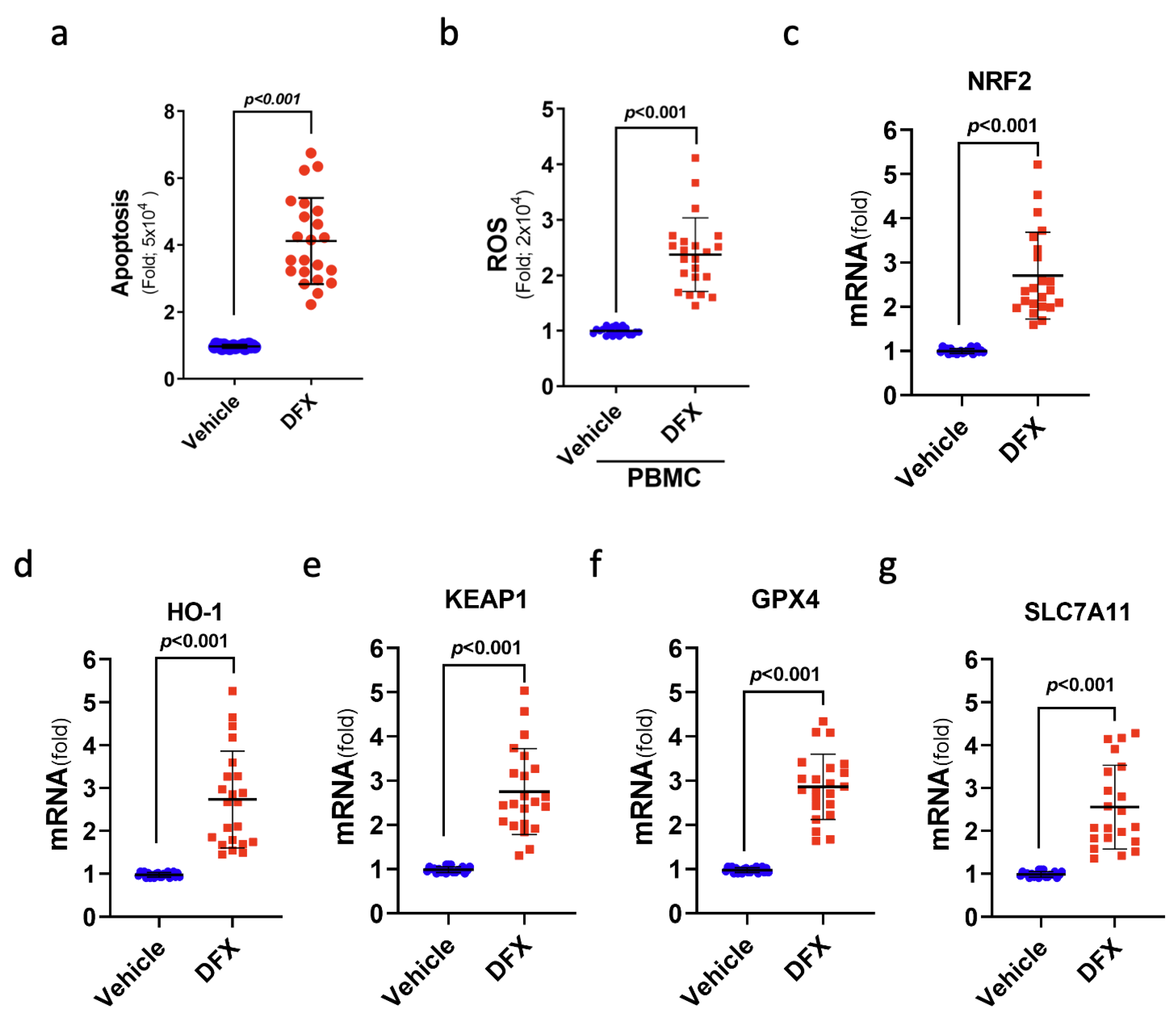
3.6. DFX Treatment Induced Ferroptosis-Mediated Cell Death in the Leukaemia Cells of Patients with ALL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Chen, Y.; Li, Z.; Huang, B.; Xu, L.; Lai, J.; Lu, Y.; Zha, X.; Liu, B.; Lan, Y. Single-Cell RNA-seq of T cells in B-ALL patients reveals an exhausted subset with remarkable heterogeneity. Adv. Sci. 2021, 8, 2101447. [Google Scholar] [CrossRef]
- Rogers, P.J.; Coccia, P.; Siegel, S.; Bleyer, W.A.; Lukens, J.; Sather, H.; Hammond, D. Yield of unpredicted bone-marrow relapse diagnosed by routine marrow aspiration in children with acute lymphoblastic leukaemia: A report from the Children’s Cancer Study Group. Lancet 1984, 323, 1320–1322. [Google Scholar] [CrossRef]
- Habel, A.; Hughes, G.; Durrant, S. CNS relapse in children with acute lymphoblastic leukaemia. Br. J. Haematol. 1985, 59, 737–738. [Google Scholar] [CrossRef]
- Moafi, A.; Ziaie, M.; Abedi, M.; Rahgozar, S.; Reisi, N.; Nematollahi, P.; Moafi, H. The relationship between iron bone marrow stores and response to treatment in pediatric acute lymphoblastic leukemia. Medicine 2017, 96, e8511. [Google Scholar] [CrossRef]
- Abedi, M.; Rahgozar, S.; Esmaeili, A. Iron protects childhood acute lymphoblastic leukemia cells from methotrexate cytotoxicity. Cancer Med. 2020, 9, 3537–3550. [Google Scholar] [CrossRef]
- Franke, G.-N.; Kubasch, A.S.; Cross, M.; Vucinic, V.; Platzbecker, U. Iron overload and its impact on outcome of patients with hematological diseases. Mol. Asp. Med. 2020, 75, 100868. [Google Scholar] [CrossRef]
- Belotti, A.; Duca, L.; Borin, L.; Realini, S.; Renso, R.; Parma, M.; Pioltelli, P.; Pogliani, E.; Cappellini, M.D. Non transferrin bound iron (NTBI) in acute leukemias throughout conventional intensive chemotherapy: Kinetics of its appearance and potential predictive role in infectious complications. Leuk. Res. 2015, 39, 88–91. [Google Scholar] [CrossRef]
- Bradley, S.; Gosriwitana, I.; Srichairatanakool, S.; Hider, R.; Porter, J. Non-transferrin-bound iron induced by myeloablative chemotherapy. Br. J. Haematol. 1997, 99, 337–343. [Google Scholar] [CrossRef]
- Eng, J.; Fish, J.D. Insidious iron burden in pediatric patients with acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 368–371. [Google Scholar] [CrossRef]
- Nair, M.; Kuttath, V.; Nair, A.R.; Rajeswari, B.; Chellappan, G.; Thankamony, P.; Parukkutty, K. Iron overload in children with leukemia receiving multiple blood transfusions. Indian Pediatr. 2018, 55, 962–965. [Google Scholar] [CrossRef]
- Wang, F.; Lv, H.; Zhao, B.; Zhou, L.; Wang, S.; Luo, J.; Liu, J.; Shang, P. Iron and leukemia: New insights for future treatments. J. Exp. Clin. Cancer Res. 2019, 38, 406. [Google Scholar] [CrossRef]
- Isidori, A.; Borin, L.; Elli, E.; Latagliata, R.; Martino, B.; Palumbo, G.; Pilo, F.; Loscocco, F.; Visani, G.; Cianciulli, P. Iron toxicity–Its effect on the bone marrow. Blood Rev. 2018, 32, 473–479. [Google Scholar] [CrossRef]
- Pilo, F.; Angelucci, E. A storm in the niche: Iron, oxidative stress and haemopoiesis. Blood Rev. 2018, 32, 29–35. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 2022, 13, 1039241. [Google Scholar] [CrossRef]
- Yang, M.; Shen, Z.; Zhang, X.; Song, Z.; Zhang, Y.; Lin, Z.; Chen, L. Ferroptosis of macrophages facilitates bone loss in apical periodontitis via NRF2/FSP1/ROS pathway. Free Radic. Biol. Med. 2023, 208, 334–347. [Google Scholar] [CrossRef]
- Basit, F.; Van Oppen, L.M.; Schöckel, L.; Bossenbroek, H.M.; Van Emst-de Vries, S.E.; Hermeling, J.C.; Grefte, S.; Kopitz, C.; Heroult, M.; HGM Willems, P. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017, 8, e2716. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Z.; Wang, S.; Ma, Q.; Li, L.; Wu, X.; Guo, Q.; Tao, L.; Shen, X. Boosting ROS-Mediated Lysosomal Membrane Permeabilization for Cancer Ferroptosis Therapy. Adv. Healthc. Mater. 2023, 12, 2202150. [Google Scholar] [CrossRef]
- Jiménez-Solas, T.; López-Cadenas, F.; Aires-Mejía, I.; Caballero-Berrocal, J.C.; Ortega, R.; Redondo, A.M.; Sánchez-Guijo, F.; Muntión, S.; García-Martín, L.; Albarrán, B. Deferasirox reduces oxidative DNA damage in bone marrow cells from myelodysplastic patients and improves their differentiation capacity. Br. J. Haematol. 2019, 187, 93–104. [Google Scholar] [CrossRef]
- Tataranni, T.; Mazzoccoli, C.; Agriesti, F.; De Luca, L.; Laurenzana, I.; Simeon, V.; Ruggieri, V.; Pacelli, C.; Della Sala, G.; Musto, P. Deferasirox drives ROS-mediated differentiation and induces interferon-stimulated gene expression in human healthy haematopoietic stem/progenitor cells and in leukemia cells. Stem Cell Res. Ther. 2019, 10, 171. [Google Scholar] [CrossRef]
- Ford, S.; Obeidy, P.; Lovejoy, D.; Bedford, M.; Nichols, L.; Chadwick, C.; Tucker, O.; Lui, G.; Kalinowski, D.; Jansson, P. Deferasirox (ICL670A) effectively inhibits oesophageal cancer growth in vitro and in vivo. Br. J. Pharmacol. 2013, 168, 1316–1328. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed]
- Boo, Y.C. Natural Nrf2 modulators for skin protection. Antioxidants 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Sanchez, J.M.H.; Lumbreras, E.; Diez-Campelo, M.; Gonzalez, T.; Lopez, D.A.; Abaigar, M.; Del Rey, M.; Martin, A.A.; de Paz, R.; Erquiaga, S. Genome-wide transcriptomics leads to the identification of deregulated genes after deferasirox therapy in low-risk MDS patients. Pharmacogenomics J. 2020, 20, 664–671. [Google Scholar] [CrossRef]
- Yi, Q.; Liang, Q.; Liu, Y.; Gong, Z.; Yan, Y. Application of genomic selection and experimental techniques to predict cell death and immunotherapeutic efficacy of ferroptosis-related CXCL2 in hepatocellular carcinoma. Front. Oncol. 2022, 12, 998736. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.-J.; Zhi-Min, Z.; Xu, X.-N.; Shi, F.; Fu, X.-L. Targeting critical pathways in ferroptosis and enhancing antitumor therapy of Platinum drugs for colorectal cancer. Sci. Prog. 2023, 106, 00368504221147173. [Google Scholar] [CrossRef]
- Jiang, F.; Jia, K.; Chen, Y.; Ji, C.; Chong, X.; Li, Z.; Zhao, F.; Bai, Y.; Ge, S.; Gao, J. ANO1-Mediated Inhibition of Cancer Ferroptosis Confers Immunotherapeutic Resistance through Recruiting Cancer-Associated Fibroblasts. Adv. Sci. 2023, 10, e2300881. [Google Scholar] [CrossRef]
- Kishore, M.; Pradeep, M.; Narne, P.; Jayalakshmi, S.; Panigrahi, M.; Patil, A.; Babu, P.P. Regulation of Keap1-Nrf2 axis in temporal lobe epilepsy—Hippocampal sclerosis patients may limit the seizure outcomes. Neurol. Sci. 2023, 44, 4441–4450. [Google Scholar] [CrossRef]
- Mukherjee, A.G.; Gopalakrishnan, A.V. The mechanistic insights of the antioxidant Keap1-Nrf2 pathway in oncogenesis: A deadly scenario. Med. Oncol. 2023, 40, 248. [Google Scholar] [CrossRef]
- von Mikecz, A.; Zhang, S.; Montminy, M.; Tan, E.M.; Hemmerich, P. Creb-binding protein (Cbp/P300) and RNA polymerase II Colocalize in transcriptionally active domains in the nucleus. J. Cell Biol. 2000, 150, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Tiu, B.; Sakamoto, K.M. CBP/p300 acetyltransferase activity in hematologic malignancies. Mol. Genet. Metab. 2016, 119, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation-deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef]
- Gao, Q.; Zhao, Y.; Luo, R.; Su, M.; Zhang, C.; Li, C.; Liu, B.; Zhou, X. Intrathecal umbilical cord mesenchymal stem cells injection alleviates neuroinflammation and oxidative stress in the cyclophosphamide-induced interstitial cystitis rats through the Sirt1/Nrf2/HO-1 pathway. Life Sci. 2023, 331, 122045. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, W.-Y.; Wang, L.-T.; Lin, P.-C.; Liao, Y.-M.; Hsu, S.-H.; Chiou, S.-S. Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis. Antioxidants 2024, 13, 424. https://doi.org/10.3390/antiox13040424
Hsu W-Y, Wang L-T, Lin P-C, Liao Y-M, Hsu S-H, Chiou S-S. Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis. Antioxidants. 2024; 13(4):424. https://doi.org/10.3390/antiox13040424
Chicago/Turabian StyleHsu, Wan-Yi, Li-Ting Wang, Pei-Chin Lin, Yu-Mei Liao, Shih-Hsien Hsu, and Shyh-Shin Chiou. 2024. "Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis" Antioxidants 13, no. 4: 424. https://doi.org/10.3390/antiox13040424
APA StyleHsu, W.-Y., Wang, L.-T., Lin, P.-C., Liao, Y.-M., Hsu, S.-H., & Chiou, S.-S. (2024). Deferasirox Causes Leukaemia Cell Death through Nrf2-Induced Ferroptosis. Antioxidants, 13(4), 424. https://doi.org/10.3390/antiox13040424