
Proteome Analysis Related to Unsaturated Fatty Acid Synthesis by Interfering with Bovine Adipocyte ACSL1 Gene
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Sample Collection and Cell Culture
2.3. Temporal Expression and siRNA Screening of ACSL1
2.4. Total Protein Extraction
2.5. Protein Inspection
2.6. Enzymatic Hydrolysis of Protein
2.7. Liquid Mass Detection
2.8. Identification and Quantification of Proteins
2.9. Cluster and Functional Enrichment Analysis of DEPs
2.10. Construction of DEP Interaction Network
2.11. Combination of Transcriptome Data to Construct DEL-DEP Regulatory Network
3. Results
3.1. ACSL1 Temporal Expression and Effective siRNA Screening
3.2. Determination of Protein Extracted from Bovine Adipocytes
3.3. Overview of Proteomics Information
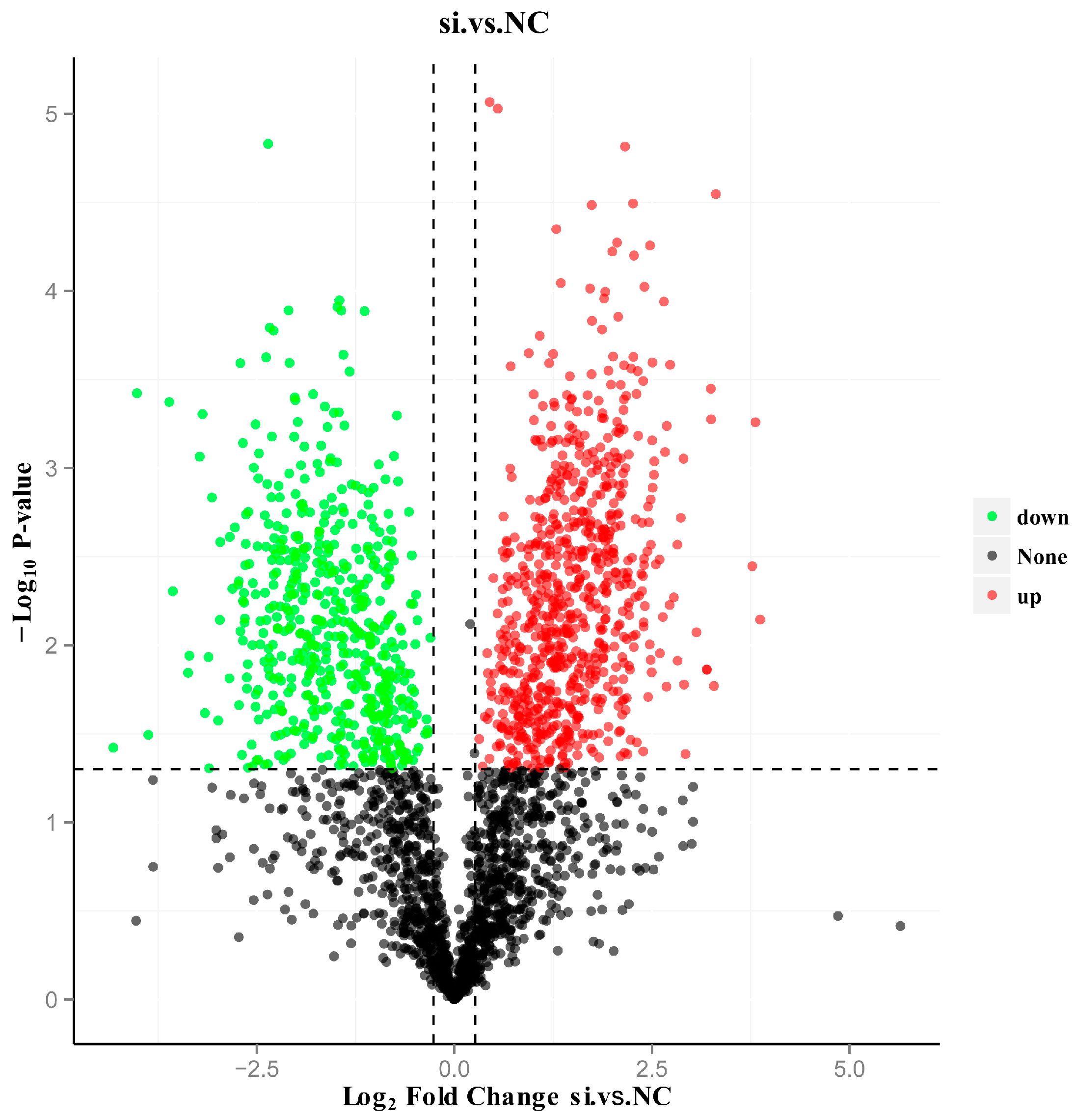
3.4. Screening of DEPs
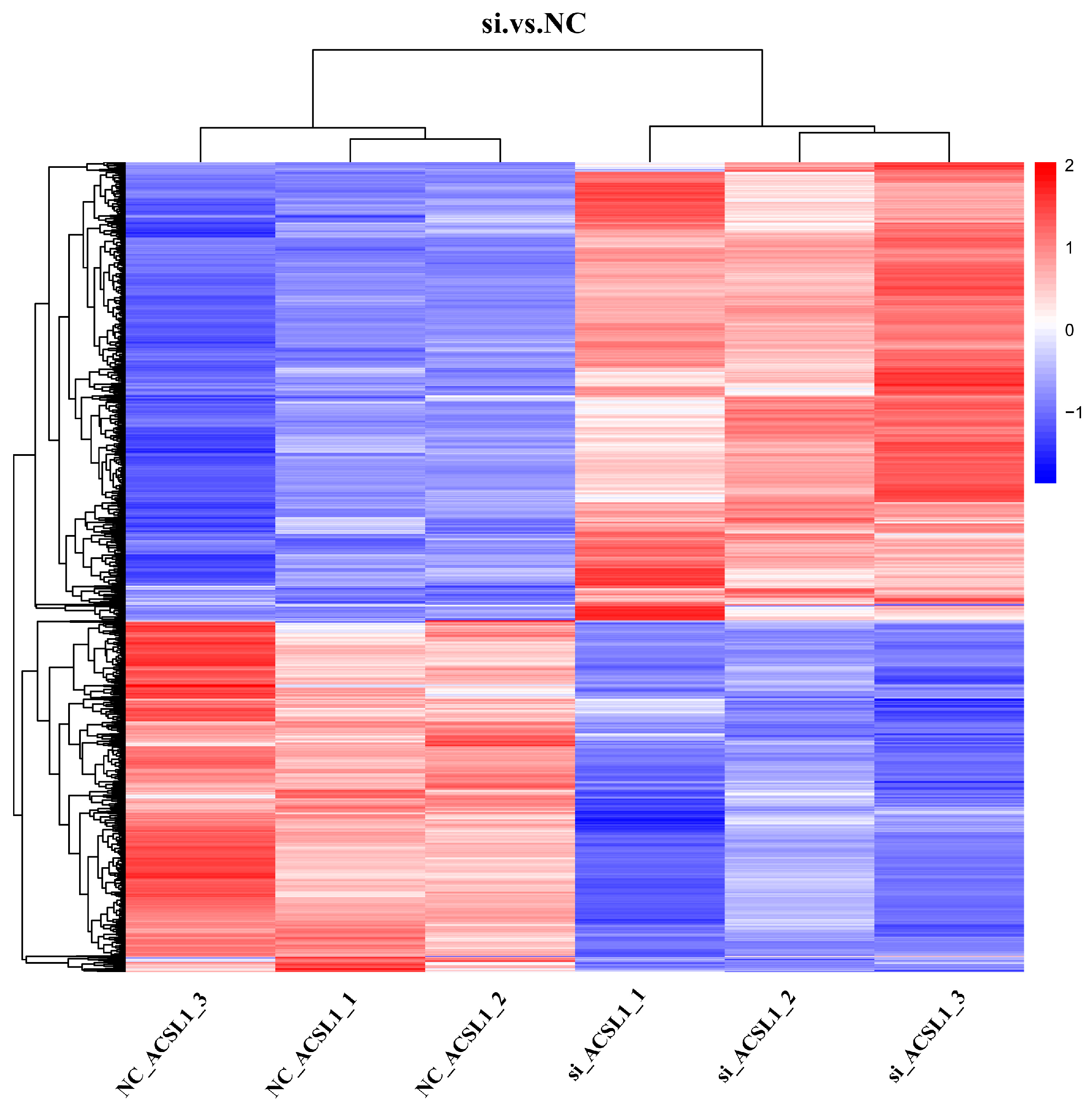
3.5. Cluster Analysis of DEPs
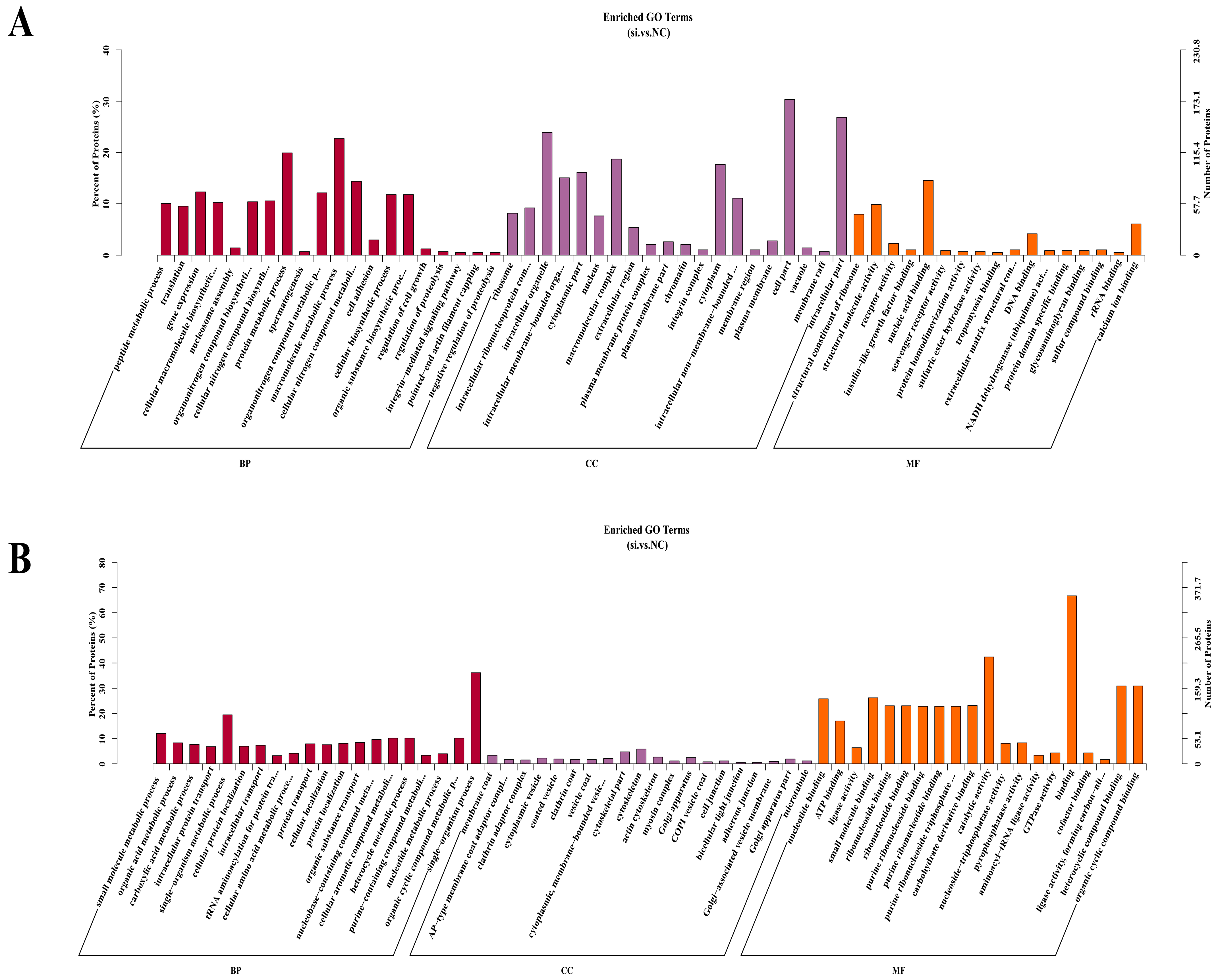
3.6. GO Enrichment Analysis of DEPs
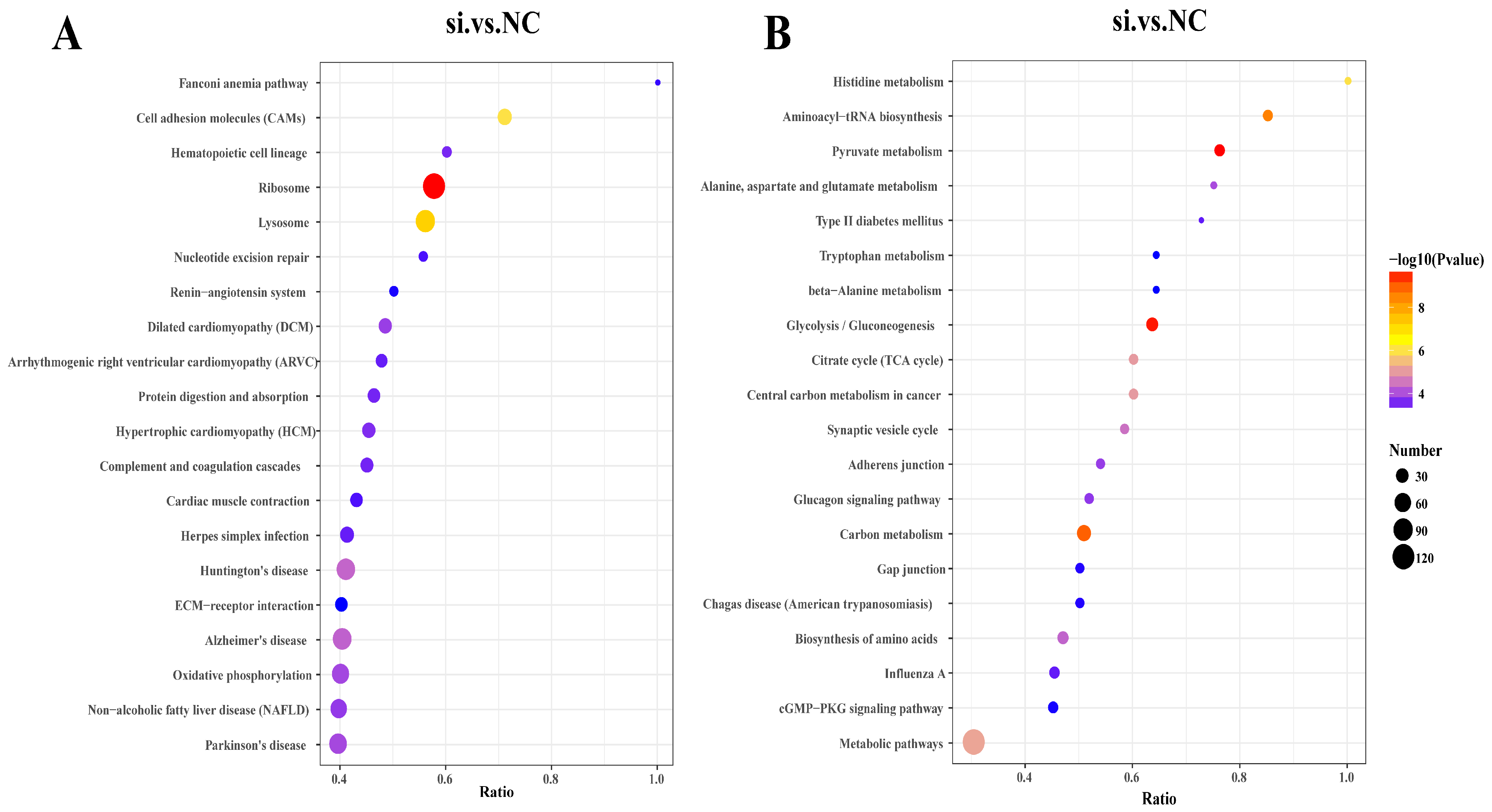
3.7. KEGG Enrichment Analysis of DEPs
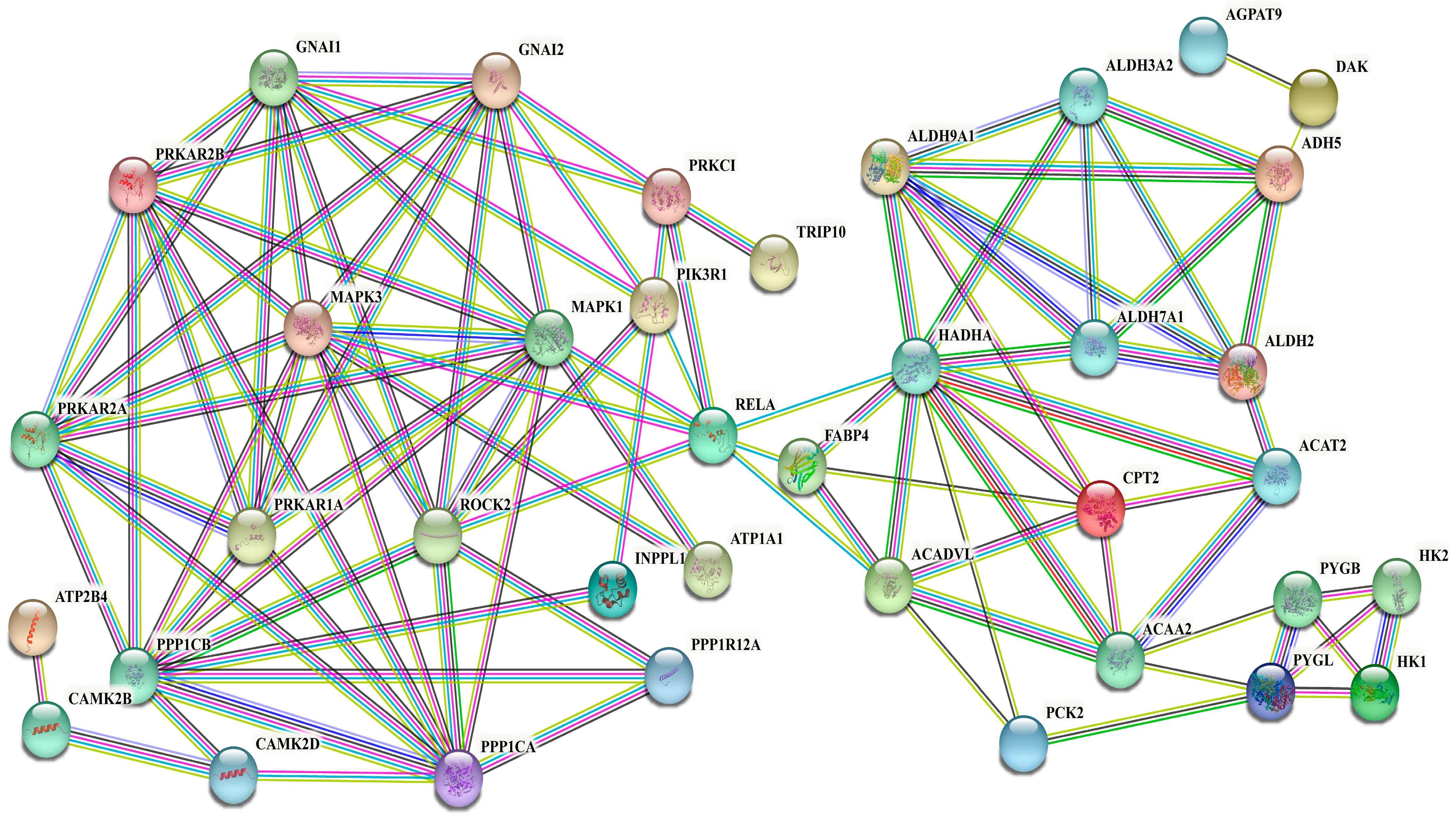
3.8. Interaction Networks of DEPs in Signaling Pathways Related to Synthesis of UFAs
3.9. lncRNA–Protein Regulatory Network Construction Combined with Transcriptome Data
4. Discussion
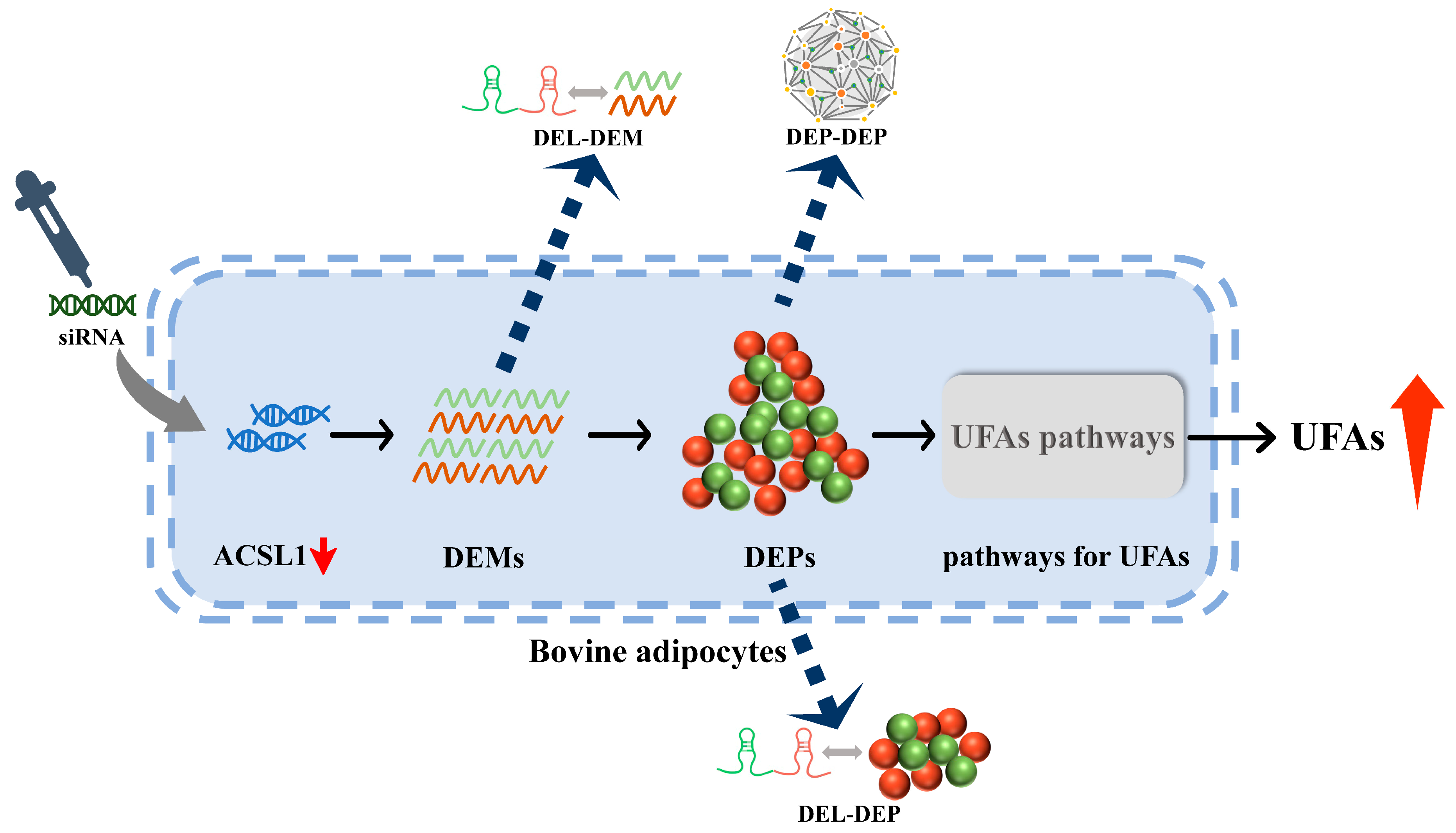
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lopez-Huertas, E. Health effects of oleic acid and long chain omega-3 fatty acids (EPA and DHA) enriched milks. A review of intervention studies. Pharmacol. Res. 2010, 61, 200–207. [Google Scholar] [CrossRef]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-3 fatty acids in health and disease and in growth and development. Am. J. Clin. Nutr. 1991, 54, 438–463. [Google Scholar] [CrossRef]
- Barcelo-Coblijn, G.; Murphy, E.J. Alpha-linolenic acid and its conversion to longer chain n-3 fatty acids: Benefits for human health and a role in maintaining tissue n-3 fatty acid levels. Prog. Lipid Res. 2009, 48, 355–374. [Google Scholar] [CrossRef]
- Gawron-Skarbek, A.; Guligowska, A.; Prymont-Przyminska, A.; Nowak, D.; Kostka, T. The Anti-Inflammatory and Antioxidant Impact of Dietary Fatty Acids in Cardiovascular Protection in Older Adults May Be Related to Vitamin C Intake. Antioxidants 2023, 12, 267. [Google Scholar] [CrossRef]
- Gonzalez-Benjumea, A.; Linde, D.; Carro, J.; Ullrich, R.; Hofrichter, M.; Martinez, A.T.; Gutierrez, A. Regioselective and Stereoselective Epoxidation of n-3 and n-6 Fatty Acids by Fungal Peroxygenases. Antioxidants 2021, 10, 1888. [Google Scholar] [CrossRef]
- Soupene, E.; Kuypers, F.A. Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. 2008, 233, 507–521. [Google Scholar] [CrossRef]
- Coleman, R.A.; Lewin, T.M.; Muoio, D.M. Physiological and nutritional regulation of enzymes of triacylglycerol synthesis. Annu. Rev. Nutr. 2000, 20, 77–103. [Google Scholar] [CrossRef]
- Zhan, T.; Poppelreuther, M.; Ehehalt, R.; Fullekrug, J. Overexpressed FATP1, ACSVL4/FATP4 and ACSL1 increase the cellular fatty acid uptake of 3T3-L1 adipocytes but are localized on intracellular membranes. PLoS ONE 2012, 7, e45087. [Google Scholar] [CrossRef]
- Ellis, J.M.; Li, L.O.; Wu, P.C.; Koves, T.R.; Ilkayeva, O.; Stevens, R.D.; Watkins, S.M.; Muoio, D.M.; Coleman, R.A. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. 2010, 12, 53–64. [Google Scholar] [CrossRef]
- Widmann, P.; Nuernberg, K.; Kuehn, C.; Weikard, R. Association of an ACSL1 gene variant with polyunsaturated fatty acids in bovine skeletal muscle. BMC Genet. 2011, 12, 96. [Google Scholar] [CrossRef] [PubMed]
- Li, L.O.; Mashek, D.G.; An, J.; Doughman, S.D.; Newgard, C.B.; Coleman, R.A. Overexpression of rat long chain acyl-coa synthetase 1 alters fatty acid metabolism in rat primary hepatocytes. J. Biol. Chem. 2006, 281, 37246–37255. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, S.; Liu, S.; Wang, Y.; Jin, H.; Ma, H.; Luo, X.; Cao, Y.; Lian, Z. Effects of Long-Chain Fatty Acyl-CoA Synthetase 1 on Diglyceride Synthesis and Arachidonic Acid Metabolism in Sheep Adipocytes. Int. J. Mol. Sci. 2020, 21, 2044. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.D.; Zan, L.S.; Li, A.N.; Cheng, G.; Li, S.J.; Zhang, Y.R.; Wang, X.Y.; Zhang, Y.Y. Characterization of the promoter region of the bovine long-chain acyl-CoA synthetase 1 gene: Roles of E2F1, Sp1, KLF15, and E2F4. Sci. Rep. 2016, 6, 19661. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Su, X.; Han, X.; Zan, L.; Hu, J.; Luo, Y.; Wang, J.; Bai, Y.; Zhao, Z. Effects of Silencing ACSL1 Gene by siRNA on the Synthesis of Unsaturated Fatty Acids in Adipocytes of Qinchuan Beef Cattle. J. Agric. Biotechnol. 2020, 28, 1722–1732. [Google Scholar]
- Zhao, Z.; Abbas, R.S.; Tian, H.; Shi, B.; Luo, Y.; Wang, J.; Liu, X.; Li, S.; Bai, Y.; Hu, J. Effects of overexpression of ACSL1 gene on the synthesis of unsaturated fatty acids in adipocytes of bovine. Arch. Biochem. Biophys. 2020, 695, 108648. [Google Scholar] [CrossRef]
- Zhu, C.; Cheng, H.; Li, N.; Liu, T.; Ma, Y. Isobaric Tags for Relative and Absolute Quantification-Based Proteomics Reveals Candidate Proteins of Fat Deposition in Chinese Indigenous Sheep with Morphologically Different Tails. Front. Genet. 2021, 12, 710449. [Google Scholar] [CrossRef]
- Han, J.; Guo, T.; Yue, Y.; Lu, Z.; Liu, J.; Yuan, C.; Niu, C.; Yang, M.; Yang, B. Quantitative proteomic analysis identified differentially expressed proteins with tail/rump fat deposition in Chinese thin- and fat-tailed lambs. PLoS ONE 2021, 16, e246279. [Google Scholar] [CrossRef]
- Wu, C.Y.; Wu, Y.Y.; Liu, C.D.; Wang, Y.X.; Na, W.; Wang, N.; Li, H. Comparative proteome analysis of abdominal adipose tissues between fat and lean broilers. Proteome Sci. 2016, 14, 9. [Google Scholar] [CrossRef]
- Wang, L.; Leng, L.; Ding, R.; Gong, P.; Liu, C.; Wang, N.; Li, H.; Du, Z.Q.; Cheng, B. Integrated transcriptome and proteome analysis reveals potential mechanisms for differential abdominal fat deposition between divergently selected chicken lines. J. Proteom. 2021, 241, 104242. [Google Scholar] [CrossRef]
- Na, W.; Wu, Y.Y.; Gong, P.F.; Wu, C.Y.; Cheng, B.H.; Wang, Y.X.; Wang, N.; Du, Z.Q.; Li, H. Embryonic transcriptome and proteome analyses on hepatic lipid metabolism in chickens divergently selected for abdominal fat content. BMC Genom. 2018, 19, 384. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Wang, W.; Wang, Y.; Sun, Y.; Kang, L.; Zhang, Q.; Jiang, Y. TMT-labeled quantitative proteomic analyses on the longissimus dorsi to identify the proteins underlying intramuscular fat content in pigs. J. Proteom. 2020, 213, 103630. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Shang, P.; Li, Q.; Wang, L.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. iTRAQ-based proteomic analysis reveals key proteins affecting muscle growth and lipid deposition in pigs. Sci. Rep. 2017, 7, 46717. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.H.; Wang, Z.; Yue, Y.X.; Li, H.; Li, Z.J.; Han, R.L.; Tian, Y.D.; Kang, X.T.; Liu, X.J. miR-34a-5p Increases Hepatic Triglycerides and Total Cholesterol Levels by Regulating ACSL1 Protein Expression in Laying Hens. Int. J. Mol. Sci. 2019, 20, 4420. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Gao, Q.; Tian, Z.; Lu, X.; Sun, Y.; Chen, Z.; Zhang, H.; Mao, Y.; Yang, Z. MIR221HG Is a Novel Long Noncoding RNA that Inhibits Bovine Adipocyte Differentiation. Genes 2019, 11, 29. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Liu, L.; Li, H.; Sun, Y.; Luo, H.; Li, T.; Wang, S.; Dalton, S.; Zhao, R.C.; Chen, R. Long noncoding RNA ADINR regulates adipogenesis by transcriptionally activating C/EBPalpha. Stem Cell Rep. 2021, 16, 1006–1008. [Google Scholar] [CrossRef]
- Poleti, M.D.; Regitano, L.; Souza, G.; Cesar, A.; Simas, R.C.; Silva-Vignato, B.; Oliveira, G.B.; Andrade, S.; Cameron, L.C.; Coutinho, L.L. Longissimus dorsi muscle label-free quantitative proteomic reveals biological mechanisms associated with intramuscular fat deposition. J. Proteom. 2018, 179, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Wang, J.; Li, H.; Gao, L.; Geng, J.; Ma, Z.; Liu, J.; Zhang, J.; Xie, P.; Chen, L. Combined transcriptome and proteome analyses reveal differences in the longissimus dorsi muscle between Kazakh cattle and Xinjiang brown cattle. Anim. Biosci. 2021, 34, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.N.; Kim, S.H.; Yoon, D.H.; Lee, H.G.; Kang, H.S.; Seo, K.S. Proteome Analysis of Bovine Longissimus dorsi Muscle Associated with the Marbling Score. Asian-Australas. J. Anim. Sci. 2012, 25, 1083–1088. [Google Scholar] [CrossRef]
- Bai, Y.; Li, X.; Chen, Z.; Li, J.; Tian, H.; Ma, Y.; Raza, S.; Shi, B.; Han, X.; Luo, Y.; et al. Interference with ACSL1 Gene in Bovine Adipocytes: Transcriptome Profiling of mRNA and lncRNA Related to Unsaturated Fatty Acid Synthesis. Front. Vet. Sci. 2021, 8, 788316. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Reis, B.S.; Lund, P.J.; Paul, M.R.; Leboeuf, M.; Djomo, A.M.; Nadeem, Z.; Lopes, M.; Vitorino, F.N.; Unlu, G.; et al. Histone butyrylation in the mouse intestine is mediated by the microbiota and associated with regulation of gene expression. Nat. Metab. 2024, 6, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Shuman, S. Transcriptional interference at tandem lncRNA and protein-coding genes: An emerging theme in regulation of cellular nutrient homeostasis. Nucleic Acids Res. 2020, 48, 8243–8254. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, C.; Zhang, C.; Zhou, Y.; Zhao, E.; Zhang, Y.; Pan, X.; Huang, H.; Liao, W.; Wang, X. LncRNA MIR200CHG inhibits EMT in gastric cancer by stabilizing miR-200c from target-directed miRNA degradation. Nat. Commun. 2023, 14, 8141. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Hu, Z.; Chen, F.; Zhu, R.; Deng, Y.; Shao, X.; Li, Y.; Zhao, J.; Li, H.; Zhang, B.; et al. Transcriptome analysis of long non-coding RNAs of the nucleus accumbens in cocaine-conditioned mice. J. Neurochem. 2012, 123, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhan, Y.; Lang, Z.; Li, Y.; Zhang, W.; Zheng, J. LncRNA-SNHG5 mediates activation of hepatic stellate cells by regulating NF2 and Hippo pathway. Commun. Biol. 2024, 7, 266. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Gillette, M.A.; Satpathy, S.; Cao, S.; Dhanasekaran, S.M.; Vasaikar, S.V.; Krug, K.; Petralia, F.; Li, Y.; Liang, W.W.; Reva, B.; et al. Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 2020, 182, 200–225. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, K.; Shi, Z.; Zhang, J.; Zhu, J.; Zhu, S.; Zhang, A.; Jia, Z.; Wang, G.; Yu, S.; et al. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int. J. Oncol. 2012, 40, 2004–2012. [Google Scholar] [CrossRef]
- Hocquette, J.F.; Gondret, F.; Baeza, E.; Medale, F.; Jurie, C.; Pethick, D.W. Intramuscular fat content in meat-producing animals: Development, genetic and nutritional control, and identification of putative markers. Animal 2010, 4, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Chung, E.R. Identification of differentially expressed genes between high and low marbling score grades of the longissimus lumborum muscle in Hanwoo (Korean cattle). Meat Sci. 2016, 121, 114–118. [Google Scholar] [CrossRef]
- Vogel, C.; Abreu, R.S.; Ko, D.; Le, S.Y.; Shapiro, B.A.; Burns, S.C.; Sandhu, D.; Boutz, D.R.; Marcotte, E.M.; Penalva, L.O. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400. [Google Scholar] [CrossRef]
- Chang, X.; Xu, Y.; Cheng, L.; Yi, K.; Gu, X.; Luo, Z.; Zhang, J.; Wang, J.; Geng, F. Quantitative proteomic analysis of cattle-yak and yak longissimus thoracis provides insights into the differential mechanisms of meat quality. Food Res. Int. 2023, 173, 113253. [Google Scholar] [CrossRef] [PubMed]
- Golick, L.; Han, Y.; Kim, Y.; Park, S.W. BRD7 regulates the insulin-signaling pathway by increasing phosphorylation of GSK3beta. Cell. Mol. Life Sci. 2018, 75, 1857–1869. [Google Scholar] [CrossRef]
- Cheatham, B.; Vlahos, C.J.; Cheatham, L.; Wang, L.; Blenis, J.; Kahn, C.R. Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis, and glucose transporter translocation. Mol. Cell. Biol. 1994, 14, 4902–4911. [Google Scholar] [CrossRef]
- Shepherd, P.R.; Nave, B.T.; Siddle, K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3-L1 adipocytes: Evidence for the involvement of phosphoinositide 3-kinase and p70 ribosomal protein-S6 kinase. Biochem. J. 1995, 305 Pt 1, 25–28. [Google Scholar] [CrossRef]
- Pessin, J.E.; Saltiel, A.R. Signaling pathways in insulin action: Molecular targets of insulin resistance. J. Clin. Investig. 2000, 106, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kim, H.J.; Rhyu, D.Y. Caulerpa lentillifera extract ameliorates insulin resistance and regulates glucose metabolism in C57BL/KsJ-db/db mice via PI3K/AKT signaling pathway in myocytes. J. Transl. Med. 2015, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.P.; Ha, J.M.; Yun, S.J.; Kim, E.K.; Chung, S.W.; Hong, K.W.; Kim, C.D.; Bae, S.S. Transcriptional activation of peroxisome proliferator-activated receptor-gamma requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2010, 399, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Jin, H.; Cao, Y.; Zhang, L.; Zhao, Y.; Jin, X.; Yu, Y. RNA-Seq Analysis Reveals a Positive Role of HTR2A in Adipogenesis in Yan Yellow Cattle. Int. J. Mol. Sci. 2018, 19, 1760. [Google Scholar] [CrossRef] [PubMed]
- Kohn, A.D.; Summers, S.A.; Birnbaum, M.J.; Roth, R.A. Expression of a constitutively active Akt Ser/Thr kinase in 3T3-L1 adipocytes stimulates glucose uptake and glucose transporter 4 translocation. J. Biol. Chem. 1996, 271, 31372–31378. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Mariman, E.C.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef]
- Lee, H.J.; Jang, M.; Kim, H.; Kwak, W.; Park, W.; Hwang, J.Y.; Lee, C.K.; Jang, G.W.; Park, M.N.; Kim, H.C.; et al. Comparative Transcriptome Analysis of Adipose Tissues Reveals that ECM-Receptor Interaction Is Involved in the Depot-Specific Adipogenesis in Cattle. PLoS ONE 2013, 8, e66267. [Google Scholar] [CrossRef]
- Ivessa, N.E.; Rehberg, E.; Kienzle, B.; Seif, F.; Hermann, R.; Hermann, M.; Schneider, W.J.; Gordon, D.A. Molecular cloning, expression, and hormonal regulation of the chicken microsomal triglyceride transfer protein. Gene 2013, 523, 1–9. [Google Scholar] [CrossRef]
- Sophos, N.A.; Pappa, A.; Ziegler, T.L.; Vasiliou, V. Aldehyde dehydrogenase gene superfamily: The 2000 update. Chem. Biol. Interact. 2001, 130–132, 323–337. [Google Scholar] [CrossRef]
- Zhang, H.M.; Xia, H.L.; Jiang, H.R.; Mao, Y.J.; Qu, K.X.; Huang, B.Z.; Gong, Y.C.; Yang, Z.P. Longissimus dorsi muscle transcriptomic analysis of Yunling and Chinese simmental cattle differing in intramuscular fat content and fatty acid composition. Genome 2018, 61, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Bosma, M. Lipid homeostasis in exercise. Drug Discov. Today 2014, 19, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Dunning, K.R.; Cashman, K.; Russell, D.L.; Thompson, J.G.; Norman, R.J.; Robker, R.L. Beta-oxidation is essential for mouse oocyte developmental competence and early embryo development. Biol. Reprod. 2010, 83, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Herrera, E.; Amusquivar, E. Lipid metabolism in the fetus and the newborn. Diabetes Metab. Res. Rev. 2000, 16, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Park, B.O.; Ahrends, R.; Teruel, M.N. Consecutive positive feedback loops create a bistable switch that controls preadipocyte-to-adipocyte conversion. Cell Rep. 2012, 2, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.W.; Klemm, D.J.; Vinson, C.; Lane, M.D. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein beta gene during adipogenesis. J. Biol. Chem. 2004, 279, 4471–4478. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.E.; Fankell, D.M.; Erickson, P.F.; Majka, S.M.; Crossno, J.J.; Klemm, D.J. Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J. Biol. Chem. 2006, 281, 40341–40353. [Google Scholar] [CrossRef]
- Niehof, M.; Manns, M.P.; Trautwein, C. CREB controls LAP/C/EBP beta transcription. Mol. Cell. Biol. 1997, 17, 3600–3613. [Google Scholar] [CrossRef]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008, 7, 339–347. [Google Scholar] [CrossRef]
- Weihua, L.; Chunli, M.; Bin, Y.; Chunyan, Y.; Beining, Z.; Yanfeng, X. LncRNA Gm15290 sponges miR-27b to promote PPARγ-induced fat deposition and contribute to body weight gain in mice. Biochem. Biophys. Res. Commun. 2017, 493, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Tang, G.; Zhang, Q.; Yong, W.; Zhang, W.; Xiao, J.; Wei, C.; He, C.; Yang, G.; Pang, W. A Novel lnc-RNA, Named lnc-ORA, Is Identified by RNA-Seq Analysis, and Its Knockdown Inhibits Adipogenesis by Regulating the PI3K/AKT/mTOR Signaling Pathway. Cells 2019, 8, 477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, F.; Sun, J.W.; Li, D.H.; Li, W.T.; Jiang, R.R.; Li, Z.J.; Liu, X.J.; Han, R.L.; Li, G.X.; et al. LncRNA IMFNCR Promotes Intramuscular Adipocyte Differentiation by Sponging miR-128-3p and miR-27b-3p. Front. Genet. 2019, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Bai, Z.; Xu, D.; Yuan, B.; Lo, K.A.; Yoon, M.J.; Lim, Y.C.; Knoll, M.; Slavov, N.; Chen, S.; et al. De Novo Reconstruction of Adipose Tissue Transcriptomes Reveals Long Non-coding RNA Regulators of Brown Adipocyte Development. Cell Metab. 2015, 21, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Li, M.; Jian, W.; Song, C.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. A novel lncRNA BADLNCR1 inhibits bovine adipogenesis by repressing GLRX5 expression. J. Cell. Mol. Med. 2020, 24, 7175–7186. [Google Scholar] [CrossRef]
- Cai, R.; Zhang, Q.; Wang, Y.; Yong, W.; Zhao, R.; Pang, W. Lnc-ORA interacts with microRNA-532-3p and IGF2BP2 to inhibit skeletal muscle myogenesis. J. Biol. Chem. 2021, 296, 100376. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACSL1 GenBank No. | siRNA No. | Target Sequence |
|---|---|---|
| NM_001076085.1 | ACSL1 siRNA-1 | GGATAGAGGAGTACCTGTA |
| NM_001076085.1 | ACSL1 siRNA-2 | CCCTATGAATGGCTTTCAT |
| NM_001076085.1 | ACSL1 siRNA-3 | ACTCTTCTCTATCGACAAT |
| Time (min) | Flow Rate (nL/min) | Mobile Phase A (%) | Mobile Phase B (%) |
|---|---|---|---|
| 0 | 600 | 94 | 6 |
| 2 | 600 | 90 | 10 |
| 45 | 600 | 70 | 30 |
| 48 | 600 | 65 | 35 |
| 50 | 600 | 50 | 50 |
| 51 | 600 | 0 | 100 |
| 60.5 | 600 | 95 | 5 |
| 61.5 | 600 | 95 | 5 |
| 62 | 600 | 5 | 95 |
| 67 | 600 | 5 | 95 |
| 70 | 600 | 95 | 5 |
| Sample | Protein Concentration | Total Protein |
|---|---|---|
| NC-ACSL1-1 | 1.10 | 132.00 |
| NC-ACSL1-2 | 0.99 | 118.80 |
| NC-ACSL1-3 | 1.05 | 126.00 |
| si-ACSL1-1 | 0.46 | 55.20 |
| si-ACSL1-2 | 0.42 | 50.40 |
| si-ACSL1-3 | 0.39 | 42.90 |
| Classification | Number |
|---|---|
| Total spectra | 400,282 |
| Matched spectrum | 140,710 |
| Peptide | 35,802 |
| Identified protein | 3558 |
| Map ID | Map Title | p-Value | Annotated DEPs for This Pathway |
|---|---|---|---|
| map04024 | cAMP signaling pathway | 0.015 | E1BC59, A0A3Q1LUJ8, A7MBH9, F1N319, Q3SWW9, Q3T0E7, P46196, A0A3Q1MNN7, A0A3Q1M647, P63097, A0A3Q1M066, A1XG22, D3K0R6, P23727 |
| map04921 | Oxytocin signaling pathway | 0.019 | P60712, Q3SYU2, E1BC59, A7MBH9, F1N319, Q3SWW9, Q3T0E7, P46196, A0A3Q1MNN7, A0A3Q1M647, P63097, E1BA29, A0A3Q1MB84, A0A3Q1M066, A5D7T5 |
| map00071 | Fatty acid degradation | 0.0228 | P48818, Q17QI3, A0A3S5ZP98, A0A3Q1NDR1, Q3T0R7, F1N1M7, Q3ZC42, A0A3Q1LZB8, P20000, Q2KJH9 |
| map00561 | Glycerol metabolism | 0.0289 | A0A3S5ZP98, A0A3Q1LZB8, P20000, Q2KJH9, A0A3Q1MGZ4, E1BGF8 |
| map04910 | Insulin signaling pathway | 0.0416 | A0A3Q1M168, A0A3Q1M0F0, F1MDS3, Q3SWW9, P00515, Q3T0E7, P46196, A0A3Q1MNN7, E1BME6, F1MU24, P00514, B0JYK4, A2VDU0, I7CLV3, F1MQ96, E1BBJ7, P23727 |
| map04923 | Regulation of lipolysis in adipocytes | 0.0459 | A7MBH9, P63097, I7CLV3, F1MHQ4, P23727 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, Y.; Li, J.; Wei, Y.; Chen, Z.; Liu, Z.; Guo, D.; Jia, X.; Niu, Y.; Shi, B.; Zhang, X.; et al. Proteome Analysis Related to Unsaturated Fatty Acid Synthesis by Interfering with Bovine Adipocyte ACSL1 Gene. Antioxidants 2024, 13, 641. https://doi.org/10.3390/antiox13060641
Bai Y, Li J, Wei Y, Chen Z, Liu Z, Guo D, Jia X, Niu Y, Shi B, Zhang X, et al. Proteome Analysis Related to Unsaturated Fatty Acid Synthesis by Interfering with Bovine Adipocyte ACSL1 Gene. Antioxidants. 2024; 13(6):641. https://doi.org/10.3390/antiox13060641
Chicago/Turabian StyleBai, Yanbin, Jingsheng Li, Yali Wei, Zongchang Chen, Zhanxin Liu, Dashan Guo, Xue Jia, Yanmei Niu, Bingang Shi, Xiaolan Zhang, and et al. 2024. "Proteome Analysis Related to Unsaturated Fatty Acid Synthesis by Interfering with Bovine Adipocyte ACSL1 Gene" Antioxidants 13, no. 6: 641. https://doi.org/10.3390/antiox13060641
APA StyleBai, Y., Li, J., Wei, Y., Chen, Z., Liu, Z., Guo, D., Jia, X., Niu, Y., Shi, B., Zhang, X., Zhao, Z., Hu, J., Han, X., Wang, J., Liu, X., & Li, S. (2024). Proteome Analysis Related to Unsaturated Fatty Acid Synthesis by Interfering with Bovine Adipocyte ACSL1 Gene. Antioxidants, 13(6), 641. https://doi.org/10.3390/antiox13060641