

Advance in Iron Metabolism, Oxidative Stress and Cellular Dysfunction in Experimental and Human Kidney Diseases
Abstract
:1. Introduction
2. The Regulation of Iron Metabolism
2.1. Iron Absorption from Guts to Plasma
2.2. Plasma Iron Transportation and Uptake by Other Cells
2.3. Cellular Iron Transfer to Mitochondria and Its Utilization
2.4. Cellular Iron Storage
2.5. Cellular Iron Export
2.6. The Regulatory Mechanisms of Iron Hemostasis
2.6.1. The Iron-Regulatory Protein (IRP) and Iron-Responsive Element (IRE) Regulatory System
2.6.2. The Hypoxia-Inducible Factor (HIF) Regulatory System
2.6.3. Hepcidin–Ferroportin Regulatory System
3. Renal Iron Homeostasis and Cellular Dysfunction in Kidney Diseases
3.1. Renal Iron Homeostasis
3.2. Abnormal Iron Metabolism and Cellular Dysfunction in Renal Injury and Disease
4. The Regulation of Redox Homeostasis in the Kidneys
4.1. ROS and Oxidative Stress in the Kidneys
4.2. Antioxidant Defense Systems in the Kidneys
5. Oxidative Stress and Cellular Dysfunction in Kidney Diseases
5.1. Oxidative Stress and Renal Tubular Cell Dysfunction in Kidney Disease
5.2. Oxidative Stress and Podocyte Dysfunction in Kidney Diseases
5.3. Oxidative Stress and Mesangial Cell and Interstitial Fibroblast Dysfunction in Kidney Disease
5.4. Oxidative Stress and Endothelial Cell Dysfunction in Kidney Disease
6. Oxidative Stress-Related Molecules in Kidney Disease
6.1. Nrf2
6.2. NF-κB
6.3. Sirtuin 1
7. The Crosstalk of Abnormal Iron Metabolism and Oxidative Stress in Kidney Diseases
7.1. How Oxidative Stress Affects Iron Metabolism
7.2. Abnormal Iron Metabolism Leads to Oxidative Stress and Ferroptosis
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xie, H.; Yang, N.; Yu, C.; Lu, L. Uremic toxins mediate kidney diseases: The role of aryl hydrocarbon receptor. Cell. Mol. Biol. Lett. 2024, 29, 38. [Google Scholar] [CrossRef]
- Chawla, L.S.; Bellomo, R.; Bihorac, A.; Goldstein, S.L.; Siew, E.D.; Bagshaw, S.M.; Bittleman, D.; Cruz, D.; Endre, Z.; Fitzgerald, R.L.; et al. Acute kidney disease and renal recovery: Consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat. Rev. Nephrol. 2017, 13, 241–257. [Google Scholar] [CrossRef]
- André, C.; Bodeau, S.; Kamel, S.; Bennis, Y.; Caillard, P. The AKI-to-CKD Transition: The Role of Uremic Toxins. Int. J. Mol. Sci. 2023, 24, 16152. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, C. Oxidative Stress: A Culprit in the Progression of Diabetic Kidney Disease. Antioxidants 2024, 13, 455. [Google Scholar] [CrossRef]
- Hassler, J.R. IgA nephropathy: A brief review. Semin. Diagn. Pathol. 2020, 37, 143–147. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Synthesis and breakdown of universal metabolic precursors promoted by iron. Nature 2019, 569, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, T.; Abe, M.; Kobayashi, H. Iron Metabolism and Inflammatory Mediators in Patients with Renal Dysfunction. Int. J. Mol. Sci. 2024, 25, 3745. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Hasuike, Y.; Otaki, Y.; Nanami, M.; Kuragano, T. Dysregulated iron metabolism in patients on hemodialysis. Contrib. Nephrol. 2015, 185, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. JASN 2012, 23, 1631–1634. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.; Zhang, H.; Chen, H.; Hao, J.; Liu, H.; Li, X. Mitochondrial metabolism and targeted treatment strategies in ischemic-induced acute kidney injury. Cell Death Discov. 2024, 10, 69. [Google Scholar] [CrossRef]
- Kimura, T.; Suzuki, K. Components of the electron transport system in adrenal steroid hydroxylase. Isolation and properties of non-heme iron protein (adrenodoxin). J. Biol. Chem. 1967, 242, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Monsen, E.R.; Hallberg, L.; Layrisse, M.; Hegsted, D.M.; Cook, J.D.; Mertz, W.; Finch, C.A. Estimation of available dietary iron. Am. J. Clin. Nutr. 1978, 31, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.B.; Hurrell, R.F. Nutritional iron deficiency. Lancet 2007, 370, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Riedel, H.D.; Remus, A.J.; Fitscher, B.A.; Stremmel, W. Characterization and partial purification of a ferrireductase from human duodenal microvillus membranes. Biochem. J. 1995, 309 Pt 3, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef]
- Dunn, L.L.; Suryo Rahmanto, Y.; Richardson, D.R. Iron uptake and metabolism in the new millennium. Trends Cell Biol. 2007, 17, 93–100. [Google Scholar] [CrossRef]
- Schneider, C.; Owen, M.J.; Banville, D.; Williams, J.G. Primary structure of human transferrin receptor deduced from the mRNA sequence. Nature 1984, 311, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar] [CrossRef] [PubMed]
- Bartnikas, T.B. Known and potential roles of transferrin in iron biology. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2012, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Enns, C.A.; Sussman, H.H. Physical characterization of the transferrin receptor in human placentae. J. Biol. Chem. 1981, 256, 9820–9823. [Google Scholar] [CrossRef] [PubMed]
- Ponka, P.; Beaumont, C.; Richardson, D.R. Function and regulation of transferrin and ferritin. Semin. Hematol. 1998, 35, 35–54. [Google Scholar] [PubMed]
- Andrews, N.C. Forging a field: The golden age of iron biology. Blood 2008, 112, 219–230. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like protein 14 (ZIP14) promotes the cellular assimilation of iron from transferrin. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef]
- Levy, J.E.; Jin, O.; Fujiwara, Y.; Kuo, F.; Andrews, N.C. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 1999, 21, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Montemiglio, L.C.; Testi, C.; Ceci, P.; Falvo, E.; Pitea, M.; Savino, C.; Arcovito, A.; Peruzzi, G.; Baiocco, P.; Mancia, F.; et al. Cryo-EM structure of the human ferritin-transferrin receptor 1 complex. Nat. Commun. 2019, 10, 1121. [Google Scholar] [CrossRef]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed]
- van Raaij, S.E.G.; Srai, S.K.S.; Swinkels, D.W.; van Swelm, R.P.L. Iron uptake by ZIP8 and ZIP14 in human proximal tubular epithelial cells. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2019, 32, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A. Low molecular weight intracellular iron transport compounds. Blood 1977, 50, 433–439. [Google Scholar] [CrossRef]
- Vyoral, D.; Hradilek, A.; Neuwirt, J. Transferrin and iron distribution in subcellular fractions of K562 cells in the early stages of transferrin endocytosis. Biochim. Biophys. Acta 1992, 1137, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef]
- Jain, C.; Shah, Y.M. PCBP1 is essential for proper iron absorption. Blood 2023, 142, 1585–1587. [Google Scholar] [CrossRef]
- Ponka, P. Tissue-specific regulation of iron metabolism and heme synthesis: Distinct control mechanisms in erythroid cells. Blood 1997, 89, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. Intracellular kinetics of iron in reticulocytes: Evidence for endosome involvement in iron targeting to mitochondria. Blood 2005, 105, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Provance, D.W., Jr.; Gourley, C.R.; Silan, C.M.; Cameron, L.C.; Shokat, K.M.; Goldenring, J.R.; Shah, K.; Gillespie, P.G.; Mercer, J.A. Chemical-genetic inhibition of a sensitized mutant myosin Vb demonstrates a role in peripheral-pericentriolar membrane traffic. Proc. Natl. Acad. Sci. USA 2004, 101, 1868–1873. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B.J.; Morgan, E.H. The kinetics of transferrin endocytosis and iron uptake from transferrin in rabbit reticulocytes. J. Biol. Chem. 1983, 258, 9108–9115. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.E.; Jin, O.; Bennett, C.; Morgan, K.; Wang, F.; Trenor, C.C., 3rd; Fleming, M.D.; Andrews, N.C. A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat. Genet. 2005, 37, 1270–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. The anemia of “haemoglobin-deficit” (hbd/hbd) mice is caused by a defect in transferrin cycling. Exp. Hematol. 2006, 34, 593–598. [Google Scholar] [CrossRef]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Seguin, A.; Jia, X.; Earl, A.M.; Li, L.; Wallace, J.; Qiu, A.; Bradley, T.; Shrestha, R.; Troadec, M.B.; Hockin, M.; et al. The mitochondrial metal transporters mitoferrin1 and mitoferrin2 are required for liver regeneration and cell proliferation in mice. J. Biol. Chem. 2020, 295, 11002–11020. [Google Scholar] [CrossRef]
- Troadec, M.B.; Warner, D.; Wallace, J.; Thomas, K.; Spangrude, G.J.; Phillips, J.; Khalimonchuk, O.; Paw, B.H.; Ward, D.M.; Kaplan, J. Targeted deletion of the mouse Mitoferrin1 gene: From anemia to protoporphyria. Blood 2011, 117, 5494–5502. [Google Scholar] [CrossRef]
- Napier, I.; Ponka, P.; Richardson, D.R. Iron trafficking in the mitochondrion: Novel pathways revealed by disease. Blood 2005, 105, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Mühlenhoff, U. Maturation of iron-sulfur proteins in eukaryotes: Mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 2008, 77, 669–700. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat. Commun. 2020, 11, 6310. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Fischbach, F.A.; Hoy, T.G.; Haggis, G.H. Ferric oxyhydroxide core of ferritin. Nature 1967, 216, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar] [CrossRef]
- Koorts, A.M.; Viljoen, M. Ferritin and ferritin isoforms I: Structure-function relationships, synthesis, degradation and secretion. Arch. Physiol. Biochem. 2007, 113, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Garringer, H.J.; Goodwin, C.B.; Richine, B.; Acton, A.; VanDuyn, N.; Muhoberac, B.B.; Irimia-Dominguez, J.; Chan, R.J.; Peacock, M.; et al. Systemic and cerebral iron homeostasis in ferritin knock-out mice. PLoS ONE 2015, 10, e0117435. [Google Scholar] [CrossRef]
- Arosio, P.; Yokota, M.; Drysdale, J.W. Structural and immunological relationships of isoferritins in normal and malignant cells. Cancer Res. 1976, 36, 1735–1739. [Google Scholar]
- Li, J.Y.; Feng, Y.H.; Li, Y.X.; He, P.Y.; Zhou, Q.Y.; Tian, Y.P.; Yao, R.Q.; Yao, Y.M. Ferritinophagy: A novel insight into the double-edged sword in ferritinophagy-ferroptosis axis and human diseases. Cell Prolif. 2024, e13621. [Google Scholar]
- Pan, Y.; Ren, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.; Zhang, H.; Fan, X.; et al. Structural basis of ion transport and inhibition in ferroportin. Nat. Commun. 2020, 11, 5686. [Google Scholar] [CrossRef] [PubMed]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of hepcidin-bound ferroportin reveals iron homeostatic mechanisms. Nature 2020, 586, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Yanatori, I.; Richardson, D.R.; Imada, K.; Kishi, F. Iron Export through the Transporter Ferroportin 1 Is Modulated by the Iron Chaperone PCBP2. J. Biol. Chem. 2016, 291, 17303–17318. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, F.; Guo, X.; An, P.; Tao, Y.; Wang, F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology 2012, 56, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin deficiency in erythroid cells causes serum iron deficiency and promotes hemolysis due to oxidative stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Kato, K.; Yanatori, I.; Murohara, T.; Toyokuni, S. Ferroptosis-dependent extracellular vesicles from macrophage contribute to asbestos-induced mesothelial carcinogenesis through loading ferritin. Redox Biol. 2021, 47, 102174. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; Farzam, F.; et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017, 36, 42–60. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e574. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Jin, Z.; Bandyopadhyay, G.; Wang, G.; Zhang, D.; Rocha, K.C.E.; Liu, X.; Zhao, H.; Kisseleva, T.; Brenner, D.A.; et al. Aberrant iron distribution via hepatocyte-stellate cell axis drives liver lipogenesis and fibrosis. Cell Metab. 2022, 34, 1201–1213.e1205. [Google Scholar] [CrossRef]
- Kenny, T.C.; Khan, A.; Son, Y.; Yue, L.; Heissel, S.; Sharma, A.; Pasolli, H.A.; Liu, Y.; Gamazon, E.R.; Alwaseem, H.; et al. Integrative genetic analysis identifies FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab. 2023, 35, 1057–1071.e1012. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.; Gray, N.K.; Hentze, M.W. IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol. Cell 1998, 2, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, M.; Nakatsuka, Y.; Vandenbon, A.; Ori, D.; Uehata, T.; Tsujimura, T.; Suzuki, Y.; Mino, T.; Takeuchi, O. Regnase-1 Maintains Iron Homeostasis via the Degradation of Transferrin Receptor 1 and Prolyl-Hydroxylase-Domain-Containing Protein 3 mRNAs. Cell Rep. 2017, 19, 1614–1630. [Google Scholar] [CrossRef] [PubMed]
- Corral, V.M.; Schultz, E.R.; Eisenstein, R.S.; Connell, G.J. Roquin is a major mediator of iron-regulated changes to transferrin receptor-1 mRNA stability. iScience 2021, 24, 102360. [Google Scholar] [CrossRef]
- Muto, Y.; Nishiyama, M.; Nita, A.; Moroishi, T.; Nakayama, K.I. Essential role of FBXL5-mediated cellular iron homeostasis in maintenance of hematopoietic stem cells. Nat. Commun. 2017, 8, 16114. [Google Scholar] [CrossRef]
- Smith, S.R.; Ghosh, M.C.; Ollivierre-Wilson, H.; Hang Tong, W.; Rouault, T.A. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol. Dis. 2006, 36, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Kaden, S.; Gröne, H.J.; Hentze, M.W. Iron regulatory proteins are essential for intestinal function and control key iron absorption molecules in the duodenum. Cell Metab. 2008, 7, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Ferring-Appel, D.; Becker, C.; Gretz, N.; Gröne, H.J.; Schümann, K.; Hentze, M.W. Iron regulatory proteins control a mucosal block to intestinal iron absorption. Cell Rep. 2013, 3, 844–857. [Google Scholar] [CrossRef]
- Nairz, M.; Ferring-Appel, D.; Casarrubea, D.; Sonnweber, T.; Viatte, L.; Schroll, A.; Haschka, D.; Fang, F.C.; Hentze, M.W.; Weiss, G.; et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe 2015, 18, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, N.; Pantopoulos, K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood 2013, 122, 1658–1668. [Google Scholar] [CrossRef] [PubMed]
- Cooperman, S.S.; Meyron-Holtz, E.G.; Olivierre-Wilson, H.; Ghosh, M.C.; McConnell, J.P.; Rouault, T.A. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood 2005, 106, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J. Biol. Chem. 1993, 268, 21513–21518. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Tsuzuki, Y.; Fukumura, D.; Oosthuyse, B.; Koike, C.; Carmeliet, P.; Jain, R.K. Vascular endothelial growth factor (VEGF) modulation by targeting hypoxia-inducible factor-1alpha--> hypoxia response element--> VEGF cascade differentially regulates vascular response and growth rate in tumors. Cancer Res. 2000, 60, 6248–6252. [Google Scholar] [PubMed]
- Firth, J.D.; Ebert, B.L.; Pugh, C.W.; Ratcliffe, P.J. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: Similarities with the erythropoietin 3’ enhancer. Proc. Natl. Acad. Sci. USA 1994, 91, 6496–6500. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Gu, X.; Hoestje, S.; Epner, D.E. Identification of an additional hypoxia responsive element in the glyceraldehyde-3-phosphate dehydrogenase gene promoter. Biochim. Biophys. Acta 2002, 1574, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.H.; Gonzalez, F.J. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Davis, M.R.; Shawron, K.M.; Rendina, E.; Peterson, S.K.; Lucas, E.A.; Smith, B.J.; Clarke, S.L. Hypoxia inducible factor-2 α is translationally repressed in response to dietary iron deficiency in Sprague-Dawley rats. J. Nutr. 2011, 141, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef]
- Abboud, S.; Haile, D.J. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef]
- Qiao, B.; Sugianto, P.; Fung, E.; Del-Castillo-Rueda, A.; Moran-Jimenez, M.J.; Ganz, T.; Nemeth, E. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012, 15, 918–924. [Google Scholar] [CrossRef]
- D’Alessio, F.; Hentze, M.W.; Muckenthaler, M.U. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef]
- Varga, E.; Pap, R.; Jánosa, G.; Sipos, K.; Pandur, E. IL-6 Regulates Hepcidin Expression Via the BMP/SMAD Pathway by Altering BMP6, TMPRSS6 and TfR2 Expressions at Normal and Inflammatory Conditions in BV2 Microglia. Neurochem. Res. 2021, 46, 1224–1238. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Gochanour, E.M.; Sundaram, V.; Shah, R.A.; Handa, P. Hepcidin Signaling in Health and Disease: Ironing Out the Details. Hepatol. Commun. 2021, 5, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Wolff, N.A. Iron transport in the kidney: Implications for physiology and cadmium nephrotoxicity. Met. Integr. Biometal Sci. 2016, 8, 17–42. [Google Scholar] [CrossRef] [PubMed]
- Martines, A.M.; Masereeuw, R.; Tjalsma, H.; Hoenderop, J.G.; Wetzels, J.F.; Swinkels, D.W. Iron metabolism in the pathogenesis of iron-induced kidney injury. Nat. Rev. Nephrol. 2013, 9, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Kozyraki, R.; Fyfe, J.; Verroust, P.J.; Jacobsen, C.; Dautry-Varsat, A.; Gburek, J.; Willnow, T.E.; Christensen, E.I.; Moestrup, S.K. Megalin-dependent cubilin-mediated endocytosis is a major pathway for the apical uptake of transferrin in polarized epithelia. Proc. Natl. Acad. Sci. USA 2001, 98, 12491–12496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Meyron-Holtz, E.; Rouault, T.A. Renal iron metabolism: Transferrin iron delivery and the role of iron regulatory proteins. J. Am. Soc. Nephrol. JASN 2007, 18, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Spektor, L.; Cohen, L.A.; Lifshitz, L.; Magid Gold, I.; Zhang, D.L.; Truman-Rosentsvit, M.; Leichtmann-Bardoogo, Y.; Nyska, A.; Addadi, S.; et al. Orchestrated regulation of iron trafficking proteins in the kidney during iron overload facilitates systemic iron retention. PLoS ONE 2018, 13, e0204471. [Google Scholar] [CrossRef]
- Smith, C.P.; Lee, W.K.; Haley, M.; Poulsen, S.B.; Thévenod, F.; Fenton, R.A. Proximal tubule transferrin uptake is modulated by cellular iron and mediated by apical membrane megalin-cubilin complex and transferrin receptor 1. J. Biol. Chem. 2019, 294, 7025–7036. [Google Scholar] [CrossRef]
- Gburek, J.; Verroust, P.J.; Willnow, T.E.; Fyfe, J.C.; Nowacki, W.; Jacobsen, C.; Moestrup, S.K.; Christensen, E.I. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J. Am. Soc. Nephrol. JASN 2002, 13, 423–430. [Google Scholar] [CrossRef]
- van Swelm, R.P.L.; Vos, M.; Verhoeven, F.; Thévenod, F.; Swinkels, D.W. Endogenous hepcidin synthesis protects the distal nephron against hemin and hemoglobin mediated necroptosis. Cell Death Dis. 2018, 9, 550. [Google Scholar] [CrossRef]
- van Raaij, S.; van Swelm, R.; Bouman, K.; Cliteur, M.; van den Heuvel, M.C.; Pertijs, J.; Patel, D.; Bass, P.; van Goor, H.; Unwin, R.; et al. Publisher Correction: Tubular iron deposition and iron handling proteins in human healthy kidney and chronic kidney disease. Sci. Rep. 2018, 8, 13390. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Girijashanker, K.; Dalton, T.P.; Reed, J.; Li, H.; Soleimani, M.; Nebert, D.W. ZIP8, member of the solute-carrier-39 (SLC39) metal-transporter family: Characterization of transporter properties. Mol. Pharmacol. 2006, 70, 171–180. [Google Scholar] [CrossRef]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [PubMed]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Wolff, N.A.; Lee, W.K.; Thévenod, F. Lipocalin-2 (24p3/neutrophil gelatinase-associated lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J. Biol. Chem. 2012, 287, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.; Donovan, A.; Ned-Sykes, R.; Andrews, N.C. Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 11714–11719. [Google Scholar] [CrossRef]
- Rubio-Navarro, A.; Sanchez-Niño, M.D.; Guerrero-Hue, M.; García-Caballero, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; Román, E.; et al. Podocytes are new cellular targets of haemoglobin-mediated renal damage. J. Pathol. 2018, 244, 296–310. [Google Scholar] [CrossRef]
- Slotki, I.; Cabantchik, Z.I. The Labile Side of Iron Supplementation in CKD. J. Am. Soc. Nephrol. JASN 2015, 26, 2612–2619. [Google Scholar] [CrossRef] [PubMed]
- Moulouel, B.; Houamel, D.; Delaby, C.; Tchernitchko, D.; Vaulont, S.; Letteron, P.; Thibaudeau, O.; Puy, H.; Gouya, L.; Beaumont, C.; et al. Hepcidin regulates intrarenal iron handling at the distal nephron. Kidney Int. 2013, 84, 756–766. [Google Scholar] [CrossRef]
- Nakayama, M.; Kaizu, Y.; Uesugi, N.; Nakashita, S.; Suehiro, T. A case of IgA nephropathy and renal hemosiderosis associated with primary hemochromatosis. Ren. Fail. 2008, 30, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Ozkurt, S.; Acikalin, M.F.; Temiz, G.; Akay, O.M.; Soydan, M. Renal hemosiderosis and rapidly progressive glomerulonephritis associated with primary hemochromatosis. Ren. Fail. 2014, 36, 814–816. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.C.; Jeffrey, G.; Ryan, J. Haemochromatosis. Lancet 2023, 401, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Olynyk, J.K.; Ramm, G.A. Hemochromatosis. N. Engl. J. Med. 2022, 387, 2159–2170. [Google Scholar] [CrossRef]
- Sponsel, H.T.; Alfrey, A.C.; Hammond, W.S.; Durr, J.A.; Ray, C.; Anderson, R.J. Effect of iron on renal tubular epithelial cells. Kidney Int. 1996, 50, 436–444. [Google Scholar] [CrossRef]
- Hassan, R.H.; Kandil, S.M.; Zeid, M.S.; Zaki, M.E.; Fouda, A.E. Kidney injury in infants and children with iron-deficiency anemia before and after iron treatment. Hematology 2017, 22, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Norden, A.G.; Lapsley, M.; Lee, P.J.; Pusey, C.D.; Scheinman, S.J.; Tam, F.W.; Thakker, R.V.; Unwin, R.J.; Wrong, O. Glomerular protein sieving and implications for renal failure in Fanconi syndrome. Kidney Int. 2001, 60, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.A.; Sampson, B.; Muller, B.R.; Curtis, J.R. Urinary iron loss in the nephrotic syndrome--an unusual cause of iron deficiency with a note on urinary copper losses. Postgrad. Med. J. 1984, 60, 125–128. [Google Scholar] [CrossRef]
- Ellis, D. Anemia in the course of the nephrotic syndrome secondary to transferrin depletion. J. Pediatr. 1977, 90, 953–955. [Google Scholar] [CrossRef]
- Howard, R.L.; Buddington, B.; Alfrey, A.C. Urinary albumin, transferrin and iron excretion in diabetic patients. Kidney Int. 1991, 40, 923–926. [Google Scholar] [CrossRef]
- Prinsen, B.; Velden, M.; Kaysen, G.A.; Straver, H.; Rijn, H.; Stellaard, F.; Berger, R.; Rabelink, T.J. Transferrin synthesis is increased in nephrotic patients insufficiently to replace urinary losses. J. Am. Soc. Nephrol. JASN 2001, 12, 1017–1025. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Baud, L.; Hagege, J.; Sraer, J.; Rondeau, E.; Perez, J.; Ardaillou, R. Reactive oxygen production by cultured rat glomerular mesangial cells during phagocytosis is associated with stimulation of lipoxygenase activity. J. Exp. Med. 1983, 158, 1836–1852. [Google Scholar] [CrossRef]
- Ohsaki, Y.; O’Connor, P.; Mori, T.; Ryan, R.P.; Dickinson, B.C.; Chang, C.J.; Lu, Y.; Ito, S.; Cowley, A.W., Jr. Increase of sodium delivery stimulates the mitochondrial respiratory chain H2O2 production in rat renal medullary thick ascending limb. Am. J. Physiol. Ren. Physiol. 2012, 302, F95–F102. [Google Scholar] [CrossRef]
- Radeke, H.H.; Cross, A.R.; Hancock, J.T.; Jones, O.T.; Nakamura, M.; Kaever, V.; Resch, K. Functional expression of NADPH oxidase components (alpha- and beta-subunits of cytochrome b558 and 45-kDa flavoprotein) by intrinsic human glomerular mesangial cells. J. Biol. Chem. 1991, 266, 21025–21029. [Google Scholar] [CrossRef]
- Pfaller, W.; Rittinger, M. Quantitative morphology of the rat kidney. Int. J. Biochem. 1980, 12, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z. Hydroxyl radical generations form the physiologically relevant Fenton-like reactions. Free Radic. Biol. Med. 2023, 208, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 7779–7788. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat. Rev. Nephrol. 2021, 17, 575–590. [Google Scholar] [CrossRef]
- Baylis, C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat. Clin. Pract. Nephrol. 2006, 2, 209–220. [Google Scholar] [CrossRef]
- Nishimura, K.; Taguchi, K.; Kishi, S.; Brooks, C.R.; Ochi, A.; Kadoya, H.; Ikeda, Y.; Miyoshi, M.; Tamaki, M.; Abe, H.; et al. Dual disruption of eNOS and ApoE gene accelerates kidney fibrosis and senescence after injury. Biochem. Biophys. Res. Commun. 2021, 556, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Knowles, J.W.; Reddick, R.L.; Jennette, J.C.; Shesely, E.G.; Smithies, O.; Maeda, N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-)Apoe(-/-) mice are ameliorated by enalapril treatment. J. Clin. Investig. 2000, 105, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Larsen, F.J.; Nyström, T.; Hezel, M.; Borniquel, S.; Weitzberg, E.; Lundberg, J.O. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc. Natl. Acad. Sci. USA 2010, 107, 17716–17720. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Haruna, Y.; Arakawa, S.; Horike, H.; Komai, N.; Sasaki, T.; Tsujioka, K.; Makino, H.; Kashihara, N. NAD(P)H oxidase and uncoupled nitric oxide synthase are major sources of glomerular superoxide in rats with experimental diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2005, 288, F1144–F1152. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.L.; Hooker, E.; Larrivée, B. Diabetic Kidney Disease, Endothelial Damage, and Podocyte-Endothelial Crosstalk. Kidney Med. 2021, 3, 105–115. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell. Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef] [PubMed]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Nishio, Y.; Okamura, T.; Yoshida, Y.; Maegawa, H.; Kojima, H.; Masada, M.; Toda, N.; Kikkawa, R.; Kashiwagi, A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ. Res. 2000, 87, 566–573. [Google Scholar] [CrossRef]
- Yamamoto, E.; Hirata, Y.; Tokitsu, T.; Kusaka, H.; Sakamoto, K.; Yamamuro, M.; Kaikita, K.; Watanabe, H.; Hokimoto, S.; Sugiyama, S.; et al. The pivotal role of eNOS uncoupling in vascular endothelial dysfunction in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2015, 190, 335–337. [Google Scholar] [CrossRef]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transpl. 2008, 23, 3806–3813. [Google Scholar] [CrossRef]
- Fujita, H.; Fujishima, H.; Chida, S.; Takahashi, K.; Qi, Z.; Kanetsuna, Y.; Breyer, M.D.; Harris, R.C.; Yamada, Y.; Takahashi, T. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J. Am. Soc. Nephrol. JASN 2009, 20, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.J.; Reichardt-Pascal, S.Y.; Vaughan, D.; Russell, G.I. Differential effect of ischaemia-reperfusion injury on anti-oxidant enzyme activity in the rat kidney. Exp. Nephrol. 1995, 3, 348–354. [Google Scholar]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; He, W.; Liou, Y.C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Tamura, S.; Honsho, M.; Yada, H.; Yagita, Y.; Kosako, H.; Fujiki, Y. Mitotic phosphorylation of Pex14p regulates peroxisomal import machinery. J. Cell Biol. 2020, 219, e202001003. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. CMLS 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Chae, H.Z.; Chung, S.J.; Rhee, S.G. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994, 269, 27670–27678. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H₂O₂, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Sandalova, T.; Lindqvist, Y.; Arnér, E.S. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Holmgren, A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 2000, 275, 18121–18128. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. CCS 2015, 13, 39. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Pham-Huy, L.A.; He, H.; Pham-Huy, C. Free radicals, antioxidants in disease and health. Int. J. Biomed. Sci. IJBS 2008, 4, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, V.; Valko, L.; Stolc, S.; Valko, M.; Ondrejicková, O.; Horáková, L.; Placek, J.; Troncone, A. Free radicals in rabbit spinal cord ischemia: Electron spin resonance spectroscopy and correlation with SOD activity. Cell. Mol. Neurobiol. 1998, 18, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Stolc, S.; Valko, L.; Valko, M.; Lombardi, V. A technique for the fast sampling of biological tissues for electron paramagnetic resonance spectroscopy. Free Radic. Biol. Med. 1996, 20, 89–91. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Kensler, T.W. Oxidative mechanisms in carcinogenesis. Br. Med. Bull. 1993, 49, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Zhou, S.; Zhou, Z.; Liu, Y.; Yang, L.; Liu, J.; Zhang, Y.; Li, H.; Liu, Y.; Hou, F.F.; et al. Wnt9a Promotes Renal Fibrosis by Accelerating Cellular Senescence in Tubular Epithelial Cells. J. Am. Soc. Nephrol. JASN 2018, 29, 1238–1256. [Google Scholar] [CrossRef]
- Kim, S.R.; Puranik, A.S.; Jiang, K.; Chen, X.; Zhu, X.Y.; Taylor, I.; Khodadadi-Jamayran, A.; Lerman, A.; Hickson, L.J.; Childs, B.G.; et al. Progressive Cellular Senescence Mediates Renal Dysfunction in Ischemic Nephropathy. J. Am. Soc. Nephrol. JASN 2021, 32, 1987–2004. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Loo, T.M.; Okada, R.; Kamachi, F.; Watanabe, Y.; Wakita, M.; Watanabe, S.; Kawamoto, S.; Miyata, K.; Barber, G.N.; et al. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat. Commun. 2018, 9, 1249. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, L.; Liu, Y. Cellular Senescence in Kidney Fibrosis: Pathologic Significance and Therapeutic Strategies. Front. Pharmacol. 2020, 11, 601325. [Google Scholar] [CrossRef]
- Kanwar, Y.S.; Sun, L.; Xie, P.; Liu, F.Y.; Chen, S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 2011, 6, 395–423. [Google Scholar] [CrossRef]
- Kumar, D.; Robertson, S.; Burns, K.D. Evidence of apoptosis in human diabetic kidney. Mol. Cell. Biochem. 2004, 259, 67–70. [Google Scholar] [CrossRef]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Ajay, A.K.; Chang, J.H.; Mou, S.; Zhao, H.; Kishi, S.; Li, J.; Brooks, C.R.; Xiao, S.; Woo, H.M.; et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021, 33, 1042–1061.e1047. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochemist. Rev. 2016, 37, 85–98. [Google Scholar]
- Munshi, R.; Hsu, C.; Himmelfarb, J. Advances in understanding ischemic acute kidney injury. BMC Med. 2011, 9, 11. [Google Scholar] [CrossRef]
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181. [Google Scholar] [CrossRef]
- Hall, A.M.; Rhodes, G.J.; Sandoval, R.M.; Corridon, P.R.; Molitoris, B.A. In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury. Kidney Int. 2013, 83, 72–83. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Zweier, J.L.; Flaherty, J.T.; Weisfeldt, M.L. Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc. Natl. Acad. Sci. USA 1987, 84, 1404–1407. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Shanley, P.F.; Rosen, M.D.; Brezis, M.; Silva, P.; Epstein, F.H.; Rosen, S. Topography of focal proximal tubular necrosis after ischemia with reflow in the rat kidney. Am. J. Pathol. 1986, 122, 462–468. [Google Scholar]
- Friedewald, J.J.; Rabb, H. Inflammatory cells in ischemic acute renal failure. Kidney Int. 2004, 66, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Wu, X.; Pan, T.; Xu, S.; Hu, J.; Ding, X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. eBioMedicine 2019, 49, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Pavlakou, P.; Liakopoulos, V.; Eleftheriadis, T.; Mitsis, M.; Dounousi, E. Oxidative Stress and Acute Kidney Injury in Critical Illness: Pathophysiologic Mechanisms-Biomarkers-Interventions, and Future Perspectives. Oxid. Med. Cell. Longev. 2017, 2017, 6193694. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Gomez, H.; Kellum, J.A. Sepsis-induced acute kidney injury revisited: Pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care 2014, 20, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Safirstein, R.L. Cisplatin nephrotoxicity. Semin. Nephrol. 2003, 23, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Nagase, M.; Ayuzawa, N.; Kawarazaki, W.; Ishizawa, K.; Ueda, K.; Yoshida, S.; Fujita, T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: Role of small GTPase Rac1. Hypertension 2012, 59, 500–506. [Google Scholar] [CrossRef]
- Papaharalambus, C.; Sajjad, W.; Syed, A.; Zhang, C.; Bergo, M.O.; Alexander, R.W.; Ahmad, M. Tumor necrosis factor alpha stimulation of Rac1 activity. Role of isoprenylcysteine carboxylmethyltransferase. J. Biol. Chem. 2005, 280, 18790–18796. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Li, Y.; Li, Y.; Shan, L.; Zhu, H.; Feng, Q.; Arnold, J.M.; Peng, T. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes 2009, 58, 2386–2395. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Fujita, T. Role of Rac1-mineralocorticoid-receptor signalling in renal and cardiac disease. Nat. Rev. Nephrol. 2013, 9, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Reiser, J. Proteinuria: An enzymatic disease of the podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.H.; Hoffmann, S.; Endlich, N.; Velic, A.; Schwab, A.; Weide, T.; Schlatter, E.; Pavenstädt, H. Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J. Mol. Med. 2008, 86, 1379–1394. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum--new mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef]
- Inagi, R.; Nangaku, M.; Onogi, H.; Ueyama, H.; Kitao, Y.; Nakazato, K.; Ogawa, S.; Kurokawa, K.; Couser, W.G.; Miyata, T. Involvement of endoplasmic reticulum (ER) stress in podocyte injury induced by excessive protein accumulation. Kidney Int. 2005, 68, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Alpers, C.E.; Pichler, R.; Johnson, R.J. Phenotypic features of cortical interstitial cells potentially important in fibrosis. Kidney Int. Suppl. 1996, 54, S28–S31. [Google Scholar]
- Kliem, V.; Johnson, R.J.; Alpers, C.E.; Yoshimura, A.; Couser, W.G.; Koch, K.M.; Floege, J. Mechanisms involved in the pathogenesis of tubulointerstitial fibrosis in 5/6-nephrectomized rats. Kidney Int. 1996, 49, 666–678. [Google Scholar] [CrossRef]
- Chai, Q.; Krag, S.; Chai, S.; Ledet, T.; Wogensen, L. Localisation and phenotypical characterisation of collagen-producing cells in TGF-beta 1-induced renal interstitial fibrosis. Histochem. Cell Biol. 2003, 119, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Hastings, R.R.; De la Garza, M.A. Sequential expression of cellular fibronectin by platelets, macrophages, and mesangial cells in proliferative glomerulonephritis. Am. J. Pathol. 1994, 145, 585–597. [Google Scholar] [PubMed]
- Tang, W.W.; Van, G.Y.; Qi, M. Myofibroblast and alpha 1 (III) collagen expression in experimental tubulointerstitial nephritis. Kidney Int. 1997, 51, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Block, K.; Hernandez, J.; Bhandari, B.; Wagner, B.; Barnes, J.L.; Abboud, H.E. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005, 280, 39616–39626. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. JASN 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Cucoranu, I.; Clempus, R.; Dikalova, A.; Phelan, P.J.; Ariyan, S.; Dikalov, S.; Sorescu, D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ. Res. 2005, 97, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Eid, A.; Griendling, K.K.; Lee, D.Y.; Wittrant, Y.; Gorin, Y. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: Role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 2008, 283, 24061–24076. [Google Scholar] [CrossRef] [PubMed]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004, 24, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Pollock, J.S. Coupled and uncoupled NOS: Separate but equal? Uncoupled NOS in endothelial cells is a critical pathway for intracellular signaling. Circ. Res. 2006, 98, 717–719. [Google Scholar] [CrossRef]
- Aldámiz-Echevarría, L.; Andrade, F. Asymmetric dimethylarginine, endothelial dysfunction and renal disease. Int. J. Mol. Sci. 2012, 13, 11288–11311. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Mao, X.; Guo, H.; Wang, L.; Li, Z.; Zhang, Y.; Wang, Y.; Wang, H.; Zhang, X.; Peng, W. Indoxyl sulfate potentiates endothelial dysfunction via reciprocal role for reactive oxygen species and RhoA/ROCK signaling in 5/6 nephrectomized rats. Free Radic. Res. 2017, 51, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Hue, M.; Rayego-Mateos, S.; Vázquez-Carballo, C.; Palomino-Antolín, A.; García-Caballero, C.; Opazo-Rios, L.; Morgado-Pascual, J.L.; Herencia, C.; Mas, S.; Ortiz, A.; et al. Protective Role of Nrf2 in Renal Disease. Antioxidants 2020, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Nrf2, cellular redox regulation, and neurologic implications. Neurology 2017, 88, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.W.; Hsu, Y.C.; Chang, C.C.; Hsieh, C.C.; Lin, C.L. Insights into the Molecular Mechanisms of NRF2 in Kidney Injury and Diseases. Int. J. Mol. Sci. 2023, 24, 6053. [Google Scholar] [CrossRef]
- Kong, W.; Fu, J.; Liu, N.; Jiao, C.; Guo, G.; Luan, J.; Wang, H.; Yao, L.; Wang, L.; Yamamoto, M.; et al. Nrf2 deficiency promotes the progression from acute tubular damage to chronic renal fibrosis following unilateral ureteral obstruction. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2018, 33, 771–783. [Google Scholar] [CrossRef]
- Dandona, P.; Thusu, K.; Cook, S.; Snyder, B.; Makowski, J.; Armstrong, D.; Nicotera, T. Oxidative damage to DNA in diabetes mellitus. Lancet 1996, 347, 444–445. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells Devoted Mol. Cell. Mech. 2008, 13, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharmacol. Exp. Ther. 2010, 335, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Xu, Y.; Yuan, Y.; Tian, L.; Wang, Q.; Xie, Y.; Shao, X.; Zhang, M.; Ni, Z.; Mou, S. Renoprotective mechanisms of Astragaloside IV in cisplatin-induced acute kidney injury. Free Radic. Res. 2017, 51, 669–683. [Google Scholar] [CrossRef] [PubMed]
- Atilano-Roque, A.; Aleksunes, L.M.; Joy, M.S. Bardoxolone methyl modulates efflux transporter and detoxifying enzyme expression in cisplatin-induced kidney cell injury. Toxicol. Lett. 2016, 259, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ma, N.; Fan, X.; Yu, Q.; Ci, X. The role of Nrf2 in acute kidney injury: Novel molecular mechanisms and therapeutic approaches. Free Radic. Biol. Med. 2020, 158, 1–12. [Google Scholar] [CrossRef]
- Qiu, Z.; He, J.; Shao, G.; Hu, J.; Li, X.; Zhou, H.; Li, M.; Yang, B. Obacunone Retards Renal Cyst Development in Autosomal Dominant Polycystic Kidney Disease by Activating NRF2. Antioxidants 2021, 11, 38. [Google Scholar] [CrossRef]
- Tang, P.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Panizo, S.; Martínez-Arias, L.; Alonso-Montes, C.; Cannata, P.; Martín-Carro, B.; Fernández-Martín, J.L.; Naves-Díaz, M.; Carrillo-López, N.; Cannata-Andía, J.B. Fibrosis in Chronic Kidney Disease: Pathogenesis and Consequences. Int. J. Mol. Sci. 2021, 22, 408. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; Sanchez-Niño, M.D.; Ramos, A.M.; Moreno, J.A.; Santamaria, B.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. NF-kappaB in renal inflammation. J. Am. Soc. Nephrol. JASN 2010, 21, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Liu, T.; Qiao, Y.; Liu, D.; Yang, L.; Mao, H.; Ma, F.; Wang, Y.; Peng, L.; Zhan, Y. Oxidative stress and inflammation in diabetic nephropathy: Role of polyphenols. Front. Immunol. 2023, 14, 1185317. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Tang, B.; Zhang, C. Signaling pathways of chronic kidney diseases, implications for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, Y.; Wang, D.; Zhang, L. Effects of Qingshen Granules on the Oxidative Stress-NF/kB Signal Pathway in Unilateral Ureteral Obstruction Rats. Evid.-Based Complement. Altern. Med. Ecam 2018, 2018, 4761925. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Kim, Y.; Park, C.W. Adenosine monophosphate-activated protein kinase in diabetic nephropathy. Kidney Res. Clin. Pract. 2016, 35, 69–77. [Google Scholar] [CrossRef]
- Yao, L.; Liang, X.; Qiao, Y.; Chen, B.; Wang, P.; Liu, Z. Mitochondrial dysfunction in diabetic tubulopathy. Metab. Clin. Exp. 2022, 131, 155195. [Google Scholar] [CrossRef]
- Yuan, T.; Yang, T.; Chen, H.; Fu, D.; Hu, Y.; Wang, J.; Yuan, Q.; Yu, H.; Xu, W.; Xie, X. New insights into oxidative stress and inflammation during diabetes mellitus-accelerated atherosclerosis. Redox Biol. 2019, 20, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Aboolian, A.; Urner, S.; Roden, M.; Jha, J.C.; Jandeleit-Dahm, K. Diabetic Kidney Disease: From Pathogenesis to Novel Treatment Possibilities. Handb. Exp. Pharmacol. 2022, 274, 269–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Z.; Wang, X.; Hu, Y.; Kong, J.; Lai, J.; Li, T.; Hu, B.; Zhang, Y.; Zheng, X.; et al. Sappanone a prevents diabetic kidney disease by inhibiting kidney inflammation and fibrosis via the NF-κB signaling pathway. Front. Pharmacol. 2022, 13, 953004. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Singla, S.K.; Puri, V.; Puri, S. The restrained expression of NF-kB in renal tissue ameliorates folic acid induced acute kidney injury in mice. PLoS ONE 2015, 10, e115947. [Google Scholar] [CrossRef] [PubMed]
- Johnson, F.L.; Patel, N.S.A.; Purvis, G.S.D.; Chiazza, F.; Chen, J.; Sordi, R.; Hache, G.; Merezhko, V.V.; Collino, M.; Yaqoob, M.M.; et al. Inhibition of IκB Kinase at 24 Hours After Acute Kidney Injury Improves Recovery of Renal Function and Attenuates Fibrosis. J. Am. Heart Assoc. 2017, 6, e005092. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-κB signaling suppression. Cell. Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef]
- Lv, L.L.; Tang, P.M.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602. [Google Scholar] [CrossRef]
- Yang, L.; Brooks, C.R.; Xiao, S.; Sabbisetti, V.; Yeung, M.Y.; Hsiao, L.L.; Ichimura, T.; Kuchroo, V.; Bonventre, J.V. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J. Clin. Investig. 2015, 125, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Markó, L.; Vigolo, E.; Hinze, C.; Park, J.K.; Roël, G.; Balogh, A.; Choi, M.; Wübken, A.; Cording, J.; Blasig, I.E.; et al. Tubular Epithelial NF-κB Activity Regulates Ischemic AKI. J. Am. Soc. Nephrol. JASN 2016, 27, 2658–2669. [Google Scholar] [CrossRef]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Kume, S.; Kanasaki, K.; Takeda-Watanabe, A.; Koya, D. Sirtuins as possible drug targets in type 2 diabetes. Curr. Drug Targets 2013, 14, 622–636. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, D.Y.; Sha, W.G.; Shen, L.; Lu, G.Y.; Yin, X.; Wang, M.J. High glucose induces renal tubular epithelial injury via Sirt1/NF-kappaB/microR-29/Keap1 signal pathway. J. Transl. Med. 2015, 13, 352. [Google Scholar] [CrossRef]
- Lempiäinen, J.; Finckenberg, P.; Mervaala, E.E.; Sankari, S.; Levijoki, J.; Mervaala, E.M. Caloric restriction ameliorates kidney ischaemia/reperfusion injury through PGC-1α-eNOS pathway and enhanced autophagy. Acta Physiol. 2013, 208, 410–421. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sánchez-Ramos, C.; Prieto-Arroyo, I.; Urbánek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]
- Kao, C.L.; Chen, L.K.; Chang, Y.L.; Yung, M.C.; Hsu, C.C.; Chen, Y.C.; Lo, W.L.; Chen, S.J.; Ku, H.H.; Hwang, S.J. Resveratrol protects human endothelium from H2O2-induced oxidative stress and senescence via SirT1 activation. J. Atheroscler. Thromb. 2010, 17, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Finkel, T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 2002, 295, 2450–2452. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Zhang, M.Z.; You, L.; Davis, L.S.; Fan, H.; Yang, H.C.; Fogo, A.B.; Zent, R.; Harris, R.C.; et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J. Clin. Investig. 2010, 120, 1056–1068. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Ricardo, S.D.; Bertram, J.F.; Nikolic-Paterson, D.J. Resveratrol inhibits renal fibrosis in the obstructed kidney: Potential role in deacetylation of Smad3. Am. J. Pathol. 2010, 177, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Connelly, K.A.; Thai, K.; Wu, X.; Kapus, A.; Kepecs, D.; Gilbert, R.E. Sirtuin 1 Activation Reduces Transforming Growth Factor-β1-Induced Fibrogenesis and Affords Organ Protection in a Model of Progressive, Experimental Kidney and Associated Cardiac Disease. Am. J. Pathol. 2017, 187, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.Y.; Dai, Y.; Liu, R.; He, H.; Kretzler, M.; Jim, B.; Cohen, C.D.; He, J.C. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS ONE 2011, 6, e23566. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhong, Y.; Li, X.; Chen, H.; Jim, B.; Zhou, M.M.; Chuang, P.Y.; He, J.C. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 2014, 63, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.Y.; Cai, W.; Li, X.; Fang, L.; Xu, J.; Yacoub, R.; He, J.C.; Lee, K. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am. J. Physiol. Ren. Physiol. 2017, 313, F621–F628. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; He, J.; Sun, X.; Li, L.; Zhang, X.; Gan, H. Activation of sirtuin1 protects against ischemia/reperfusion-induced acute kidney injury. Biomed. Pharmacother. 2020, 125, 110021. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R.; Raineri, I.; Epstein, L.B.; White, C.W. Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 1995, 270, 13399–13405. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Klausner, R.D. Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem. Sci. 1996, 21, 174–177. [Google Scholar] [CrossRef]
- Cairo, G.; Recalcati, S.; Pietrangelo, A.; Minotti, G. The iron regulatory proteins: Targets and modulators of free radical reactions and oxidative damage. Free Radic. Biol. Med. 2002, 32, 1237–1243. [Google Scholar] [CrossRef]
- Cairo, G.; Castrusini, E.; Minotti, G.; Bernelli-Zazzera, A. Superoxide and hydrogen peroxide-dependent inhibition of iron regulatory protein activity: A protective stratagem against oxidative injury. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 1326–1335. [Google Scholar] [CrossRef]
- Pantopoulos, K.; Mueller, S.; Atzberger, A.; Ansorge, W.; Stremmel, W.; Hentze, M.W. Differences in the regulation of iron regulatory protein-1 (IRP-1) by extra- and intracellular oxidative stress. J. Biol. Chem. 1997, 272, 9802–9808. [Google Scholar] [CrossRef] [PubMed]
- Andriopoulos, B.; Hegedüsch, S.; Mangin, J.; Riedel, H.D.; Hebling, U.; Wang, J.; Pantopoulos, K.; Mueller, S. Sustained hydrogen peroxide induces iron uptake by transferrin receptor-1 independent of the iron regulatory protein/iron-responsive element network. J. Biol. Chem. 2007, 282, 20301–20308. [Google Scholar] [CrossRef] [PubMed]
- Mehlhase, J.; Sandig, G.; Pantopoulos, K.; Grune, T. Oxidation-induced ferritin turnover in microglial cells: Role of proteasome. Free Radic. Biol. Med. 2005, 38, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Niecknig, H.; Tug, S.; Reyes, B.D.; Kirsch, M.; Fandrey, J.; Berchner-Pfannschmidt, U. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic. Res. 2012, 46, 705–717. [Google Scholar] [CrossRef]
- Kuragano, T.; Shimonaka, Y.; Kida, A.; Furuta, M.; Nanami, M.; Otaki, Y.; Hasuike, Y.; Nonoguchi, H.; Nakanishi, T. Determinants of hepcidin in patients on maintenance hemodialysis: Role of inflammation. Am. J. Nephrol. 2010, 31, 534–540. [Google Scholar] [CrossRef]
- Ashby, D.R.; Gale, D.P.; Busbridge, M.; Murphy, K.G.; Duncan, N.D.; Cairns, T.D.; Taube, D.H.; Bloom, S.R.; Tam, F.W.; Chapman, R.S.; et al. Plasma hepcidin levels are elevated but responsive to erythropoietin therapy in renal disease. Kidney Int. 2009, 75, 976–981. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J. Ferroptosis, a Rising Force against Renal Fibrosis. Oxid. Med. Cell. Longev. 2022, 2022, 7686956. [Google Scholar] [CrossRef]
- Gong, S.; Zhang, A.; Yao, M.; Xin, W.; Guan, X.; Qin, S.; Liu, Y.; Xiong, J.; Yang, K.; Xiong, L.; et al. REST contributes to AKI-to-CKD transition through inducing ferroptosis in renal tubular epithelial cells. JCI Insight 2023, 8, e166001. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, H.; Sheng, J.; Zhang, W.; Lei, J.; Gan, W.; Cai, F.; Yang, Y. Vitexin attenuates chronic kidney disease by inhibiting renal tubular epithelial cell ferroptosis via NRF2 activation. Mol. Med. 2023, 29, 147. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, J.; Zhu, B.; Fan, J.; Hu, Q.; Wang, L. Tectorigenin protects against unilateral ureteral obstruction by inhibiting Smad3-mediated ferroptosis and fibrosis. Phytother. Res. PTR 2022, 36, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Ni, Y.; Gong, Y.; Kang, X.; Guo, H.; Liu, X.; Li, J.; Wang, L. Formononetin ameliorates ferroptosis-associated fibrosis in renal tubular epithelial cells and in mice with chronic kidney disease by suppressing the Smad3/ATF3/SLC7A11 signaling. Life Sci. 2023, 315, 121331. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kang, S.W.; Joo, J.; Han, S.H.; Shin, H.; Nam, B.Y.; Park, J.; Yoo, T.H.; Kim, G.; Lee, P.; et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, S.; Sun, Z.; Dong, H.; Yu, H.; Huang, M.; Gao, X. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1α/HO-1 Pathway in db/db Mice. Front. Endocrinol. 2021, 12, 626390. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bi, R.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Cui, X.; Yang, H.; Gao, X.; Zhang, D. Ferroptosis involves in renal tubular cell death in diabetic nephropathy. Eur. J. Pharmacol. 2020, 888, 173574. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Fei, L.; Wang, X.; Lei, Y.; Yu, L.; Xu, W.; Chen, J.; Zhu, E.; Zhong, M.; Huang, M.; et al. ZIP14 is involved in iron deposition and triggers ferroptosis in diabetic nephropathy. Met. Integr. Biometal Sci. 2022, 14, mfac034. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Li, Q.; Liao, J.; Chen, W.; Zhang, K.; Li, H.; Ma, F.; Zhang, H.; Han, Q.; Guo, J.; Li, Y.; et al. NAC alleviative ferroptosis in diabetic nephropathy via maintaining mitochondrial redox homeostasis through activating SIRT3-SOD2/Gpx4 pathway. Free Radic. Biol. Med. 2022, 187, 158–170. [Google Scholar] [CrossRef]
- Agborbesong, E.; Li, L.X.; Li, L.; Li, X. Molecular Mechanisms of Epigenetic Regulation, Inflammation, and Cell Death in ADPKD. Front. Mol. Biosci. 2022, 9, 922428. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid Peroxidation Drives Renal Cyst Growth In Vitro through Activation of TMEM16A. J. Am. Soc. Nephrol. JASN 2019, 30, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.X.; Ding, H.; Torres, V.E.; Yu, C.; Li, X. Ferroptosis Promotes Cyst Growth in Autosomal Dominant Polycystic Kidney Disease Mouse Models. J. Am. Soc. Nephrol. JASN 2021, 32, 2759–2776. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, S.; Hidaka, S.; Yanai, M.; Ishioka, K.; Matsui, K.; Mochida, Y.; Moriya, H.; Ohtake, T.; Kobayashi, S. Renal hemosiderosis presenting with acute kidney Injury and macroscopic hematuria in Immunoglobulin A nephropathy: A case report. BMC Nephrol. 2021, 22, 132. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, J.H.; Li, Q.; He, Q.; Lin, W.Q. Lipid peroxidation in IgA nephropathy and the effect of lipo-prostaglandin E1. J. Nephrol. 2005, 18, 243–248. [Google Scholar] [PubMed]
- Wu, J.; Shao, X.; Shen, J.; Lin, Q.; Zhu, X.; Li, S.; Li, J.; Zhou, W.; Qi, C.; Ni, Z. Downregulation of PPARα mediates FABP1 expression, contributing to IgA nephropathy by stimulating ferroptosis in human mesangial cells. Int. J. Biol. Sci. 2022, 18, 5438–5458. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, J.; Xu, H.; Zhou, C.; Han, B.; Zhu, H.; Hu, Z.; Ma, Z.; Ming, Z.; Yao, Y.; et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020, 11, 629. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Mässenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, M.; Bi, R.; Su, Y.; Quan, F.; Lin, Y.; Yue, C.; Cui, X.; Zhao, Q.; Liu, S.; et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022, 51, 102262. [Google Scholar] [CrossRef]
- Qi, Y.; Hu, M.; Qiu, Y.; Zhang, L.; Yan, Y.; Feng, Y.; Feng, C.; Hou, X.; Wang, Z.; Zhang, D.; et al. Mitoglitazone ameliorates renal ischemia/reperfusion injury by inhibiting ferroptosis via targeting mitoNEET. Toxicol. Appl. Pharmacol. 2023, 465, 116440. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, H. CHAC1 exacerbates LPS-induced ferroptosis and apoptosis in HK-2 cells by promoting oxidative stress. Allergol. Immunopathol. 2023, 51, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; An, S.; Wang, T.; Li, J.; Yu, B.; Zeng, Z.; Chen, Z.; Lin, B.; Lin, X.; Gao, Y. Melatonin suppresses ferroptosis via activation of the Nrf2/HO-1 signaling pathway in the mouse model of sepsis-induced acute kidney injury. Int. Immunopharmacol. 2022, 112, 109162. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Yu, X.; Qiao, Y.; Pan, S.; Wang, R.; Zheng, B.; Wang, H.; Ren, K.D.; Liu, H.; Yang, Y. Ferroptosis and Acute Kidney Injury (AKI): Molecular Mechanisms and Therapeutic Potentials. Front. Pharmacol. 2022, 13, 858676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Yu, P.; Wang, T.T.; Du, Y.W.; Chen, Y.; Li, Z.; He, M.L.; Feng, L.; Li, H.R.; Han, X.; et al. Polydatin Attenuates Cisplatin-Induced Acute Kidney Injury by Inhibiting Ferroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 9947191. [Google Scholar] [CrossRef]

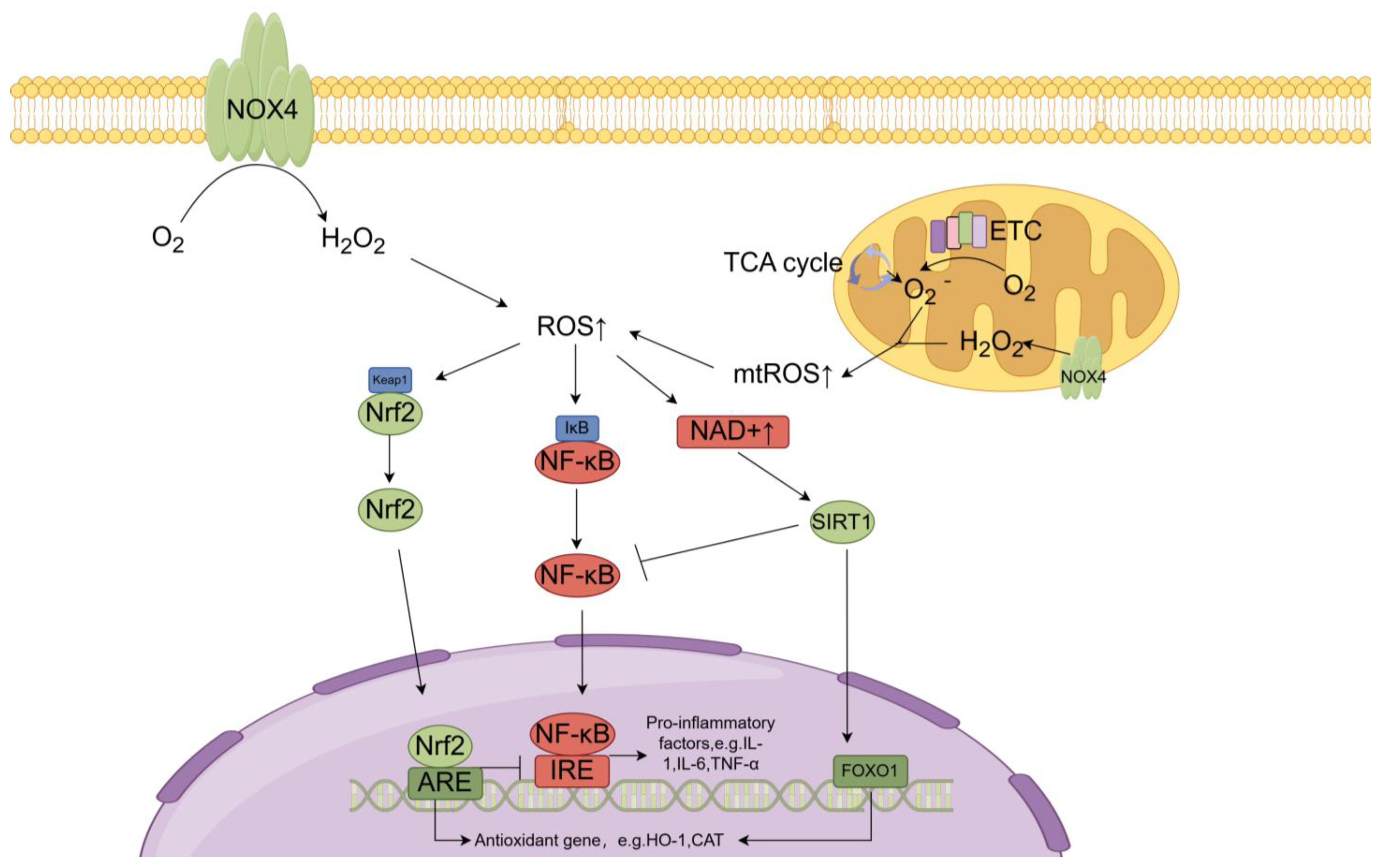
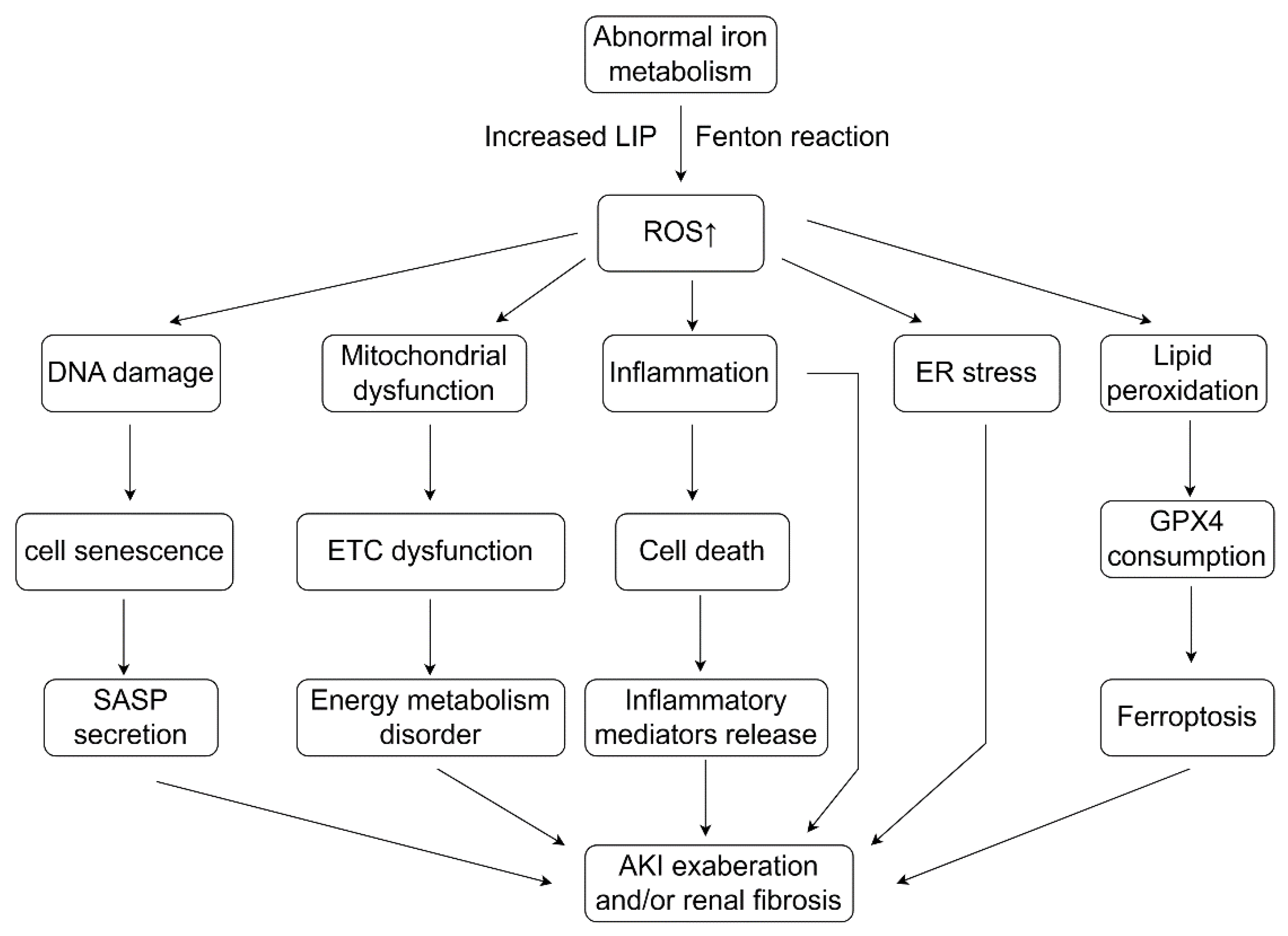

{kind=link}
{kind=link}
{kind=link}
| Reactive Oxygen Species | Properties | |
|---|---|---|
| Non- radical | Hydrogen peroxide (H2O2) | Low oxidative activity, participates in many physiological processes as a signal molecule |
| Organic hydroperoxides (ROOH) | Lipid peroxides derived from polyunsaturated acids (PUFAs) in ferroptosis | |
| Singlet oxygen | High oxidative activity involved in many biological processes | |
| Electronically excited carbonyl (R–C=O) | High oxidative activity | |
| Peroxynitrite (ONOO−) | Formed by the reaction of superoxide with nitric oxide | |
| Ozone (O3) | In atmosphere, but toxic to humans | |
| Free radical | Superoxide (O2−) | Relatively low oxidative activity, participates in the synthesis of H2O2 |
| Hydroxyl radicals (HO∙) | High oxidative activity and unstable, react with various cellular proteins, DNA, lipids | |
| Peroxyl radical (ROO∙) | High oxidative activity, involved in the spread of lipid peroxidation | |
| Nitric oxide (NO∙) | Relatively low oxidative activity, participates in the synthesis of H2O2 |
| Enzymatic System | Non-Enzymatic System |
|---|---|
| Superoxide dismutase (SOD) | Glutathione (GSH) |
| Catalase (CAT) | Antioxidant vitamins: vitamin A/C/E |
| Glutathione peroxidase (GPX) | Antioxidant minerals: copper, zinc, manganese |
| Thioredoxin (Trx) | Hormones: melatonin, flavenoids, |
| coenzyme Q |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, T.; Yao, L.; Li, X. Advance in Iron Metabolism, Oxidative Stress and Cellular Dysfunction in Experimental and Human Kidney Diseases. Antioxidants 2024, 13, 659. https://doi.org/10.3390/antiox13060659
Xie T, Yao L, Li X. Advance in Iron Metabolism, Oxidative Stress and Cellular Dysfunction in Experimental and Human Kidney Diseases. Antioxidants. 2024; 13(6):659. https://doi.org/10.3390/antiox13060659
Chicago/Turabian StyleXie, Tiancheng, Li Yao, and Xiaogang Li. 2024. "Advance in Iron Metabolism, Oxidative Stress and Cellular Dysfunction in Experimental and Human Kidney Diseases" Antioxidants 13, no. 6: 659. https://doi.org/10.3390/antiox13060659