
Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers

 ,
,  , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Oxidative Modifications
2.1. Sulfenic, Sulfinic and Sulfonic Acids
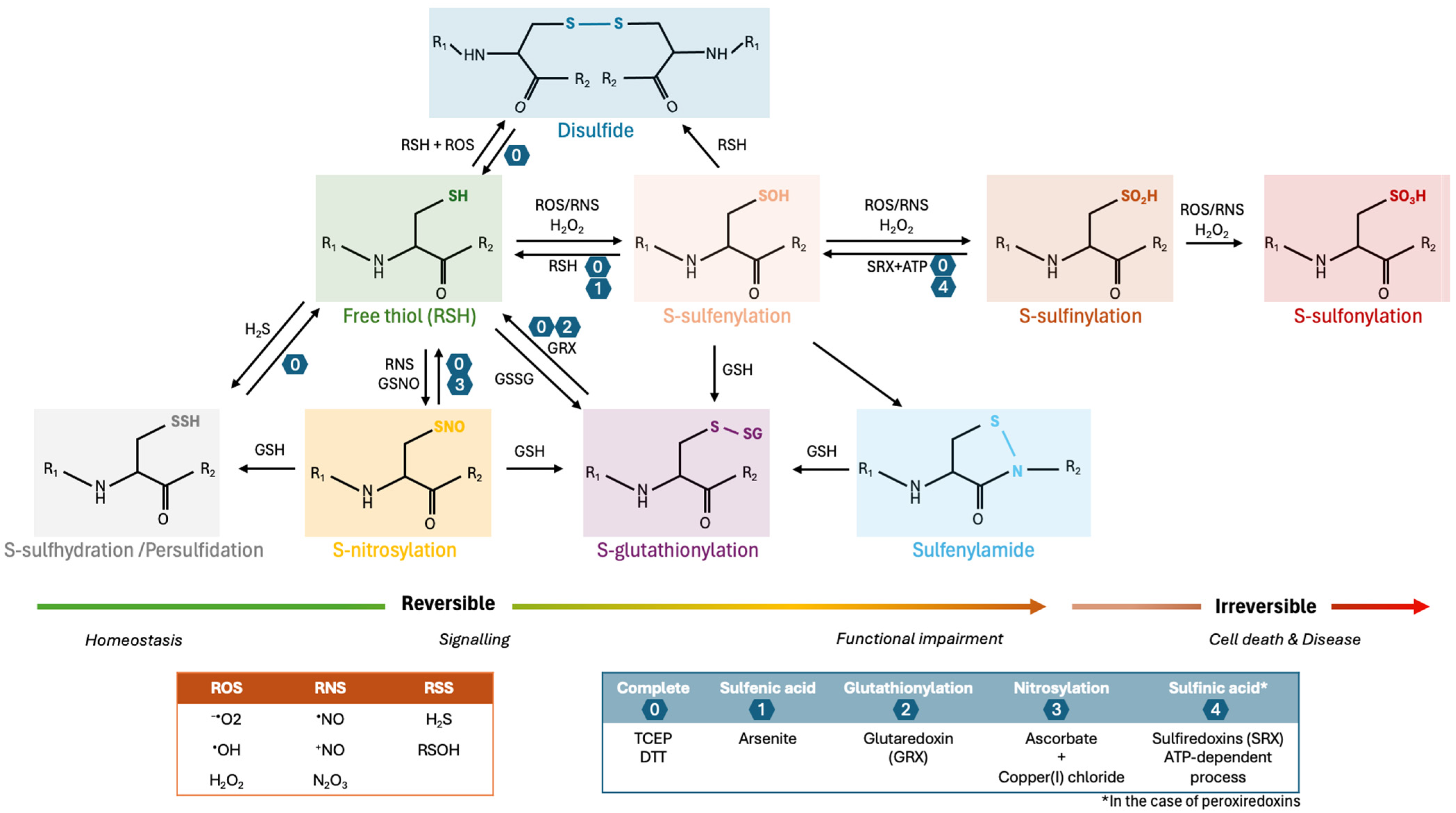
2.2. Disulfide Bonds
2.3. S-Sulfhydration
2.4. S-Glutathionylation
2.5. S-Nitrosylation and Nitration
2.6. Carbonylation
3. Methods Used to Identify and Quantify Redox-PTMs of Proteins
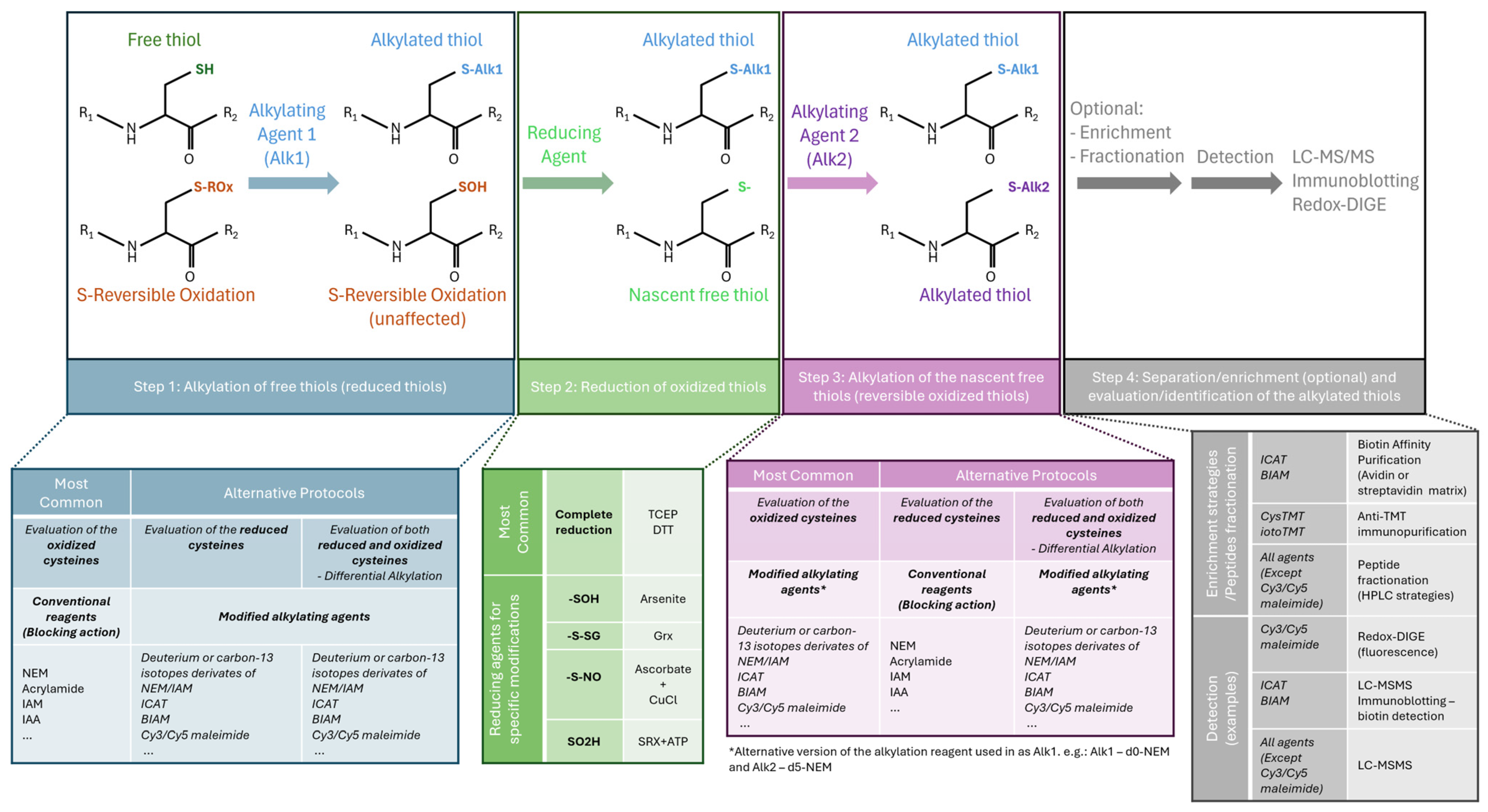
3.1. Detection of Cys Oxidation
3.2. Methodological Considerations and Limitations
4. Structural and Functional Effects of Redox-PTMs: Computational Modelling Contribution to Improve Mechanistic Understanding
4.1. Molecular Dynamics of Protein Conformation Changes
4.2. Molecular Dynamics to Explore the Effects of PTMs on Protein Conformation
4.3. Current Challenges in Molecular Dynamics Applied to Redox-PTMs
5. Redox-PTMs in the Pathophysiology of NDDs
5.1. Redox-PTMs and Their Crosstalk in NDDs
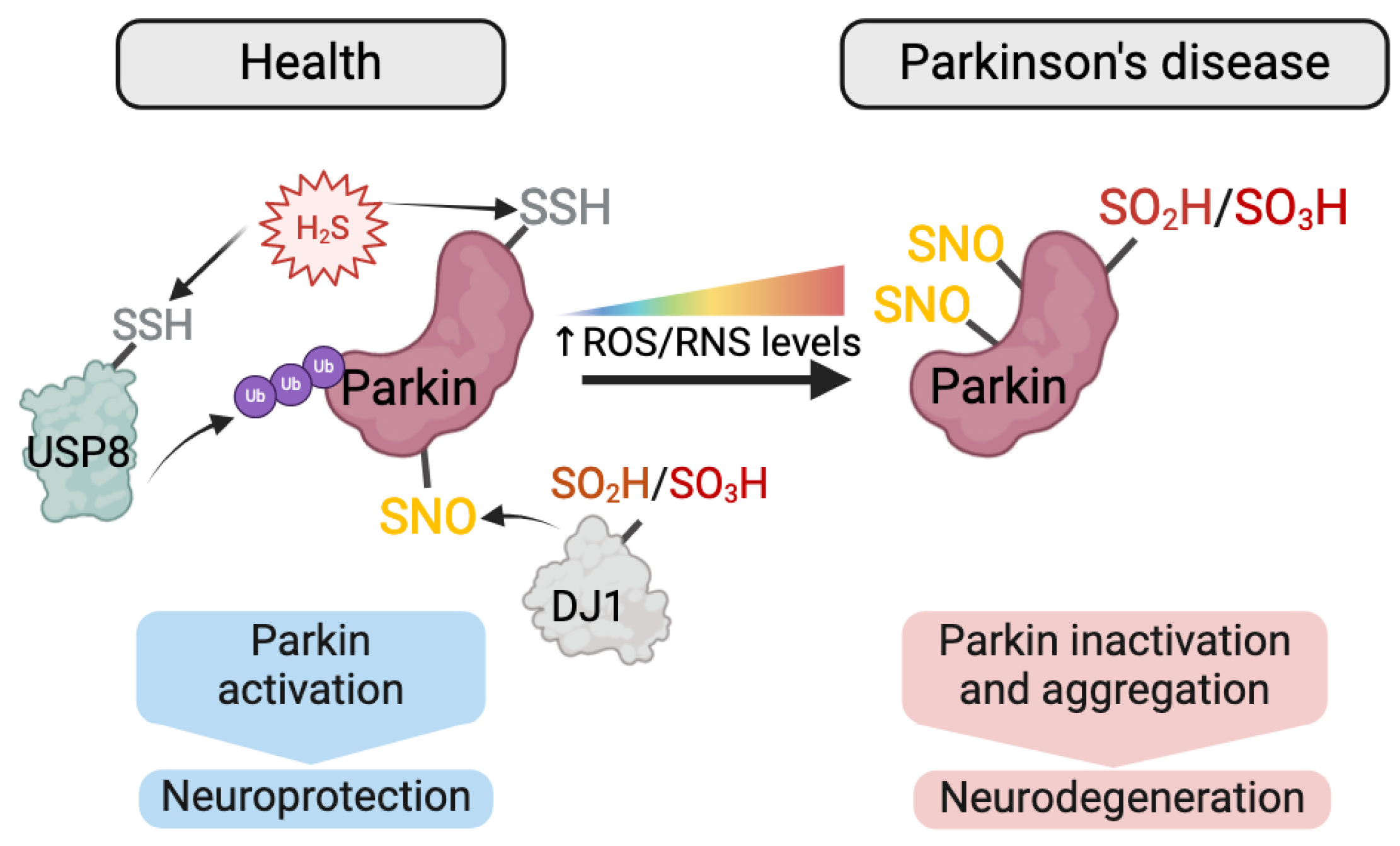
5.2. Redox-PTMs as Biomarkers of NDDs
6. Future Directions
6.1. Redox-PTMs of Disease-Relevant Proteins to Diagnose NDDs
6.2. Redox-PTMs of Disease-Relevant Proteins to Follow Disease Progression
6.3. From Discovery of Redox-PTMs of Proteins to Implementation in the Clinic
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wong, C.M.; Cheema, A.K.; Zhang, L.; Suzuki, Y.J. Protein carbonylation as a novel mechanism in redox signaling. Circ. Res. 2008, 102, 310–318. [Google Scholar] [CrossRef]
- Stair, E.R.; Hicks, L.M. Recent advances in mass spectrometry-based methods to investigate reversible cysteine oxidation. Curr. Opin. Chem. Biol. 2023, 77, 102389. [Google Scholar] [CrossRef]
- Miki, H.; Funato, Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012, 151, 255–261. [Google Scholar] [CrossRef]
- Bin, P.; Huang, R.; Zhou, X. Oxidation Resistance of the Sulfur Amino Acids: Methionine and Cysteine. Biomed Res. Int. 2017, 2017, 9584932. [Google Scholar] [CrossRef]
- Jones, D.P.; Go, Y.M. Mapping the cysteine proteome: Analysis of redox-sensing thiols. Curr. Opin. Chem. Biol. 2011, 15, 103–112. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 2018, 14, 244–252. [Google Scholar] [CrossRef]
- Finelli, M.J. Redox Post-translational Modifications of Protein Thiols in Brain Aging and Neurodegenerative Conditions-Focus on S-Nitrosation. Front. Aging Neurosci. 2020, 12, 254. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef]
- Zuo, J.; Zhang, Z.; Luo, M.; Zhou, L.; Nice, E.C.; Zhang, W.; Wang, C.; Huang, C. Redox signaling at the crossroads of human health and disease. MedComm 2022, 3, e127. [Google Scholar] [CrossRef]
- Ryan, B.J.; Nissim, A.; Winyard, P.G. Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2014, 2, 715–724. [Google Scholar] [CrossRef]
- Nakamura, T.; Oh, C.K.; Zhang, X.; Lipton, S.A. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577. [Google Scholar] [CrossRef]
- Ju, Y.J.; Lee, H.W.; Choi, J.W.; Choi, M.S. The Role of Protein S-Nitrosylation in Protein Misfolding-Associated Diseases. Life 2021, 11, 705. [Google Scholar] [CrossRef]
- Kehm, R.; Baldensperger, T.; Raupbach, J.; Hohn, A. Protein oxidation—Formation mechanisms, detection and relevance as biomarkers in human diseases. Redox Biol. 2021, 42, 101901. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef]
- Fedorova, M.; Bollineni, R.C.; Hoffmann, R. Protein carbonylation as a major hallmark of oxidative damage: Update of analytical strategies. Mass Spectrom. Rev. 2014, 33, 79–97. [Google Scholar] [CrossRef]
- Miseta, A.; Csutora, P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef]
- Zhong, Q.; Xiao, X.; Qiu, Y.; Xu, Z.; Chen, C.; Chong, B.; Zhao, X.; Hai, S.; Li, S.; An, Z.; et al. Protein posttranslational modifications in health and diseases: Functions, regulatory mechanisms, and therapeutic implications. MedComm 2023, 4, e261. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004. [Google Scholar] [CrossRef]
- Wojdyla, K.; Rogowska-Wrzesinska, A. Differential alkylation-based redox proteomics—Lessons learnt. Redox Biol. 2015, 6, 240–252. [Google Scholar] [CrossRef]
- Lee, Y.M.; He, W.; Liou, Y.C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef]
- Roos, G.; Messens, J. Protein sulfenic acid formation: From cellular damage to redox regulation. Free Radic. Biol. Med. 2011, 51, 314–326. [Google Scholar] [CrossRef]
- Lo Conte, M.; Carroll, K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef]
- Groitl, B.; Jakob, U. Thiol-based redox switches. Biochim. Biophys. Acta 2014, 1844, 1335–1343. [Google Scholar] [CrossRef]
- Vignane, T.; Hugo, M.; Hoffmann, C.; Katsouda, A.; Petric, J.; Wang, H.; Miler, M.; Comas, F.; Petrovic, D.; Chen, S.; et al. Protein thiol alterations drive aberrant phase separation in aging. bioRxiv 2023. [Google Scholar] [CrossRef]
- Woo, H.A.; Kang, S.W.; Kim, H.K.; Yang, K.S.; Chae, H.Z.; Rhee, S.G. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J. Biol. Chem. 2003, 278, 47361–47364. [Google Scholar] [CrossRef]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef]
- Chang, T.S.; Jeong, W.; Woo, H.A.; Lee, S.M.; Park, S.; Rhee, S.G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004, 279, 50994–51001. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef]
- Mishra, M.; Jiang, H.; Wu, L.; Chawsheen, H.A.; Wei, Q. The sulfiredoxin-peroxiredoxin (Srx-Prx) axis in cell signal transduction and cancer development. Cancer Lett. 2015, 366, 150–159. [Google Scholar] [CrossRef]
- Rudyk, O.; Eaton, P. Biochemical methods for monitoring protein thiol redox states in biological systems. Redox Biol. 2014, 2, 803–813. [Google Scholar] [CrossRef]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2018, 47, 231–268. [Google Scholar] [CrossRef]
- Pham, T.K.; Buczek, W.A.; Mead, R.J.; Shaw, P.J.; Collins, M.O. Proteomic Approaches to Study Cysteine Oxidation: Applications in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 678837. [Google Scholar] [CrossRef]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef]
- Bechtel, T.J.; Weerapana, E. From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics 2017, 17, 1600391. [Google Scholar] [CrossRef]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar] [CrossRef]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.H.; Inaba, K.; Okumura, M. The Protein Disulfide Isomerase Family: From proteostasis to pathogenesis. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129338. [Google Scholar] [CrossRef]
- Mossuto, M.F. Disulfide bonding in neurodegenerative misfolding diseases. Int. J. Cell Biol. 2013, 2013, 318319. [Google Scholar] [CrossRef]
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6. [Google Scholar] [CrossRef]
- Mitra, A.; Sarkar, N. The role of intra and inter-molecular disulfide bonds in modulating amyloidogenesis: A review. Arch. Biochem. Biophys. 2022, 716, 109113. [Google Scholar] [CrossRef]
- Cumming, R.C.; Schubert, D. Amyloid-beta induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. Faseb j 2005, 19, 2060–2062. [Google Scholar] [CrossRef]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.T.; Hata, F.; Inui, T.; Takeuchi, T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J. Biol. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef]
- Tripathi, S.J.; Chakraborty, S.; Miller, E.; Pieper, A.A.; Paul, B.D. Hydrogen sulfide signalling in neurodegenerative diseases. Br. J. Pharmacol. 2023. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Goodwin, L.R.; Benishin, C.G.; Reiffenstein, R.J.; Francom, D.M.; Taylor, J.D.; Dieken, F.P. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem. Pharmacol. 1989, 38, 973–981. [Google Scholar] [CrossRef]
- Warenycia, M.W.; Smith, K.A.; Blashko, C.S.; Kombian, S.B.; Reiffenstein, R.J. Monoamine oxidase inhibition as a sequel of hydrogen sulfide intoxication: Increases in brain catecholamine and 5-hydroxytryptamine levels. Arch. Toxicol. 1989, 63, 131–136. [Google Scholar] [CrossRef]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef]
- Petrovic, D.; Kouroussis, E.; Vignane, T.; Filipovic, M.R. The Role of Protein Persulfidation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2021, 13, 674135. [Google Scholar] [CrossRef]
- Aschner, M.; Skalny, A.V.; Ke, T.; da Rocha, J.B.; Paoliello, M.M.; Santamaria, A.; Bornhorst, J.; Rongzhu, L.; Svistunov, A.A.; Djordevic, A.B.; et al. Hydrogen Sulfide (H2S) Signaling as a Protective Mechanism against Endogenous and Exogenous Neurotoxicants. Curr. Neuropharmacol. 2022, 20, 1908–1924. [Google Scholar] [CrossRef]
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma hydrogen sulfide: A biomarker of Alzheimer’s disease and related dementias. Alzheimer Dement. 2021, 17, 1391–1402. [Google Scholar] [CrossRef]
- Giuliani, D.; Ottani, A.; Zaffe, D.; Galantucci, M.; Strinati, F.; Lodi, R.; Guarini, S. Hydrogen sulfide slows down progression of experimental Alzheimer’s disease by targeting multiple pathophysiological mechanisms. Neurobiol. Learn. Mem. 2013, 104, 82–91. [Google Scholar] [CrossRef]
- Giovinazzo, D.; Bursac, B.; Sbodio, J.I.; Nalluru, S.; Vignane, T.; Snowman, A.M.; Albacarys, L.M.; Sedlak, T.W.; Torregrossa, R.; Whiteman, M.; et al. Hydrogen sulfide is neuroprotective in Alzheimer’s disease by sulfhydrating GSK3beta and inhibiting Tau hyperphosphorylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017225118. [Google Scholar] [CrossRef]
- Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. [Google Scholar] [CrossRef]
- Kalinina, E.; Novichkova, M. Glutathione in Protein Redox Modulation through S-Glutathionylation and S-Nitrosylation. Molecules 2021, 26, 435. [Google Scholar] [CrossRef]
- Musaogullari, A.; Chai, Y.C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases—Focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef]
- Tew, K.D.; Townsend, D.M. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metab. Rev. 2011, 43, 179–193. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Morris, T.E.; Fraser, J.P.; He, L.; Tew, K.D. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006, 66, 6800–6806. [Google Scholar] [CrossRef]
- Shelton, M.D.; Chock, P.B.; Mieyal, J.J. Glutaredoxin: Role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005, 7, 348–366. [Google Scholar] [CrossRef]
- Gravina, S.A.; Mieyal, J.J. Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry 1993, 32, 3368–3376. [Google Scholar] [CrossRef]
- Nakamura, S.; Matsushima, M.; Song, H.; Kikuchi, M. A role of PDI in the reductive cleavage of mixed disulfides. J. Biochem. 1996, 120, 525–530. [Google Scholar] [CrossRef]
- Garrido, M.; Tereshchenko, Y.; Zhevtsova, Z.; Taschenberger, G.; Bahr, M.; Kugler, S. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 2011, 121, 475–485. [Google Scholar] [CrossRef]
- Haseena, P.A.; Diwakar, L.; Ravindranath, V. Protein Glutathionylation and Glutaredoxin: Role in Neurodegenerative Diseases. Antioxidants 2022, 11, 2334. [Google Scholar] [CrossRef]
- Townsend, D.M. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 2007, 7, 313–324. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef]
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; van Horssen, J.; Gama, M.J. S-Glutathionylation of Keap1: A new role for glutathione S-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466. [Google Scholar] [CrossRef]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.; Wolf, C.R.; Gama, M.J. Glutathione S-transferase pi mediates MPTP-induced c-Jun N-terminal kinase activation in the nigrostriatal pathway. Mol. Neurobiol. 2012, 45, 466–477. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef]
- Radi, R. Protein tyrosine nitration: Biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef]
- Lee, J.R.; Kim, J.K.; Lee, S.J.; Kim, K.P. Role of protein tyrosine nitration in neurodegenerative diseases and atherosclerosis. Arch. Pharmacal Res. 2009, 32, 1109–1118. [Google Scholar] [CrossRef]
- Sarchielli, P.; Galli, F.; Floridi, A.; Floridi, A.; Gallai, V. Relevance of protein nitration in brain injury: A key pathophysiological mechanism in neurodegenerative, autoimmune, or inflammatory CNS diseases and stroke. Amino Acids 2003, 25, 427–436. [Google Scholar] [CrossRef]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. Npj Park. Dis. 2022, 8, 104. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic. Res. 2011, 45, 53–58. [Google Scholar] [CrossRef]
- Akagawa, M. Protein carbonylation: Molecular mechanisms, biological implications, and analytical approaches. Free Radic. Res. 2021, 55, 307–320. [Google Scholar] [CrossRef]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparlo, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Henning, C.; Glomb, M.A. Pathways of the Maillard reaction under physiological conditions. Glycoconj. J. 2016, 33, 499–512. [Google Scholar] [CrossRef]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000, 28, 1685–1696. [Google Scholar] [CrossRef]
- Aldini, G.; Dalle-Donne, I.; Facino, R.M.; Milzani, A.; Carini, M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med. Res. Rev. 2007, 27, 817–868. [Google Scholar] [CrossRef]
- Vallet, S.D.; Ricard-Blum, S. Lysyl oxidases: From enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019, 63, 349–364. [Google Scholar] [CrossRef]
- Nystrom, T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005, 24, 1311–1317. [Google Scholar] [CrossRef]
- Curtis, J.M.; Hahn, W.S.; Long, E.K.; Burrill, J.S.; Arriaga, E.A.; Bernlohr, D.A. Protein carbonylation and metabolic control systems. Trends Endocrinol. Metab. 2012, 23, 399–406. [Google Scholar] [CrossRef]
- Shacter, E. Quantification and significance of protein oxidation in biological samples. Drug Metab. Rev. 2000, 32, 307–326. [Google Scholar] [CrossRef]
- Choi, J.; Levey, A.I.; Weintraub, S.T.; Rees, H.D.; Gearing, M.; Chin, L.S.; Li, L. Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases. J. Biol. Chem. 2004, 279, 13256–13264. [Google Scholar] [CrossRef]
- Kabuta, T.; Setsuie, R.; Mitsui, T.; Kinugawa, A.; Sakurai, M.; Aoki, S.; Uchida, K.; Wada, K. Aberrant molecular properties shared by familial Parkinson’s disease-associated mutant UCH-L1 and carbonyl-modified UCH-L1. Hum. Mol. Genet. 2008, 17, 1482–1496. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Protein Oxidation in Aging and Alzheimer’s Disease Brain. Antioxidants 2024, 13, 574. [Google Scholar] [CrossRef]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M. Mass spectrometry-based methods for identifying oxidized proteins in disease: Advances and challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef]
- Carvalho, A.N.; Firuzi, O.; Gama, M.J.; Horssen, J.V.; Saso, L. Oxidative Stress and Antioxidants in Neurological Diseases: Is There Still Hope? Curr. Drug Targets 2017, 18, 705–718. [Google Scholar] [CrossRef]
- Teixeira, D.; Fernandes, R.; Prudencio, C.; Vieira, M. 3-Nitrotyrosine quantification methods: Current concepts and future challenges. Biochimie 2016, 125, 1–11. [Google Scholar] [CrossRef]
- Gryszczyńska, B.; Formanowicz, D.; Budzyń, M.; Wanic-Kossowska, M.; Pawliczak, E.; Formanowicz, P.; Majewski, W.; Strzyżewski, K.W.; Kasprzak, M.P.; Iskra, M. Advanced Oxidation Protein Products and Carbonylated Proteins as Biomarkers of Oxidative Stress in Selected Atherosclerosis-Mediated Diseases. BioMed Res. Int. 2017, 2017, 4975264. [Google Scholar] [CrossRef]
- Hanasand, M.; Omdal, R.; Norheim, K.B.; Goransson, L.G.; Brede, C.; Jonsson, G. Improved detection of advanced oxidation protein products in plasma. Clin. Chim. Acta 2012, 413, 901–906. [Google Scholar] [CrossRef]
- Liu, B.; Hou, X.; Zhou, Q.; Tian, J.; Zhu, P.; Xu, J.; Hou, F.; Fu, N. Detection of advanced oxidation protein products in patients with chronic kidney disease by a novel monoclonal antibody. Free Radic. Res. 2011, 45, 662–671. [Google Scholar] [CrossRef]
- Selmeci, L.; Seres, L.; Antal, M.; Lukacs, J.; Regoly-Merei, A.; Acsady, G. Advanced oxidation protein products (AOPP) for monitoring oxidative stress in critically ill patients: A simple, fast and inexpensive automated technique. Clin. Chem. Lab. Med. 2005, 43, 294–297. [Google Scholar] [CrossRef]
- Capeillere-Blandin, C.; Gausson, V.; Descamps-Latscha, B.; Witko-Sarsat, V. Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim. Biophys. Acta 2004, 1689, 91–102. [Google Scholar] [CrossRef]
- Bettinger, J.Q.; Welle, K.A.; Hryhorenko, J.R.; Ghaemmaghami, S. Quantitative Analysis of in Vivo Methionine Oxidation of the Human Proteome. J. Proteome Res. 2020, 19, 624–633. [Google Scholar] [CrossRef]
- Liang, X.; Kaya, A.; Zhang, Y.; Le, D.T.; Hua, D.; Gladyshev, V.N. Characterization of methionine oxidation and methionine sulfoxide reduction using methionine-rich cysteine-free proteins. BMC Biochem. 2012, 13, 21. [Google Scholar] [CrossRef]
- Lin, F.; Ren, H.; Lin, F.; Pan, Z.; Wu, L.; Yang, N. Evaluation of the Effect of Nutritional Intervention on Patients with Nasopharyngeal Carcinoma. J. Healthc. Eng. 2022, 2022, 2531671. [Google Scholar] [CrossRef]
- Pan, K.T.; Chen, Y.Y.; Pu, T.H.; Chao, Y.S.; Yang, C.Y.; Bomgarden, R.D.; Rogers, J.C.; Meng, T.C.; Khoo, K.H. Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox Signal. 2014, 20, 1365–1381. [Google Scholar] [CrossRef]
- Schilling, B.; Yoo, C.B.; Collins, C.J.; Gibson, B.W. Determining cysteine oxidation status using differential alkylation. Int. J. Mass. Spectrom. 2004, 236, 117–127. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef]
- Yang, F.; Jia, G.; Guo, J.; Liu, Y.; Wang, C. Quantitative Chemoproteomic Profiling with Data-Independent Acquisition-Based Mass Spectrometry. J. Am. Chem. Soc. 2022, 144, 901–911. [Google Scholar] [CrossRef]
- Gu, L.; Robinson, R.A. Proteomic approaches to quantify cysteine reversible modifications in aging and neurodegenerative diseases. Proteom. Clin. Appl. 2016, 10, 1159–1177. [Google Scholar] [CrossRef]
- Hurd, T.R.; James, A.M.; Lilley, K.S.; Murphy, M.P. Chapter 19 Measuring redox changes to mitochondrial protein thiols with redox difference gel electrophoresis (redox-DIGE). Methods Enzymol. 2009, 456, 343–361. [Google Scholar] [CrossRef]
- Anjo, S.I.; Melo, M.N.; Loureiro, L.R.; Sabala, L.; Castanheira, P.; Graos, M.; Manadas, B. oxSWATH: An integrative method for a comprehensive redox-centered analysis combined with a generic differential proteomics screening. Redox Biol. 2019, 22, 101130. [Google Scholar] [CrossRef]
- Fu, C.; Hu, J.; Liu, T.; Ago, T.; Sadoshima, J.; Li, H. Quantitative analysis of redox-sensitive proteome with DIGE and ICAT. J. Proteome Res. 2008, 7, 3789–3802. [Google Scholar] [CrossRef]
- Parker, J.; Zhu, N.; Zhu, M.; Chen, S. Profiling thiol redox proteome using isotope tagging mass spectrometry. J. Vis. Exp. 2012, 24, 3766. [Google Scholar] [CrossRef]
- Capelo, J.L.; Carreira, R.J.; Fernandes, L.; Lodeiro, C.; Santos, H.M.; Simal-Gandara, J. Latest developments in sample treatment for 18O-isotopic labeling for proteomics mass spectrometry-based approaches: A critical review. Talanta 2010, 80, 1476–1486. [Google Scholar] [CrossRef]
- Doron, S.; Lampl, N.; Savidor, A.; Katina, C.; Gabashvili, A.; Levin, Y.; Rosenwasser, S. SPEAR: A proteomics approach for simultaneous protein expression and redox analysis. Free. Radic. Biol. Med. 2021, 176, 366–377. [Google Scholar] [CrossRef]
- Huang, J.; Staes, A.; Impens, F.; Demichev, V.; Van Breusegem, F.; Gevaert, K.; Willems, P. CysQuant: Simultaneous quantification of cysteine oxidation and protein abundance using data dependent or independent acquisition mass spectrometry. Redox Biol. 2023, 67, 102908. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Lu, S.; Fan, S.B.; Yang, B.; Li, Y.X.; Meng, J.M.; Wu, L.; Li, P.; Zhang, K.; Zhang, M.J.; Fu, Y.; et al. Mapping native disulfide bonds at a proteome scale. Nat. Methods 2015, 12, 329–331. [Google Scholar] [CrossRef]
- Yan, T.; Julio, A.R.; Villanueva, M.; Jones, A.E.; Ball, A.B.; Boatner, L.M.; Turmon, A.C.; Nguyen, K.B.; Yen, S.L.; Desai, H.S.; et al. Proximity-labeling chemoproteomics defines the subcellular cysteinome and inflammation-responsive mitochondrial redoxome. Cell Chem. Biol. 2023, 30, 811–827.e817. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Tsikas, D. Analytical methods for 3-nitrotyrosine quantification in biological samples: The unique role of tandem mass spectrometry. Amino Acids 2012, 42, 45–63. [Google Scholar] [CrossRef]
- Held, J.M.; Gibson, B.W. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteom. 2012, 11, R111.013037. [Google Scholar] [CrossRef]
- Rogowska-Wrzesinska, A.; Wojdyla, K.; Nedic, O.; Baron, C.P.; Griffiths, H.R. Analysis of protein carbonylation--pitfalls and promise in commonly used methods. Free Radic. Res. 2014, 48, 1145–1162. [Google Scholar] [CrossRef]
- Kramer, P.A.; Duan, J.; Qian, W.J.; Marcinek, D.J. The Measurement of Reversible Redox Dependent Post-translational Modifications and Their Regulation of Mitochondrial and Skeletal Muscle Function. Front. Physiol. 2015, 6, 347. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Heimer, N.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Redox proteomics: Methods for the identification and enrichment of redox-modified proteins and their applications. Proteomics 2016, 16, 197–213. [Google Scholar] [CrossRef]
- Mnatsakanyan, R.; Markoutsa, S.; Walbrunn, K.; Roos, A.; Verhelst, S.H.L.; Zahedi, R.P. Proteome-wide detection of S-nitrosylation targets and motifs using bioorthogonal cleavable-linker-based enrichment and switch technique. Nat. Commun. 2019, 10, 2195. [Google Scholar] [CrossRef]
- Qu, Z.; Meng, F.; Bomgarden, R.D.; Viner, R.I.; Li, J.; Rogers, J.C.; Cheng, J.; Greenlief, C.M.; Cui, J.; Lubahn, D.B.; et al. Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 2014, 13, 3200–3211. [Google Scholar] [CrossRef]
- Mnatsakanyan, R.; Shema, G.; Basik, M.; Batist, G.; Borchers, C.H.; Sickmann, A.; Zahedi, R.P. Detecting post-translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev. Proteom. 2018, 15, 515–535. [Google Scholar] [CrossRef]
- Fu, L.; Li, Z.; Liu, K.; Tian, C.; He, J.; He, J.; He, F.; Xu, P.; Yang, J. A quantitative thiol reactivity profiling platform to analyze redox and electrophile reactive cysteine proteomes. Nat. Protoc. 2020, 15, 2891–2919. [Google Scholar] [CrossRef]
- Held, J.M.; Danielson, S.R.; Behring, J.B.; Atsriku, C.; Britton, D.J.; Puckett, R.L.; Schilling, B.; Campisi, J.; Benz, C.C.; Gibson, B.W. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol. Cell Proteom. 2010, 9, 1400–1410. [Google Scholar] [CrossRef]
- Babu, M.; Snyder, M. Multi-Omics Profiling for Health. Mol. Cell Proteom. 2023, 22, 100561. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acids: An overview. J. Nutr. 2006, 136, 1636S–1640S. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Murray, C.I.; Van Eyk, J.E. Chasing cysteine oxidative modifications: Proteomic tools for characterizing cysteine redox status. Circ. Cardiovasc. Genet. 2012, 5, 591. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta General. Subj. 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Su, Z.; Burchfield, J.G.; Yang, P.; Humphrey, S.J.; Yang, G.; Francis, D.; Yasmin, S.; Shin, S.Y.; Norris, D.M.; Kearney, A.L.; et al. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 2019, 10, 5486. [Google Scholar] [CrossRef]
- Shental-Bechor, D.; Levy, Y. Effect of glycosylation on protein folding: A close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. USA 2008, 105, 8256–8261. [Google Scholar] [CrossRef]
- Theillet, F.X.; Smet-Nocca, C.; Liokatis, S.; Thongwichian, R.; Kosten, J.; Yoon, M.K.; Kriwacki, R.W.; Landrieu, I.; Lippens, G.; Selenko, P. Cell signaling, post-translational protein modifications and NMR spectroscopy. J. Biomol. NMR 2012, 54, 217–236. [Google Scholar] [CrossRef]
- Karplus, M.; Petsko, G.A. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 1994, 12, 84–89. [Google Scholar] [CrossRef]
- Barducci, A.; Bonomi, M.; Parrinello, M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef]
- Mendieta, J.; Gago, F. In silico activation of Src tyrosine kinase reveals the molecular basis for intramolecular autophosphorylation. J. Mol. Graph. Model. 2004, 23, 189–198. [Google Scholar] [CrossRef]
- Kuzmanic, A.; Sutto, L.; Saladino, G.; Nebreda, A.R.; Gervasio, F.L.; Orozco, M. Changes in the free-energy landscape of p38alpha MAP kinase through its canonical activation and binding events as studied by enhanced molecular dynamics simulations. elife 2017, 6, e22175. [Google Scholar] [CrossRef]
- Kaszuba, K.; Grzybek, M.; Orlowski, A.; Danne, R.; Rog, T.; Simons, K.; Coskun, U.; Vattulainen, I. N-Glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc. Natl. Acad. Sci. USA 2015, 112, 4334–4339. [Google Scholar] [CrossRef]
- Zagórska, A.; Pozo-Guisado, E.; Boudeau, J.; Vitari, A.C.; Rafiqi, F.H.; Thastrup, J.; Deak, M.; Campbell, D.G.; Morrice, N.A.; Prescott, A.R.; et al. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J. Cell Biol. 2007, 176, 89–100. [Google Scholar] [CrossRef]
- Jonniya, N.A.; Sk, M.F.; Kar, P. Investigating Phosphorylation-Induced Conformational Changes in WNK1 Kinase by Molecular Dynamics Simulations. ACS Omega 2019, 4, 17404–17416. [Google Scholar] [CrossRef]
- Han, S. Force field parameters for S-nitrosocysteine and molecular dynamics simulations of S-nitrosated thioredoxin. Biochem. Biophys. Res. Commun. 2008, 377, 612–616. [Google Scholar] [CrossRef]
- Petrov, D.; Margreitter, C.; Grandits, M.; Oostenbrink, C.; Zagrovic, B. A systematic framework for molecular dynamics simulations of protein post-translational modifications. PLoS Comput. Biol. 2013, 9, e1003154. [Google Scholar] [CrossRef]
- Marchesani, F.; Gianquinto, E.; Autiero, I.; Michielon, A.; Campanini, B.; Faggiano, S.; Bettati, S.; Mozzarelli, A.; Spyrakis, F.; Bruno, S. The allosteric interplay between S-nitrosylation and glycine binding controls the activity of human serine racemase. FEBS J. 2021, 288, 3034–3054. [Google Scholar] [CrossRef]
- Papaleo, E.; Tiberti, M.; Arnaudi, M.; Pecorari, C.; Faienza, F.; Cantwell, L.; Degn, K.; Pacello, F.; Battistoni, A.; Lambrughi, M.; et al. TRAP1 S-nitrosylation as a model of population-shift mechanism to study the effects of nitric oxide on redox-sensitive oncoproteins. Cell Death Dis. 2023, 14, 284. [Google Scholar] [CrossRef]
- Castellanos, M.M.; Colina, C.M. Molecular dynamics simulations of human serum albumin and role of disulfide bonds. J. Phys. Chem. B 2013, 117, 11895–11905. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Wu, Z.; Huang, R.; Yuan, L. Crosstalk of intracellular post-translational modifications in cancer. Arch. Biochem. Biophys. 2019, 676, 108138. [Google Scholar] [CrossRef]
- Melo-Braga, M.N.; Meyer, M.; Zeng, X.; Larsen, M.R. Characterization of human neural differentiation from pluripotent stem cells using proteomics/PTMomics--current state-of-the-art and challenges. Proteomics 2015, 15, 656–674. [Google Scholar] [CrossRef]
- Huang, H.; Drici, L.; Lassen, P.S.; Palmisano, G.; Larsen, M.R. Simultaneous proteomics and three PTMomics characterization of pro-inflammatory cytokines stimulated INS-1E cells using TiO2 enrichment strategy. bioRxiv 2019, 509125. [Google Scholar] [CrossRef]
- Bogetofte, H.; Ryan, B.J.; Jensen, P.; Schmidt, S.I.; Vergoossen, D.L.E.; Barnkob, M.B.; Kiani, L.N.; Chughtai, U.; Heon-Roberts, R.; Caiazza, M.C.; et al. Post-translational proteomics platform identifies neurite outgrowth impairments in Parkinson’s disease GBA-N370S dopamine neurons. Cell Rep. 2023, 42, 112180. [Google Scholar] [CrossRef]
- Wang, Y.; Sung, C.C.; Chung, K.K. Novel enhancement mechanism of tyrosine hydroxylase enzymatic activity by nitric oxide through S-nitrosylation. Sci. Rep. 2017, 7, 44154. [Google Scholar] [CrossRef]
- Blanchard-Fillion, B.; Souza, J.M.; Friel, T.; Jiang, G.C.; Vrana, K.; Sharov, V.; Barrón, L.; Schöneich, C.; Quijano, C.; Alvarez, B.; et al. Nitration and inactivation of tyrosine hydroxylase by peroxynitrite. J. Biol. Chem. 2001, 276, 46017–46023. [Google Scholar] [CrossRef]
- Chung, K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef]
- Dunkley, P.R.; Bobrovskaya, L.; Graham, M.E.; von Nagy-Felsobuki, E.I.; Dickson, P.W. Tyrosine hydroxylase phosphorylation: Regulation and consequences. J. Neurochem. 2004, 91, 1025–1043. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Pirie, E.; Oh, C.K.; Zhang, X.; Han, X.; Cieplak, P.; Scott, H.R.; Deal, A.K.; Ghatak, S.; Martinez, F.J.; Yeo, G.W.; et al. S-nitrosylated TDP-43 triggers aggregation, cell-to-cell spread, and neurotoxicity in hiPSCs and in vivo models of ALS/FTD. Proc. Natl. Acad. Sci. USA 2021, 118, e2021368118. [Google Scholar] [CrossRef]
- Wilcox, K.C.; Zhou, L.; Jordon, J.K.; Huang, Y.; Yu, Y.; Redler, R.L.; Chen, X.; Caplow, M.; Dokholyan, N.V. Modifications of Superoxide Dismutase (SOD1) in Human Erythrocytes: A possible role in amyotrophic lateral sclerosis. J. Biol. Chem. 2009, 284, 13940–13947. [Google Scholar] [CrossRef]
- Banks, C.J.; Andersen, J.L. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019, 26, 101270. [Google Scholar] [CrossRef]
- Xu, W.C.; Liang, J.Z.; Li, C.; He, Z.X.; Yuan, H.Y.; Huang, B.Y.; Liu, X.L.; Tang, B.; Pang, D.W.; Du, H.N.; et al. Pathological hydrogen peroxide triggers the fibrillization of wild-type SOD1 via sulfenic acid modification of Cys-111. Cell Death Dis. 2018, 9, 67. [Google Scholar] [CrossRef]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef]
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a stabilized cysteine sulfinic acid is critical for the mitochondrial function of the parkinsonism protein DJ-1. J. Biol. Chem. 2009, 284, 6476–6485. [Google Scholar] [CrossRef]
- Wilson, M.A. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid. Redox Signal. 2011, 15, 111–122. [Google Scholar] [CrossRef]
- Fernandez-Caggiano, M.; Schroder, E.; Cho, H.J.; Burgoyne, J.; Barallobre-Barreiro, J.; Mayr, M.; Eaton, P. Oxidant-induced Interprotein Disulfide Formation in Cardiac Protein DJ-1 Occurs via an Interaction with Peroxiredoxin 2. J. Biol. Chem. 2016, 291, 10399–10410. [Google Scholar] [CrossRef]
- Madian, A.G.; Hindupur, J.; Hulleman, J.D.; Diaz-Maldonado, N.; Mishra, V.R.; Guigard, E.; Kay, C.M.; Rochet, J.C.; Regnier, F.E. Effect of single amino acid substitution on oxidative modifications of the Parkinson’s disease-related protein, DJ-1. Mol. Cell Proteom. 2012, 11, M111.010892. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, M.; Wilson, M.A.; Petsko, G.A.; Fink, A.L. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J. Mol. Biol. 2006, 356, 1036–1048. [Google Scholar] [CrossRef]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef]
- Ozawa, K.; Tsumoto, H.; Miura, Y.; Yamaguchi, J.; Iguchi-Ariga, S.M.M.; Sakuma, T.; Yamamoto, T.; Uchiyama, Y. DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 2020, 10, 4377. [Google Scholar] [CrossRef]
- Burte, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, F.; Yu, X.; Wang, B.; Chen, J.; Lu, F.; Peng, S.; Sun, X.; Yu, M.; Chen, H.; et al. Exogenous H2S Promoted USP8 Sulfhydration to Regulate Mitophagy in the Hearts of db/db Mice. Aging Dis. 2020, 11, 269–285. [Google Scholar] [CrossRef]
- Thomas, K.J.; Cookson, M.R. The role of PTEN-induced kinase 1 in mitochondrial dysfunction and dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 2025–2035. [Google Scholar] [CrossRef]
- Agarwal, S.; Muqit, M.M.K. PTEN-induced kinase 1 (PINK1) and Parkin: Unlocking a mitochondrial quality control pathway linked to Parkinson’s disease. Curr. Opin. Neurobiol. 2022, 72, 111–119. [Google Scholar] [CrossRef]
- Schaffert, L.N.; Carter, W.G. Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci. 2020, 10, 232. [Google Scholar] [CrossRef]
- Brembati, V.; Faustini, G.; Longhena, F.; Bellucci, A. Alpha synuclein post translational modifications: Potential targets for Parkinson’s disease therapy? Front. Mol. Neurosci. 2023, 16, 1197853. [Google Scholar] [CrossRef]
- Pancoe, S.X.; Wang, Y.J.; Shimogawa, M.; Perez, R.M.; Giannakoulias, S.; Petersson, E.J. Effects of Mutations and Post-Translational Modifications on α-Synuclein In Vitro Aggregation. J. Mol. Biol. 2022, 434, 167859. [Google Scholar] [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983.e924. [Google Scholar] [CrossRef]
- Witze, E.S.; Old, W.M.; Resing, K.A.; Ahn, N.G. Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 2007, 4, 798–806. [Google Scholar] [CrossRef]
- Barbour, H.; Nkwe, N.S.; Estavoyer, B.; Messmer, C.; Gushul-Leclaire, M.; Villot, R.; Uriarte, M.; Boulay, K.; Hlayhel, S.; Farhat, B.; et al. An inventory of crosstalk between ubiquitination and other post-translational modifications in orchestrating cellular processes. iScience 2023, 26, 106276. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible oxidative post-translational modifications in heart disease. Expert Rev. Proteomics 2019, 16, 681–693. [Google Scholar] [CrossRef]
- Oliveira, P.V.S.; Laurindo, F.R.M. Implications of plasma thiol redox in disease. Clin. Sci. 2018, 132, 1257–1280. [Google Scholar] [CrossRef]
- Silva-Costa, L.C.; Smith, B.J. Post-translational Modifications in Brain Diseases: A Future for Biomarkers. Adv. Exp. Med. Biol. 2022, 1382, 129–141. [Google Scholar] [CrossRef]
- Banfi, C.; Brioschi, M.; Barcella, S.; Veglia, F.; Biglioli, P.; Tremoli, E.; Agostoni, P. Oxidized proteins in plasma of patients with heart failure: Role in endothelial damage. Eur. J. Heart Fail. 2008, 10, 244–251. [Google Scholar] [CrossRef]
- Ng, M.L.; Ang, X.; Yap, K.Y.; Ng, J.J.; Goh, E.C.H.; Khoo, B.B.J.; Richards, A.M.; Drum, C.L. Novel Oxidative Stress Biomarkers with Risk Prognosis Values in Heart Failure. Biomedicines 2023, 11, 917. [Google Scholar] [CrossRef]
- Holder, C.; Adams, A.; McGahee, E.; Xia, B.; Blount, B.C.; Wang, L. High-Throughput and Sensitive Analysis of Free and Total 8-Isoprostane in Urine with Isotope-Dilution Liquid Chromatography-Tandem Mass Spectrometry. ACS Omega 2020, 5, 10919–10926. [Google Scholar] [CrossRef]
- Zhang, Z.J. Systematic review on the association between F2-isoprostanes and cardiovascular disease. Ann. Clin. Biochem. 2013, 50, 108–114. [Google Scholar] [CrossRef]
- Costa, M.; Horrillo, R.; Ortiz, A.M.; Pérez, A.; Mestre, A.; Ruiz, A.; Boada, M.; Grancha, S. Increased Albumin Oxidation in Cerebrospinal Fluid and Plasma from Alzheimer’s Disease Patients. J. Alzheimers Dis. 2018, 63, 1395–1404. [Google Scholar] [CrossRef]
- Greco, V.; Neri, C.; Pieragostino, D.; Spalloni, A.; Persichilli, S.; Gastaldi, M.; Mercuri, N.B.; Longone, P.; Urbani, A. Investigating Different Forms of Hydrogen Sulfide in Cerebrospinal Fluid of Various Neurological Disorders. Metabolites 2021, 11, 152. [Google Scholar] [CrossRef]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef]
- Tsai, C.W.; Tsai, C.F.; Lin, K.H.; Chen, W.J.; Lin, M.S.; Hsieh, C.C.; Lin, C.C. An investigation of the correlation between the S-glutathionylated GAPDH levels in blood and Alzheimer’s disease progression. PLoS ONE 2020, 15, e0233289. [Google Scholar] [CrossRef]
- Poulsen, K.; Bahl, J.M.; Simonsen, A.H.; Hasselbalch, S.G.; Heegaard, N.H. Distinct transthyretin oxidation isoform profile in spinal fluid from patients with Alzheimer’s disease and mild cognitive impairment. Clin. Proteom. 2014, 11, 12. [Google Scholar] [CrossRef]
- Srivastava, D.; Kukkuta Sarma, G.R.; Dsouza, D.S.; Muralidharan, M.; Srinivasan, K.; Mandal, A.K. Characterization of residue-specific glutathionylation of CSF proteins in multiple sclerosis—A MS-based approach. Anal. Biochem. 2019, 564–565, 108–115. [Google Scholar] [CrossRef]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef]
- Tramutola, A.; Abate, G.; Lanzillotta, C.; Triani, F.; Barone, E.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Marziano, M.; Memo, M.; et al. Protein nitration profile of CD3+ lymphocytes from Alzheimer disease patients: Novel hints on immunosenescence and biomarker detection. Free Radic. Biol. Med. 2018, 129, 430–439. [Google Scholar] [CrossRef]
- Abe, K.; Pan, L.H.; Watanabe, M.; Konno, H.; Kato, T.; Itoyama, Y. Upregulation of protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurol. Res. 1997, 19, 124–128. [Google Scholar] [CrossRef]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Choi, J.; Malakowsky, C.A.; Talent, J.M.; Conrad, C.C.; Gracy, R.W. Identification of oxidized plasma proteins in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2002, 293, 1566–1570. [Google Scholar] [CrossRef]
- Conrad, C.C.; Marshall, P.L.; Talent, J.M.; Malakowsky, C.A.; Choi, J.; Gracy, R.W. Oxidized proteins in Alzheimer’s plasma. Biochem. Biophys. Res. Commun. 2000, 275, 678–681. [Google Scholar] [CrossRef]
- Di Domenico, F.; Pupo, G.; Giraldo, E.; Badìa, M.C.; Monllor, P.; Lloret, A.; Schininà, M.E.; Giorgi, A.; Cini, C.; Tramutola, A.; et al. Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic. Biol. Med. 2016, 91, 1–9. [Google Scholar] [CrossRef]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef]
- Vats, A.; Gourie-Devi, M.; Ahuja, K.; Sharma, A.; Wajid, S.; Ganguly, N.K.; Taneja, V. Expression analysis of protein homeostasis pathways in the peripheral blood mononuclear cells of sporadic amyotrophic lateral sclerosis patients. J. Neurol. Sci. 2018, 387, 85–91. [Google Scholar] [CrossRef]
- Sanchez-Gomez, F.J.; Espinosa-Diez, C.; Dubey, M.; Dikshit, M.; Lamas, S. S-glutathionylation: Relevance in diabetes and potential role as a biomarker. Biol. Chem. 2013, 394, 1263–1280. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Braconi, D.; Santucci, A.; Rossi, R. Membrane Skeletal Protein S-Glutathionylation in Human Red Blood Cells as Index of Oxidative Stress. Chem. Res. Toxicol. 2019, 32, 1096–1102. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Scaloni, A.; Giustarini, D.; Cavarra, E.; Tell, G.; Lungarella, G.; Colombo, R.; Rossi, R.; Milzani, A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrom. Rev. 2005, 24, 55–99. [Google Scholar] [CrossRef]
- Cadenas-Garrido, P.; Schonvandt-Alarcos, A.; Herrera-Quintana, L.; Vázquez-Lorente, H.; Santamaría-Quiles, A.; Ruiz de Francisco, J.; Moya-Escudero, M.; Martín-Oliva, D.; Martín-Guerrero, S.M.; Rodríguez-Santana, C.; et al. Using Redox Proteomics to Gain New Insights into Neurodegenerative Disease and Protein Modification. Antioxidants 2024, 13, 127. [Google Scholar] [CrossRef]
- Fernández-Espejo, E.; Rodríguez de Fonseca, F.; Suárez, J.; Tolosa, E.; Vilas, D.; Aldecoa, I.; Berenguer, J.; Damas-Hermoso, F. Native α-Synuclein, 3-Nitrotyrosine Proteins, and Patterns of Nitro-α-Synuclein-Immunoreactive Inclusions in Saliva and Submandibulary Gland in Parkinson’s Disease. Antioxidants 2021, 10, 715. [Google Scholar] [CrossRef]
- Azevedo, R.; Jacquemin, C.; Villain, N.; Fenaille, F.; Lamari, F.; Becher, F. Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications. Cells 2022, 11, 1279. [Google Scholar] [CrossRef]
- Ren, R.J.; Dammer, E.B.; Wang, G.; Seyfried, N.T.; Levey, A.I. Proteomics of protein post-translational modifications implicated in neurodegeneration. Transl. Neurodegener. 2014, 3, 23. [Google Scholar] [CrossRef]
- de Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Reyes, J.F.; Geula, C.; Vana, L.; Binder, L.I. Selective tau tyrosine nitration in non-AD tauopathies. Acta Neuropathol. 2012, 123, 119–132. [Google Scholar] [CrossRef]
- Ehrenberg, A.J.; Khatun, A.; Coomans, E.; Betts, M.J.; Capraro, F.; Thijssen, E.H.; Senkevich, K.; Bharucha, T.; Jafarpour, M.; Young, P.N.E.; et al. Relevance of biomarkers across different neurodegenerative diseases. Alzheimers Res. Ther. 2020, 12, 56. [Google Scholar] [CrossRef]
- van Steenoven, I.; Koel-Simmelink, M.J.A.; Vergouw, L.J.M.; Tijms, B.M.; Piersma, S.R.; Pham, T.V.; Bridel, C.; Ferri, G.L.; Cocco, C.; Noli, B.; et al. Identification of novel cerebrospinal fluid biomarker candidates for dementia with Lewy bodies: A proteomic approach. Mol. Neurodegener. 2020, 15, 36. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Kimble, L.; Bayoumy, S.; Bolsewig, K.; Burtscher, F.; Coppens, S.; Das, S.; Gogishvili, D.; Fernandes Gomes, B.; Gómez de San José, N.; et al. Methods to Discover and Validate Biofluid-Based Biomarkers in Neurodegenerative Dementias. Mol. Cell Proteom. 2023, 22, 100629. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Redox proteomics and amyloid β-peptide: Insights into Alzheimer disease. J. Neurochem. 2019, 151, 459–487. [Google Scholar] [CrossRef]
- Aluise, C.D.; Robinson, R.A.; Beckett, T.L.; Murphy, M.P.; Cai, J.; Pierce, W.M.; Markesbery, W.R.; Butterfield, D.A. Preclinical Alzheimer disease: Brain oxidative stress, Abeta peptide and proteomics. Neurobiol. Dis. 2010, 39, 221–228. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Cássio, R.; Correia-Guedes, L.; Gomes, M.A.; Chegão, A.; Miranda, E.; Soares, T.; Coelho, M.; Rosa, M.M.; Ferreira, J.J.; et al. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci. Rep. 2017, 7, 13713. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Bian, S.; Haque, R.U.; Carter, E.K.; Watson, C.M.; Gordon, B.A.; Ping, L.; Duong, D.M.; Epstein, M.P.; McDade, E.; et al. Cerebrospinal fluid proteomics define the natural history of autosomal dominant Alzheimer’s disease. Nat. Med. 2023, 29, 1979–1988. [Google Scholar] [CrossRef]
- Libiger, O.; Shaw, L.M.; Watson, M.H.; Nairn, A.C.; Umaña, K.L.; Biarnes, M.C.; Canet-Avilés, R.M.; Jack, C.R., Jr.; Breton, Y.A.; Cortes, L.; et al. Longitudinal CSF proteomics identifies NPTX2 as a prognostic biomarker of Alzheimer’s disease. Alzheimers Dement. 2021, 17, 1976–1987. [Google Scholar] [CrossRef]
- Rutledge, J.; Lehallier, B.; Zarifkar, P.; Losada, P.M.; Shahid-Besanti, M.; Western, D.; Gorijala, P.; Ryman, S.; Yutsis, M.; Deutsch, G.K.; et al. Comprehensive proteomics of CSF, plasma, and urine identify DDC and other biomarkers of early Parkinson’s disease. Acta Neuropathol. 2024, 147, 52. [Google Scholar] [CrossRef]
- Canever, J.B.; Soares, E.S.; de Avelar, N.C.P.; Cimarosti, H.I. Targeting α-synuclein post-translational modifications in Parkinson’s disease. Behav. Brain Res. 2023, 439, 114204. [Google Scholar] [CrossRef]
- Contini, C.; Fadda, L.; Lai, G.; Masala, C.; Olianas, A.; Castagnola, M.; Messana, I.; Iavarone, F.; Bizzarro, A.; Masullo, C.; et al. A top-down proteomic approach reveals a salivary protein profile able to classify Parkinson’s disease with respect to Alzheimer’s disease patients and to healthy controls. Proteomics 2024, 24, e2300202. [Google Scholar] [CrossRef]
- Schmid, A.W.; Fauvet, B.; Moniatte, M.; Lashuel, H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson disease and other synucleinopathies. Mol. Cell Proteom. 2013, 12, 3543–3558. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue Affected | Redox-PTM | Reversibility | Methods of Detection | |
|---|---|---|---|---|
| Cysteine residues (C) | Reduced (Free) thiols (-S-H) | --- |
| |
| Disulfide bonds (-S-S) | Reversible |
| ||
| S-sulfhydration/ Persulfidation (-S-SH) | Reversible |
| ||
| S-glutathionylation (-S-SG) | Reversible |
| ||
| S-nitrosylation/ S-nitrosation (-S-NO) | Reversible |
| ||
| S-sulfenylation (-SOH) | Reversible |
| ||
| S-sulfinylation (-SO2H) | Reversible |
| ||
| S-sulfonylation (-SO3H) | Irreversible |
| ||
| Other residues | Arg (R) Pro (P) Lys (K) Thr (T) | Carbonylation (C=O) | Irreversible |
|
| Tyr (Y) Arg (R) Lys (K) Pro (P) Glu (E) Thr (T) | Advanced Oxidation Protein Products (AOPP):
| Irreversible |
| |
| Tyr (Y) | Nitration (3-NO2-Tyr) | Irreversible |
| |
| Met (M) | Oxidation (Met=O) | Irreversible |
| |
| Redox-PTMs | Disease | Biological Sample | Method(s) Used | Variation/Alteration | Refs. |
|---|---|---|---|---|---|
| Sulfenic acid, sulfinic acid and/or sulfonic acid | AD | CSF, blood | HPLC, LC-ESI-qTOF-MS | ↑ reversible (disulphide bond) and irreversibly (SO2H) oxidized albumin, ↓ reduced (-SH) albumin (higher differences in CSF than blood) | [204] |
| ALS | CSF | WB, ELISA | ↑ SO2H/SO3H of wild-type SOD1 in sporadic ALS | [170] | |
| PD | Postmortem tissue | WB, IHC | ↑ sulfonated Parkin in insoluble fractions in PD; ↑ Parkin in insoluble fraction (aggregation) | [178] | |
| Disulfide bonds | AD | Postmortem tissue | Redox 2DE | ↑ disulfide bonds in GAPDH in AD | [45] |
| S-sulfhydration | AD | Postmortem tissue | Dimedone-switch assay; antibody array-like approach | ↓ global sulfhydration; ↓ sulfhydration of GSK3β (Tau kinase) | [55] |
| PD | Postmortem tissue | Maleimide assay | ↓ S-sulfhydration of Parkin | [177] | |
| MS/ALS/AD/PD | CSF | MALDI-TOF-MS | ↑ global H2S-protein bound and ↑ TTR-H2S in MS (no significant increase in ALS, AD, PD as compared to controls) | [205] | |
| S-glutathionylation | AD | Postmortem tissue | 2DE; Oxyblots; MALDI-TOF | ↑ S-glutathionlylation of deoxy-Hb, CRYAB, GAPDH, ENO1 in AD | [206] |
| AD | blood | ELISA | ↑ S-glutathionylated GAPDH in AD, and levels correlate with AD progression | [207] | |
| AD | CSF | IP of TTR and nanoLC-ESI-MS | ↑ S-glutahionylated TTR in AD and MCI | [208] | |
| MS | CSF | nano-LC/ESI-MS | ↑ S-glutahionylated proteins in MS patients during relapse incl. IL-18; α1-AAT | [209] | |
| S-Nitrosylation | AD | Postmortem tissue | 2D-Oxyblot; biotin switch; ESI-QTOF MS/MS; IHC against SNO-Cys | ↑ global S-nitrosylation levels in AD. 45 S-nitrosylated proteins identified in AD incl. ↑ S-nitrosylation of SOD2, ALDOC, VDAC2 in AD | [210] |
| FTD | Postmortem tissue | Biotin switch-WB | ↑ S-nitrosylation and disulfide bonds in TDP-43 in FTD | [167] | |
| PD | Postmortem tissue | Biotin switch-WB | ↑ S-nitrosylation of Parkin | [164,166] | |
| PD/AD | Postmortem tissue | Biotin switch-WB | ↑ S-nitrosylation of PRDX2 in PD brains when compared to controls, but not in AD brains | [33] | |
| Nitration | AD | Postmortem tissue | 2DE; MALDI-TOF-MS | ↑ nitration of proteins in AD incl. ATP5F1A, VDAC, GAPDH | [211] |
| AD | Lymphocytes in blood | 2DE; HPLC-ESI-MS/MS; Slot blot; IP/WB with anti-3NT antibody | ↑ global 3-NT levels in AD; ↑ 3-NT in 10 proteins incl. ATP5F1A, CAT | [212] | |
| ALS | Postmortem tissue | IHC with anti-3-NT antibody | ↑ Immunoreactivity for 3-NT-positive motor neurons in ALS | [213] | |
| PD/DLB/MSA | Postmortem tissue | ELISA; IHC; WB with anti-nitrated-α-Syn | ↑ nitrated α-Syn in PD, DLB, MSA | [214] | |
| Carbonylation | AD | plasma | 2DE; WB with anti-DNP antibody; MALDI-TOF/MS | ↑ carbonylation of seven proteins identified incl. A1AT, FGG precursor in AD | [215] |
| AD | Plasma | HPLC, WB with anti-DNP antibody | ↑ carbonylated protein levels in AD | [216] | |
| AD | CSF | 2DE; Oxyblots; MALDI-ToF MS | ↑ carbonylated protein levels in MCI and AD; 7 proteins identified with ↑ carbonylation, incl. APOE. | [217] | |
| AD | Postmortem tissue | 2DE; MALDI-TOF/MS; HPLC-ESI/MS/MS | ↑ carbonylated SOD1 in AD and PD. | [218] | |
| ALS | PBMCs in blood | DNPH assay | ↑ carbonylated proteins in ALS | [219] | |
| PD/AD | Postmortem tissue | 2DE; MALDI-TOF/MS; HPLC-ESI/MS/MS | ↑ carbonylation, cysteine and methionine oxidation of UCH-L1 | [93] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anjo, S.I.; He, Z.; Hussain, Z.; Farooq, A.; McIntyre, A.; Laughton, C.A.; Carvalho, A.N.; Finelli, M.J. Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers. Antioxidants 2024, 13, 681. https://doi.org/10.3390/antiox13060681
Anjo SI, He Z, Hussain Z, Farooq A, McIntyre A, Laughton CA, Carvalho AN, Finelli MJ. Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers. Antioxidants. 2024; 13(6):681. https://doi.org/10.3390/antiox13060681
Chicago/Turabian StyleAnjo, Sandra I., Zhicheng He, Zohaib Hussain, Aruba Farooq, Alan McIntyre, Charles A. Laughton, Andreia Neves Carvalho, and Mattéa J. Finelli. 2024. "Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers" Antioxidants 13, no. 6: 681. https://doi.org/10.3390/antiox13060681
APA StyleAnjo, S. I., He, Z., Hussain, Z., Farooq, A., McIntyre, A., Laughton, C. A., Carvalho, A. N., & Finelli, M. J. (2024). Protein Oxidative Modifications in Neurodegenerative Diseases: From Advances in Detection and Modelling to Their Use as Disease Biomarkers. Antioxidants, 13(6), 681. https://doi.org/10.3390/antiox13060681