

Emerging Roles of Xanthine Oxidoreductase in Chronic Kidney Disease

Abstract
:1. Introduction
2. XOR Biochemistry
3. XOR Biology: Products and Functions
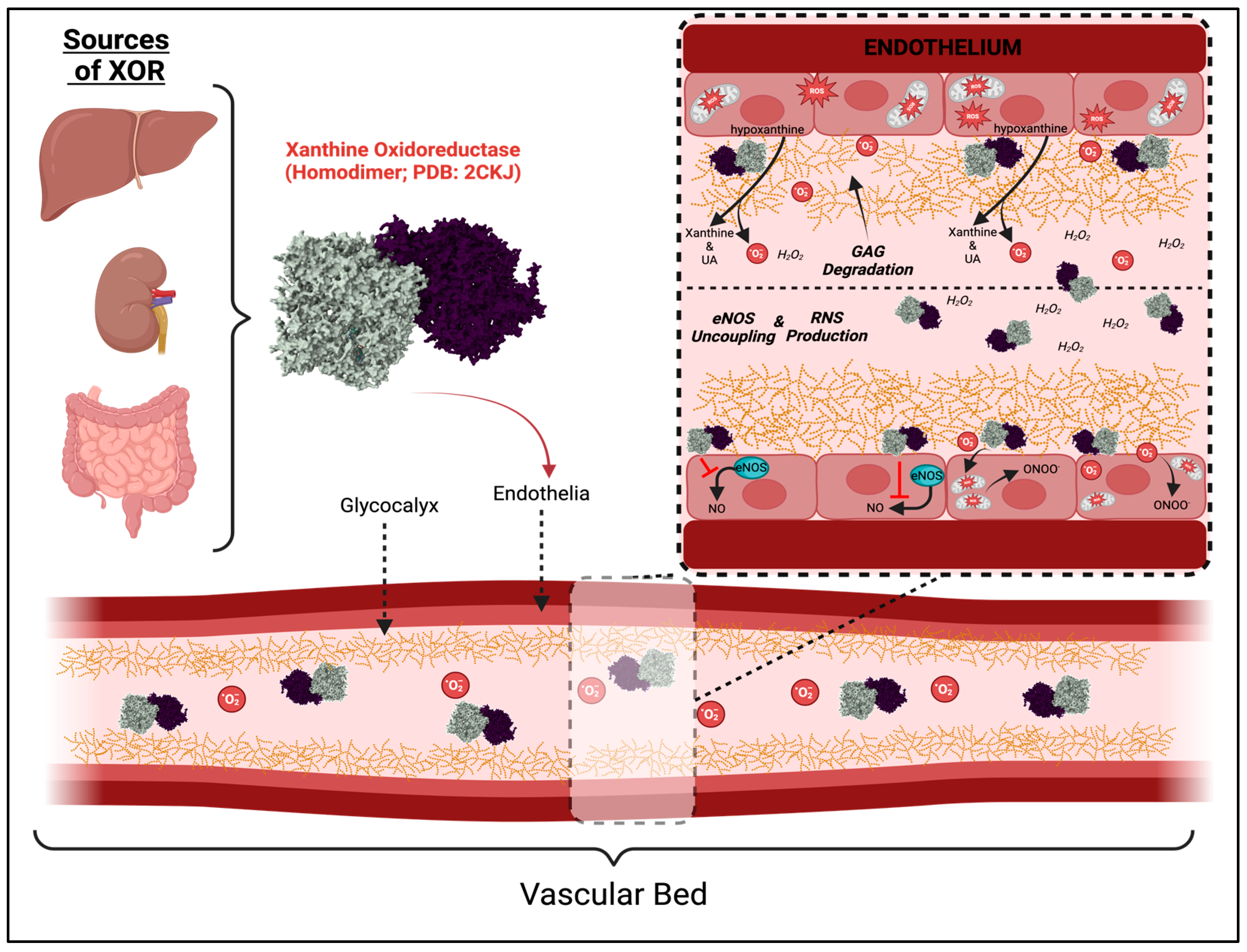
3.1. XOR Activity and Cell Damage
3.2. Role of XOR in CKD
4. XOR Activity & Diet
4.1. Diet-Induced XOR Activity in CVD and Fatty Liver
4.2. Diet, XOR Activity in CKD

{kind=link}
{kind=link}
{kind=link}
| Factor | Impact on XOR Expression/Activity | Biological Effect | Effect Size | Reference |
| Purine-rich Diet | Increase | Greater UA; elevated UA precursors promote purine catabolic activity of XOR | Odds ratio: 1.024 for animal-derived foods | [145] |
| Western Diet | Increase | Greater UA | Odds ratio: 2.15 for animal-derived and fried foods | [179] |
| Tungsten | Reduce | Reduced ROS; Prevents Molybdenum incorporation at the MoCo site thereby reducing XO-derived ROS production. | N/A | [180,181] |
| Heat | Increase | Renal function (measured in eGFR) declined with rising UA. Electrolyte hydration by sugarcane workers appeared preventative to the decline | adj β = −10.4 and 8.1, respectively | [182] |
| Xanthinuria | Non-functional | Reduced UA; elevated purines and formation of xanthine stones | 1:69,000 people (combined incidence of hereditary types I and II) | [183] |
| XDH rs207454 | Reduce | Improved survival for ‘C/C’ carriers facing gastric cancer compared to ‘A/A’ or ‘A/C’ carriers. | Hazard Ratio: 1.53 for A/A and A/C carriers | [184] |
| XDH rs185925 | Increase | Higher XOR expression was associated with acute respiratory distress syndrome in septic African American patients | Odds ratio: 1.464 for C/T carriers | [23] |
| XDH rs1884725 | Increase | Increased serum creatinine (a predictor for renal dysfunction) in septic African American patients. | β = 0.504 | [23] |
| XDH rs4952085 | Increase | Increased serum creatinine (a predictor for renal dysfunction) in septic African American patients. | β = 0.493 | [23] |
5. XOR Activity and the Environment
5.1. XOR and Heavy Metal Exposures
5.2. XOR and CKDu
6. Genetic Regulation of XOR
7. Novel Therapeutic Approaches Targeting XOR
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bobulescu, I.A.; Moe, O.W. Renal Transport of Uric Acid: Evolving Concepts and Uncertainties. Adv. Chronic Kidney Dis. 2012, 19, 358–371. [Google Scholar] [CrossRef]
- Aliciguzel, Y.; Ozen, I.; Aslan, M.; Karayalcin, U. Activities of xanthine oxidoreductase and antioxidant enzymes in different tissues of diabetic rats. J. Lab. Clin. Med. 2003, 142, 172–177. [Google Scholar] [CrossRef]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric Acid Stimulates Fructokinase and Accelerates Fructose Metabolism in the Development of Fatty Liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef]
- Wang, Q.; Qi, H.; Wu, Y.; Yu, L.; Bouchareb, R.; Li, S.; Lassén, E.; Casalena, G.; Stadler, K.; Ebefors, K.; et al. Genetic susceptibility to diabetic kidney disease is linked to promoter variants of XOR. Nat. Metab. 2023, 5, 607–625. [Google Scholar] [CrossRef]
- Nishikawa, T.; Nagata, N.; Shimakami, T.; Shirakura, T.; Matsui, C.; Ni, Y.; Zhuge, F.; Xu, L.; Chen, G.; Nagashimada, M.; et al. Xanthine oxidase inhibition attenuates insulin resistance and diet-induced steatohepatitis in mice. Sci. Rep. 2020, 10, 815. [Google Scholar] [CrossRef] [PubMed]
- Itano, S.; Kadoya, H.; Satoh, M.; Nakamura, T.; Murase, T.; Sasaki, T.; Kanwar, Y.S.; Kashihara, N. Non-purine selective xanthine oxidase inhibitor ameliorates glomerular endothelial injury in InsAkita diabetic mice. Am. J. Physiol. Physiol. 2020, 319, F765–F772. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Fukushima, T.; Usami, Y.; Hirano, K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem. J. 1993, 289, 523–527. [Google Scholar] [CrossRef]
- Yokose, C.; McCormick, N.; Choi, H.K. The role of diet in hyperuricemia and gout. Curr. Opin. Rheumatol. 2021, 33, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free. Radic. Biol. Med. 2009, 48, 493–498. [Google Scholar] [CrossRef]
- Bortolotti, M.; Polito, L.; Battelli, M.G.; Bolognesi, A. Xanthine oxidoreductase: One enzyme for multiple physiological tasks. Redox Biol. 2021, 41, 101882. [Google Scholar] [CrossRef] [PubMed]
- Duskin-Bitan, H.; Cohen, E.; Goldberg, E.; Shochat, T.; Levi, A.; Garty, M.; Krause, I. The Degree of Asymptomatic Hyperu-ricemia and the Risk of Gout. A Retrospective Analysis of a Large Cohort. Clin. Rheumatol. 2014, 33, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Cammà, C.; Cabibi, D.; Di Marco, V.; Craxì, A. Hyperuricemia is associated with histological liver damage in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2011, 34, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, C.; Zhao, Y.; Zeng, X.; Liu, F.; Fu, P. Is hyperuricemia an independent risk factor for new-onset chronic kidney disease?: A systematic review and meta-analysis based on observational cohort studies. BMC Nephrol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Butts, B.; Calhoun, D.A.; Denney, T.S.; Lloyd, S.G.; Gupta, H.; Gaddam, K.K.; Aban, I.; Oparil, S.; Sanders, P.W.; Patel, R.; et al. Plasma xanthine oxidase activity is related to increased sodium and left ventricular hypertrophy in resistant hypertension. Free Radic. Biol. Med. 2019, 134, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Taufiq, F.; Li, P.; Miake, J.; Hisatome, I. Hyperuricemia as a Risk Factor for Atrial Fibrillation Due to Soluble and Crystalized Uric Acid. Circ. Rep. 2019, 1, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Elion, G.B.; Kovensky, A.; Hitchings, G.H.; Metz, E.; Rundles, R. Metabolic studies of allopurinol, an inhibitor of xanthine oxidase. Biochem. Pharmacol. 1966, 15, 863–880. [Google Scholar] [CrossRef] [PubMed]
- Cicero, A.F.; Fogacci, F.; Cincione, R.I.; Tocci, G.; Borghi, C. Clinical Effects of Xanthine Oxidase Inhibitors in Hyperuricemic Patients. Med. Princ. Pract. 2021, 30, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Nivorozhkin, A.; Szabó, C. Therapeutic Effects of Xanthine Oxidase Inhibitors: Renaissance Half a Century after the Discovery of Allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Miesel, R.; Zuber, M. Elevated levels of xanthine oxidase in serum of patients with inflammatory and autoimmune rheumatic diseases. Inflammation 1993, 17, 551–561. [Google Scholar] [CrossRef]
- Murase, T.; Oka, M.; Nampei, M.; Miyachi, A.; Nakamura, T. A highly sensitive assay for xanthine oxidoreductase activity using a combination of [13C2,15N2]xanthine and liquid chromatography/triple quadrupole mass spectrometry. J. Label. Compd. Radiopharm. 2016, 59, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Kawachi, Y.; Fujishima, Y.; Nishizawa, H.; Nagao, H.; Nakamura, T.; Akari, S.; Murase, T.; Taya, N.; Omori, K.; Miyake, A.; et al. Plasma xanthine oxidoreductase activity in Japanese patients with type 2 diabetes across hospitalized treatment. J. Diabetes Investig. 2021, 12, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Rafaels, N.; Dudenkov, T.M.; Damarla, M.; Damico, R.; Maloney, J.P.; Moss, M.; Martin, G.S.; Sevransky, J.; Shanholtz, C.; et al. Xanthine oxidoreductase gene polymorphisms are associated with high risk of sepsis and organ failure. Respir. Res. 2023, 24, 177. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.S.; Ballou, D.P.; Palmer, G.; Massey, V. The Mechanism of Action of Xanthine Oxidase. J. Biol. Chem. 1974, 249, 4363–4382. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.M.; Massey, V. The Oxidative Half-reaction of Xanthine Dehydrogenase with NAD; Reaction Kinetics and Steady-state Mechanism. J. Biol. Chem. 1997, 272, 28335–28341. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Dempski, R.; Hille, R. The Reductive Half-reaction of Xanthine Oxidase Reaction with Aldehyde Substrates and Identification of the Catalytically Labile Oxygen*. J. Biol. Chem. 1999, 274, 3323–3330. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-C.; Velayutham, M.; Komatsu, T.; Hille, R.; Zweier, J.L. Measurement and Characterization of Superoxide Generation from Xanthine Dehydrogenase: A Redox-Regulated Pathway of Radical Generation in Ischemic Tissues. Biochemistry 2014, 53, 6615–6623. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.; Okamoto, K.; Kawaguchi, Y.; Hori, H.; Matsumura, T.; Eger, B.T.; Pai, E.F.; Nishino, T. Mechanism of the Conversion of Xanthine Dehydrogenase to Xanthine Oxidase: Identification of the Two Cysteine Disulfide Bonds and Crystal Structure of a Non-Convertible Rat Liver Xanthine Dehydrogenase Mutant. J. Biol. Chem. 2005, 280, 24888–24894. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, T.G.; Hollwarth, M.E.; Granger, D.N.; Engerson, T.D.; Landler, U.; Jones, H.P. Mechanisms of conversion of xanthine dehydrogenase to xanthine oxidase in ischemic rat liver and kidney. Am. J. Physiol. Liver Physiol. 1988, 254, G753–G760. [Google Scholar] [CrossRef]
- Weinstein, A.L.; Lalezarzadeh, F.D.; Soares, M.A.; Saadeh, P.B.; Ceradini, D.J. Normalizing dysfunctional purine metabolism accelerates diabetic wound healing. Wound Repair Regen. 2015, 23, 14–21. [Google Scholar] [CrossRef]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef] [PubMed]
- Roman, Y.M. The Role of Uric Acid in Human Health: Insights from the Uricase Gene. J. Pers. Med. 2023, 13, 1409. [Google Scholar] [CrossRef] [PubMed]
- Angermüller, S.; Bruder, G.; Völkl, A.; Wesch, H.; Fahimi, H.D. Localization of Xanthine Oxidase in Crystalline Cores of Pe-roxisomes. A Cytochemical and Biochemical Study. Eur. J. Cell Biol. 1987, 45, 137–144. [Google Scholar] [PubMed]
- Frederiks, W.M.; Vreeling-Sindelárová, H. Ultrastructural localization of xanthine oxidoreductase activity in isolated rat liver cells. Acta Histochem. 2002, 104, 29–37. [Google Scholar] [CrossRef]
- Williams, J. The Decomposition of Hydrogen Peroxide by Liver Catalase. J. Gen. Physiol. 1928, 11, 309–337. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, M.; Bonga, S.E.W. Cytochemical localization of catalase and several hydrogen peroxide-producing oxidases in the nucleoids and matrix of rat liver peroxisomes. Histochem. J. 1979, 11, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, N.; Pérez-Ruiz, F.; Lioté, F. Mechanisms and rationale for uricase use in patients with gout. Nat. Rev. Rheumatol. 2023, 19, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Jalal, D.I.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric Acid as a Mediator of Diabetic Nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef]
- Kim, I.Y.; Lee, D.W.; Lee, S.B.; Kwak, I.S. The Role of Uric Acid in Kidney Fibrosis: Experimental Evidences for the Causal Relationship. BioMed Res. Int. 2014, 2014, 638732. [Google Scholar] [CrossRef]
- Sebesta, I.; Stiburkova, B.; Krijt, J. Hereditary xanthinuria is not so rare disorder of purine metabolism. Nucleosides Nucleotides Nucleic Acids 2018, 37, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.L.; Diniz, H.; Tavares, I.; Dória, S.; Dong, J.; Kyriss, M.; Fairbanks, L.; Oliveira, J.P. Kidney Failure Secondary to Hereditary Xanthinuria due to a Homozygous Deletion of the XDH Gene in the Absence of Overt Kidney Stone Disease. Nephron 2024, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Helal, I.; McFann, K.; Reed, B.; Yan, X.-D.; Schrier, R.W.; Fick-Brosnahan, G.M. Serum uric acid, kidney volume and progression in autosomal-dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2013, 28, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Daehn, I.S.; Ekperikpe, U.S.; Stadler, K. Redox regulation in diabetic kidney disease. Am. J. Physiol. Physiol. 2023, 325, F135–F149. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, L.V.; Zietara, A.; Levchenko, V.; Spires, D.R.; Angulo, M.B.; El-Meanawy, A.; Geurts, A.M.; Dwinell, M.R.; Palygin, O.; Staruschenko, A. Lack of xanthine dehydrogenase leads to a remarkable renal decline in a novel hypouricemic rat model. iScience 2022, 25, 104887. [Google Scholar] [CrossRef] [PubMed]
- Laakso, J.; Mervaala, E.; Himberg, J.-J.; Teräväinen, T.-L.; Karppanen, H.; Vapaatalo, H.; Lapatto, R. Increased Kidney Xanthine Oxidoreductase Activity in Salt-Induced Experimental Hypertension. Hypertension 1998, 32, 902–906. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.-A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.-H. Oxidative stress with an activation of the renin–angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Dounousi, E.; Eleftheriadis, T.; Liakopoulos, V. Dietary Antioxidant Supplements and Uric Acid in Chronic Kidney Disease: A Review. Nutrients 2019, 11, 1911. [Google Scholar] [CrossRef] [PubMed]
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine Oxidoreductase-Derived Reactive Species: Physiological and Pathological Effects. Oxidative Med. Cell. Longev. 2016, 2016, 3527579. [Google Scholar] [CrossRef]
- Kadowaki, D.; Sakaguchi, S.; Miyamoto, Y.; Taguchi, K.; Muraya, N.; Narita, Y.; Sato, K.; Chuang, V.T.G.; Maruyama, T.; Otagiri, M.; et al. Direct Radical Scavenging Activity of Benzbromarone Provides Beneficial Antioxidant Properties for Hyperuricemia Treatment. Biol. Pharm. Bull. 2015, 38, 487–492. [Google Scholar] [CrossRef]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric Acid Promotes Apoptosis in Human Proximal Tubule Cells by Oxidative Stress and the Activation of NADPH Oxidase NOX 4. PLoS ONE 2014, 9, e115210. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.-H.; Liu, J.-C.; Lin, J.-W.; Chen, C.-H.; Wu, C.-H.; Cheng, T.-H. Uric acid stimulates endothelin-1 gene expression associated with NADPH oxidase in human aortic smooth muscle cells. Acta Pharmacol. Sin. 2008, 29, 1301–1312. [Google Scholar] [CrossRef]
- Houston, M.; Estevez, A.; Chumley, P.; Aslan, M.; Marklund, S.; Parks, D.A.; Freeman, B.A. Binding of Xanthine Oxidase to Vascular Endothelium: Kinetic Characterization and Oxidative Impairment of Nitric Oxide-Dependent Signaling. J. Biol. Chem. 1999, 274, 4985–4994. [Google Scholar] [CrossRef]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Sánchez-Lozada, L.G.; Lanaspa, M.A.; Cristóbal-García, M.; García-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.A.; Kang, D.-H.; Johnson, R.J. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron Exp. Nephrol. 2012, 121, e71–e78. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Hong, Q.; Zhang, X.; Xiao, W.; Wang, L.; Cui, S.; Feng, Z.; Lv, Y.; Cai, G.; Chen, X.; et al. Aldose reductase mediates endothelial cell dysfunction induced by high uric acid concentrations. Cell Commun. Signal. 2017, 15, 3. [Google Scholar] [CrossRef]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose Reductase, Oxidative Stress, and Diabetic Mellitus. Front. Pharmacol. 2012, 3, 87. [Google Scholar] [CrossRef]
- Yang, L.; Chang, B.; Guo, Y.; Wu, X.; Liu, L. The role of oxidative stress-mediated apoptosis in the pathogenesis of uric acid nephropathy. Ren. Fail. 2019, 41, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Chen, C.-J.; Ontiveros, F.; Rock, K.L. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Investig. 2010, 120, 1939–1949. [Google Scholar] [CrossRef]
- Daehn, I.; Karran, P. Immune Effector Cells Produce Lethal DNA Damage in Cells Treated with a Thiopurine. Cancer Res. 2009, 69, 2393–2399. [Google Scholar] [CrossRef]
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 963. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.A.; Wolf, M. Renal proximal tubule cells: Power and finesse. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fang, L.; Jiang, L.; Wen, P.; Cao, H.; He, W.; Dai, C.; Yang, J. Uric Acid Induces Renal Inflammation via Activating Tubular NF-κB Signaling Pathway. PLoS ONE 2012, 7, e39738. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Choi, Y.-W.; Seok, H.-Y.; Jeong, K.-H.; Lee, S.-H.; Lee, T.-W.; Ihm, C.-G.; Lim, S.J.; Moon, J.-Y. Reducing Serum Uric Acid Attenuates TGF-β1-Induced Profibrogenic Progression in Type 2 Diabetic Nephropathy. Nephron Exp. Nephrol. 2012, 121, e109–e121. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Shi, Y.; Tang, L.; Wang, Y.; Fang, L.; Jiang, W.; Lin, T.; Qiu, A.; Zhuang, S.; Liu, N. Blockade of ERK1/2 by U0126 alleviates uric acid-induced EMT and tubular cell injury in rats with hyperuricemic nephropathy. Am. J. Physiol. Physiol. 2019, 316, F660–F673. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, L.; Zhang, M.; Zhou, C.; Lin, N. Uric Acid Enhances PKC-Dependent ENOS Phosphorylationand Mediates Cellular ER Stress: A Mechanism for Uric Acid-Induced Endothelial Dysfunction. Int. J. Mol. Med. 2016, 37, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H. Hyperuricemia and Progression of Chronic Kidney Disease: Role of Phenotype Transition of Renal Tubular and Endothelial Cells. Contrib. Nephrol. 2018, 192, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Feng, Q.; Ding, G.; Zhao, M.; Che, R.; Bai, M.; Bao, H.; Zhang, A.; Huang, S. Activation of ERK1/2 by NADPH oxidase-originated reactive oxygen species mediates uric acid-induced mesangial cell proliferation. Am. J. Physiol. Physiol. 2014, 307, F396–F406. [Google Scholar] [CrossRef]
- Li, S.; Zhao, F.; Cheng, S.; Wang, X.; Hao, Y. Uric acid-induced endoplasmic reticulum stress triggers phenotypic change in rat glomerular mesangial cells. Nephrology 2013, 18, 682–689. [Google Scholar] [CrossRef]
- Daehn, I.S.; Duffield, J.S. The glomerular filtration barrier: A structural target for novel kidney therapies. Nat. Rev. Drug Discov. 2021, 20, 770–788. [Google Scholar] [CrossRef]
- Yang, K.-J.; Choi, W.J.; Chang, Y.-K.; Park, C.W.; Kim, S.Y.; Hong, Y.A. Inhibition of Xanthine Oxidase Protects against Diabetic Kidney Disease through the Amelioration of Oxidative Stress via VEGF/VEGFR Axis and NOX-FoxO3a-eNOS Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 3807. [Google Scholar] [CrossRef]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2016, 66, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kurajoh, M.; Fukumoto, S.; Murase, T.; Nakamura, T.; Yoshida, H.; Hirata, K.; Inaba, M.; Emoto, M. Association of plasma xanthine oxidoreductase activity with blood pressure affected by oxidative stress level: MedCity21 health examination registry. Sci. Rep. 2020, 10, 4437. [Google Scholar] [CrossRef]
- Maejima, I.; Takahashi, A.; Omori, H.; Kimura, T.; Takabatake, Y.; Saitoh, T.; Yamamoto, A.; Hamasaki, M.; Noda, T.; Isaka, Y.; et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J. 2013, 32, 2336–2347. [Google Scholar] [CrossRef]
- Lusco, M.A.; Fogo, A.B.; Najafian, B.; Alpers, C.E. AJKD Atlas of Renal Pathology: Gouty Nephropathy. Am. J. Kidney Dis. 2017, 69, e5–e6. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhao, T.; Xie, C.; Zheng, S.; Wan, W.; Zou, H.; Zhu, X. Performance of Ultrasound in the Clinical Evaluation of Gout and Hyperuricemia. J. Immunol. Res. 2021, 2021, 5550626. [Google Scholar] [CrossRef]
- Wu, M.; Ma, Y.; Chen, X.; Liang, N.; Qu, S.; Chen, H. Hyperuricemia causes kidney damage by promoting autophagy and NLRP3-mediated inflammation in rats with urate oxidase deficiency. Dis. Model. Mech. 2021, 14, dmm048041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sui, X.; Xu, Y.; Gu, F.; Zhang, A.; Chen, J. Association between Nod-like receptor protein 3 inflammasome and gouty nephropathy. Exp. Ther. Med. 2020, 20, 195–204. [Google Scholar] [CrossRef]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef] [PubMed]
- Nomura, J.; Kobayashi, T.; So, A.; Busso, N. Febuxostat, a Xanthine Oxidoreductase Inhibitor, Decreases NLRP3-dependent Inflammation in Macrophages by Activating the Purine Salvage Pathway and Restoring Cellular Bioenergetics. Sci. Rep. 2019, 9, 17314. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, T.; Matsumura, K.; Sakagami, K.; Fujii, K.; Tsuruya, K.; Noguchi, H.; Rovira, I.I.; Finkel, T.; Iida, M. Xanthine Oxidoreductase Depletion Induces Renal Interstitial Fibrosis Through Aberrant Lipid and Purine Accumulation in Renal Tubules. Hypertension 2009, 54, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Kang, H.; Kim, D.; Kim, M.; Ryu, E.; Lee, S.; Ryu, J.; Roncal, C.; Johnson, R.J.; Kang, D. Uric acid induced the phenotype transition of vascular endothelial cells via induction of oxidative stress and glycocalyx shedding. FASEB J. 2019, 33, 13334–13345. [Google Scholar] [CrossRef] [PubMed]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Böttinger, E.P. Glucose-Induced Reactive Oxygen Species Cause Apoptosis of Podocytes and Podocyte Depletion at the Onset of Diabetic Nephropathy. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Charlton, A.; Garzarella, J.; Jandeleit-Dahm, K.A.M.; Jha, J.C. Oxidative Stress and Inflammation in Renal and Cardiovascular Complications of Diabetes. Biology 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Jalal, D.; Sánchez-Lozada, L.G.; Kang, D.-H.; Ritz, E. Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transplant. 2013, 28, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, P.; Wang, A.Y.; Wang, X.; Wang, L.; Li, G.; Hong, D. Hyperuricemia and its related histopathological features on renal biopsy. BMC Nephrol. 2019, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, W.; McRae, S.; Marek, G.; Wymer, D.; Pannu, V.; Baylis, C.; Johnson, R.J.; Sautin, Y.Y. Hyperuricemia as a Mediator of the Proinflammatory Endocrine Imbalance in the Adipose Tissue in a Murine Model of the Metabolic Syndrome. Diabetes 2011, 60, 1258–1269. [Google Scholar] [CrossRef]
- Washio, K.; Kusunoki, Y.; Murase, T.; Nakamura, T.; Osugi, K.; Ohigashi, M.; Sukenaga, T.; Ochi, F.; Matsuo, T.; Katsuno, T.; et al. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metabolism 2017, 70, 51–56. [Google Scholar] [CrossRef]
- Takir, M.; Kostek, O.; Ozkok, A.; Elcioglu, O.C.; Bakan, A.; Erek, A.; Mutlu, H.H.; Telci, O.; Semerci, A.; Odabas, A.R.; et al. Lowering Uric Acid with Allopurinol Improves Insulin Resistance and Systemic Inflammation in Asymptomatic Hyperuricemia. J. Investig. Med. 2015, 63, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, M. Hyperuricemia, Cardiovascular Disease, and Hypertension. Pulse 2016, 3, 242–252. [Google Scholar] [CrossRef]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2021, 12, 37–71. [Google Scholar] [CrossRef]
- Kanbay, M.; Yilmaz, M.I.; Sonmez, A.; Turgut, F.; Saglam, M.; Cakir, E.; Yenicesu, M.; Covic, A.; Jalal, D.; Johnson, R.J. Serum Uric Acid Level and Endothelial Dysfunction in Patients with Nondiabetic Chronic Kidney Disease. Am. J. Nephrol. 2011, 33, 298–304. [Google Scholar] [CrossRef]
- Idigo, W.O.; Reilly, S.; Zhang, M.H.; Zhang, Y.H.; Jayaram, R.; Carnicer, R.; Crabtree, M.J.; Balligand, J.-L.; Casadei, B. Regulation of Endothelial Nitric-oxide Synthase (NOS) S-Glutathionylation by Neuronal NOS Evidence of a Functional Interaction between Myocardial Constitutive Nos Isoforms *. J. Biol. Chem. 2012, 287, 43665–43673. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Youn, J.-Y.; Cai, H. Mechanisms and consequences of ENOS dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Szabo, C. Role of the Peroxynitrite-Poly(ADP-Ribose) Polymerase Pathway in Human Disease. Am. J. Pathol. 2008, 173, 2–13. [Google Scholar] [CrossRef]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef]
- Miric, D.J.; Kisic, B.M.; Filipovic-Danic, S.; Grbic, R.; Dragojevic, I.; Miric, M.B.; Puhalo-Sladoje, D. Xanthine Oxidase Activity in Type 2 Diabetes Mellitus Patients with and without Diabetic Peripheral Neuropathy. J. Diabetes Res. 2016, 2016, 4370490. [Google Scholar] [CrossRef]
- Dehghan, A.; van Hoek, M.; Sijbrands, E.J.; Hofman, A.; Witteman, J.C. High Serum Uric Acid as a Novel Risk Factor for Type 2 Diabetes. Diabetes Care 2008, 31, 361–362. [Google Scholar] [CrossRef]
- Klisic, A.; Kocic, G.; Kavaric, N.; Jovanovic, M.; Stanisic, V.; Ninic, A. Xanthine oxidase and uric acid as independent predictors of albuminuria in patients with diabetes mellitus type 2. Clin. Exp. Med. 2018, 18, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Yamamotoya, T.; Nakatsu, Y.; Ueda, K.; Matsunaga, Y.; Inoue, M.-K.; Sakoda, H.; Fujishiro, M.; Ono, H.; Kikuchi, T.; et al. Xanthine Oxidase Inhibitor Febuxostat Exerts an Anti-Inflammatory Action and Protects against Diabetic Nephropathy Development in KK-Ay Obese Diabetic Mice. Int. J. Mol. Sci. 2019, 20, 4680. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chen, L.; Xu, Y.; Duan, Q.; Zheng, Z.; Zheng, Z.; He, D. The Effect of Allopurinol on Renal Outcomes in Patients with Diabetic Kidney Disease: A Systematic Review and Meta-Analysis. Kidney Blood Press. Res. 2022, 47, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Cai, Y.; Zhao, Q.; Tian, L.; Liu, Y.; Liu, W.J. Effects of allopurinol on renal function in patients with diabetes: A systematic review and meta-analysis. Ren. Fail. 2022, 44, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; de Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. New Engl. J. Med. 2020, 382, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.; Garcia-Arroyo, F.; Sasai, F.; Rodriguez-Iturbe, B.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Johnson, R.J. Mini Review: Reappraisal of Uric Acid in Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Haberland, A.; Luther, H.; Schimke, I. Does Allopurinol Prevent Superoxide Radical Production by Xanthine Oxidase (XOD)? Agents Actions 1991, 32, 96–97. [Google Scholar] [CrossRef] [PubMed]
- Massey, V.; Komai, H.; Palmer, G.; Elion, G.B. On the Mechanism of Inactivation of Xanthine Oxidase by Allopurinol and Other Pyrazolo[3,4-d]pyrimidines. J. Biol. Chem. 1970, 245, 2837–2844. [Google Scholar] [CrossRef]
- Horiuchi, H.; Ota, M.; Nishimura, S.-I.; Kaneko, H.; Kasahara, Y.; Ohta, T.; Komoriya, K. Allopurinol induces renal toxicity by impairing pyrimidine metabolism in mice. Life Sci. 2000, 66, 2051–2070. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Hase-Aoki, K.; Horiuchi, H.; Zhao, L.; Kasahara, Y.; Kondo, S.; Becker, M.A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005, 76, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Jeong, K.H.; Kim, Y.G.; Moon, J.Y.; Lee, S.H.; Ihm, C.G.; Sung, J.Y.; Lee, T.W. Febuxostat Ameliorates Diabetic Renal Injury in a Streptozotocin-Induced Diabetic Rat Model. Am. J. Nephrol. 2014, 40, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Sircar, D.; Chatterjee, S.; Waikhom, R.; Golay, V.; Raychaudhury, A.; Chatterjee, S.; Pandey, R. Efficacy of Febuxostat for Slowing the GFR Decline in Patients With CKD and Asymptomatic Hyperuricemia: A 6-Month, Double-Blind, Randomized, Placebo-Controlled Trial. Am. J. Kidney Dis. 2015, 66, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Hosoya, T.; Uchida, S.; Inaba, M.; Makino, H.; Ito, S.; Yamamoto, T.; Tomino, Y.; Ohno, I.; Shibagaki, Y.; et al. Febuxostat Therapy for Patients With Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am. J. Kidney Dis. 2018, 72, 798–810. [Google Scholar] [CrossRef] [PubMed]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L. Cardi-ovascular Safety of Febuxostat or Allopurinol in Patients with Gout. New Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sun, J. Febuxostat Improves Uric Acid Levels and Renal Function in Patients with Chronic Kidney Disease and Hyperuricemia: A Meta-Analysis. Appl. Bionics Biomech. 2022, 2022, 9704862. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-C.; Hung, L.Y.; Chen, Y.-C.; Lo, W.-C.; Lin, C.H.; Tam, K.-W.; Wu, M.-Y. Effects of febuxostat on renal function in patients with chronic kidney disease: A Systematic Review and Meta-Analysis. Medicine 2019, 98, e16311. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Hosoya, T.; Honda, D.; Sakamoto, R.; Narita, K.; Sasaki, T.; Okui, D.; Kimura, K. Uric acid-lowering and renoprotective effects of topiroxostat, a selective xanthine oxidoreductase inhibitor, in patients with diabetic nephropathy and hyperuricemia: A randomized, double-blind, placebo-controlled, parallel-group study (UPWARD study). Clin. Exp. Nephrol. 2018, 22, 860–870. [Google Scholar] [CrossRef]
- Mizukoshi, T.; Kato, S.; Ando, M.; Sobajima, H.; Ohashi, N.; Naruse, T.; Saka, Y.; Shimizu, H.; Nagata, T.; Maruyama, S. Renoprotective effects of topiroxostat for Hyperuricaemic patients with overt diabetic nephropathy study (ETUDE study): A prospective, randomized, multicentre clinical trial. Nephrology 2018, 23, 1023–1030. [Google Scholar] [CrossRef]
- Kato, S.; Ando, M.; Mizukoshi, T.; Nagata, T.; Katsuno, T.; Kosugi, T.; Tsuboi, N.; Maruyama, S. Randomized control trial for the assessment of the anti-albuminuric effects of topiroxostat in hyperuricemic patients with diabetic nephropathy (the ETUDE study). Nagoya J. Med. Sci. 2016, 78, 135–142. [Google Scholar] [PubMed]
- Nakamura, T.; Murase, T.; Nampei, M.; Morimoto, N.; Ashizawa, N.; Iwanaga, T.; Sakamoto, R. Effects of topiroxostat and febuxostat on urinary albumin excretion and plasma xanthine oxidoreductase activity in db/db mice. Eur. J. Pharmacol. 2016, 780, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Lozada, L.G.; Tapia, E.; Soto, V.; Ávila-Casado, C.; Franco, M.; Wessale, J.L.; Zhao, L.; Johnson, R.J. Effect of Febuxostat on the Progression of Renal Disease in 5/6 Nephrectomy Rats with and without Hyperuricemia. Nephron Physiol. 2008, 108, p69–p78. [Google Scholar] [CrossRef]
- Omori, H.; Kawada, N.; Inoue, K.; Ueda, Y.; Yamamoto, R.; Matsui, I.; Kaimori, J.; Takabatake, Y.; Moriyama, T.; Isaka, Y.; et al. Use of xanthine oxidase inhibitor febuxostat inhibits renal interstitial inflammation and fibrosis in unilateral ureteral obstructive nephropathy. Clin. Exp. Nephrol. 2012, 16, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Sakuyama, A.; Xu, L.; Qiu, J.; Namai-Takahashi, A.; Ogawa, Y.; Kohzuki, M.; Ito, O. Febuxostat ameliorates high salt intake-induced hypertension and renal damage in Dahl salt-sensitive rats. J. Hypertens. 2021, 40, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.-K.; Yamamotoya, T.; Nakatsu, Y.; Ueda, K.; Inoue, Y.; Matsunaga, Y.; Sakoda, H.; Fujishiro, M.; Ono, H.; Morii, K.; et al. The Xanthine Oxidase Inhibitor Febuxostat Suppresses the Progression of IgA Nephropathy, Possibly via Its Anti-Inflammatory and Anti-Fibrotic Effects in the gddY Mouse Model. Int. J. Mol. Sci. 2018, 19, 3967. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Fan, Y.; Xiao, W.; Chen, T.; Wen, J.; Dong, Y.; Wang, Y.; Li, S.; Xue, R.; Zheng, L.; et al. Febuxostat attenuates ER stress mediated kidney injury in a rat model of hyperuricemic nephropathy. Oncotarget 2017, 8, 111295–111308. [Google Scholar] [CrossRef]
- Lim, A.K.H.; Tesch, G.H. Inflammation in Diabetic Nephropathy. Mediat. Inflamm. 2013, 2012, 146154. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, Y.; Wang, B.; Zhang, F.; Wang, D.; Wang, Y. Allopurinol treatment improves renal function in patients with type 2 diabetes and asymptomatic hyperuricemia: 3-year randomized parallel-controlled study. Clin. Endocrinol. 2015, 83, 475–482. [Google Scholar] [CrossRef]
- Tanaka, K.; Nakayama, M.; Kanno, M.; Kimura, H.; Watanabe, K.; Tani, Y.; Hayashi, Y.; Asahi, K.; Terawaki, H.; Watanabe, T. Renoprotective effects of febuxostat in hyperuricemic patients with chronic kidney disease: A parallel-group, randomized, controlled trial. Clin. Exp. Nephrol. 2015, 19, 1044–1053. [Google Scholar] [CrossRef]
- Hosoya, T.; Ohno, I.; Nomura, S.; Hisatome, I.; Uchida, S.; Fujimori, S.; Yamamoto, T.; Hara, S. Effects of topiroxostat on the serum urate levels and urinary albumin excretion in hyperuricemic stage 3 chronic kidney disease patients with or without gout. Clin. Exp. Nephrol. 2014, 18, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Nata, N.; Ninwisut, N.; Inkong, P.; Supasyndh, O.; Satirapoj, B. Effects of febuxostat on markers of endothelial dysfunction and renal progression in patients with chronic kidney disease. Sci. Rep. 2023, 13, 13494. [Google Scholar] [CrossRef] [PubMed]
- Ejaz, A.A.; Antenor, J.A.; Kumar, V.; Roncal, C.; Garcia, G.E.; Andres-Hernando, A.; Lanaspa, M.A.; Johnson, R.J. Uric Acid: A Friend in the Past, a Foe in the Present. Integr. Med. Nephrol. Androl. 2022, 9, 8. [Google Scholar] [CrossRef]
- Yang, J.; Kamide, K.; Kokubo, Y.; Takiuchi, S.; Horio, T.; Matayoshi, T.; Yasuda, H.; Miwa, Y.; Yoshii, M.; Yoshihara, F.; et al. Assiciations of Hypertension and Its Complications with Variations in the Xanthine Dehydrogenase Gene. Hypertens. Res. 2008, 31, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, I.; Yilmaz, M.I.; Orscelik, O.; Sipahioglu, M.H.; Unal, A.; Eroglu, E.; Kalay, N.; Tokgoz, B.; Axelsson, J.; Oymak, O. Serum Uric Acid Levels and Endothelial Dysfunction in Patients with Autosomal Dominant Polycystic Kidney Disease. Nephron Clin. Pract. 2013, 123, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Desmond, R.A. Autosomal Recessive Polycystic Kidney Disease: The Clinical Experience in North America. Pediatrics 2003, 111, 1072–1080. [Google Scholar] [CrossRef]
- Torra, R.; Pérez-Gómez, M.V.; Furlano, M. Autosomal dominant polycystic kidney disease: Possibly the least silent cause of chronic kidney disease. Clin. Kidney J. 2021, 14, 2281–2284. [Google Scholar] [CrossRef]
- Menon, V.; Rudym, D.; Chandra, P.; Miskulin, D.; Perrone, R.; Sarnak, M. Inflammation, Oxidative Stress, and Insulin Resistance in Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 7–13. [Google Scholar] [CrossRef]
- Vareesangthip, K.; Tong, P.; Wilkinson, R.; Thomas, T.H. Insulin resistance in adult polycystic kidney disease. Kidney Int. 1997, 52, 503–508. [Google Scholar] [CrossRef]
- Kaehny, W.D.; Tangel, D.J.; Johnson, A.M.; Kimberling, W.J.; Schrier, R.W.; Gabow, P.A. Uric acid handling in autosomal dominant polycystic kidney disease with normal filtration rates. Am. J. Med. 1990, 89, 49–52. [Google Scholar] [CrossRef]
- Kim, H.; Koh, J.; Park, S.K.; Oh, K.H.; Kim, Y.H.; Kim, Y.; Ahn, C.; Oh, Y.K. Baseline characteristics of the autosomal-dominant polycystic kidney disease sub-cohort of the KoreaN cohort study for outcomes in patients with chronic kidney disease. Nephrology 2019, 24, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Roseland, J.M.; Haytowitz, D.B.; Pehrsson, P.R.; Ershow, A.G. Availability and quality of published data on the purine content of foods, alcoholic beverages, and dietary supplements. J. Food Compos. Anal. 2019, 84, 103281. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, H.; Cheng, X.; Li, Y.; Zhao, Y.; Mei, W.; Wei, X.; Zhou, H.; Du, Y.; Zeng, C. Dietary intake of fructose increases purine de novo synthesis: A crucial mechanism for hyperuricemia. Front. Nutr. 2022, 9, 1045805. [Google Scholar] [CrossRef] [PubMed]
- Vedder, D.; Walrabenstein, W.; Heslinga, M.; de Vries, R.; Nurmohamed, M.; van Schaardenburg, D.; Gerritsen, M. Dietary Interventions for Gout and Effect on Cardiovascular Risk Factors: A Systematic Review. Nutrients 2019, 11, 2955. [Google Scholar] [CrossRef] [PubMed]
- Aihemaitijiang, S.; Zhang, Y.; Zhang, L.; Yang, J.; Ye, C.; Halimulati, M.; Zhang, W.; Zhang, Z. The Association between Purine-Rich Food Intake and Hyperuricemia: A Cross-Sectional Study in Chinese Adult Residents. Nutrients 2020, 12, 3835. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; McFaline, J.L.; Burgis, N.E.; Dong, M.; Taghizadeh, K.; Sullivan, M.R.; Elmquist, C.E.; Cunningham, R.P.; Dedon, P.C. Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Pareek, V.; Pedley, A.M.; Benkovic, S.J. Human de novo purine biosynthesis. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Major, T.J.; Topless, R.K.; Dalbeth, N.; Merriman, T.R. Evaluation of the diet wide contribution to serum urate levels: Meta-analysis of population based cohorts. BMJ 2018, 363, k3951. [Google Scholar] [CrossRef] [PubMed]
- Rai, S.K.; Fung, T.T.; Lu, N.; Keller, S.F.; Curhan, G.C.; Choi, H.K. The Dietary Approaches to Stop Hypertension (DASH) diet, Western diet, and risk of gout in men: Prospective cohort study. BMJ 2017, 357, j1794. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Beltrán-Velasco, A.I.; Redondo-Flórez, L.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients 2023, 15, 2749. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef]
- Argueta, D.A.; DiPatrizio, N.V. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 2017, 171, 32–39. [Google Scholar] [CrossRef]
- Stevenson, R.J.; Francis, H.M.; Attuquayefio, T.; Gupta, D.; Yeomans, M.R.; Oaten, M.J.; Davidson, T. Hippocampal-dependent appetitive control is impaired by experimental exposure to a Western-style diet. R. Soc. Open Sci. 2020, 7, 191338. [Google Scholar] [CrossRef]
- Webb, A.; Bond, R.; McLean, P.; Uppal, R.; Benjamin, N.; Ahluwalia, A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia–reperfusion damage. Proc. Natl. Acad. Sci. USA 2004, 101, 13683–13688. [Google Scholar] [CrossRef]
- Godber, B.L.J.; Doel, J.J.; Sapkota, G.P.; Blake, D.R.; Stevens, C.R.; Eisenthal, R.; Harrison, R. Reduction of Nitrite to Nitric Oxide Catalyzed by Xanthine Oxidoreductase. J. Biol. Chem. 2000, 275, 7757–7763. [Google Scholar] [CrossRef]
- Peleli, M.; Zollbrecht, C.; Montenegro, M.F.; Hezel, M.; Zhong, J.; Persson, E.G.; Holmdahl, R.; Weitzberg, E.; Lundberg, J.O.; Carlström, M. Enhanced XOR activity in eNOS-deficient mice: Effects on the Nitrate-Nitrite-NO Pathway and ROS Homeostasis. Free Radic. Biol. Med. 2016, 99, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Singh, G.M.; Paciorek, C.J.; Lin, J.K.; Cowan, M.J.; Finucane, M.M.; Farzadfar, F.; Stevens, G.A.; Riley, L.M.; Lu, Y.; et al. The Global Cardiovascular Risk Transition: Associations of Four Metabolic Risk Factors with National Income, Urbanization, and Western Diet in 1980 and 2008. Circulation 2013, 127, 1493–1502. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C.; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowski, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M.; et al. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E323–E330. [Google Scholar] [CrossRef] [PubMed]
- Ballal, K.; Wilson, C.R.; Harmancey, R.; Taegtmeyer, H. Obesogenic high fat western diet induces oxidative stress and apoptosis in rat heart. Mol. Cell. Biochem. 2010, 344, 221–230. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Habibi, J.; Sun, Z.; Ramirez-Perez, F.I.; Brady, B.; Chen, D.; Martinez-Lemus, L.A.; Manrique, C.; Nistala, R.; et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metabolism 2017, 74, 32–40. [Google Scholar] [CrossRef]
- Akki, A.; Seymour, A.-M.L. Western diet impairs metabolic remodelling and contractile efficiency in cardiac hypertrophy. Cardiovasc. Res. 2009, 81, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Vecchione, C.; Jung, O.; Schreiber, J.G.; Shiri-Sverdlov, R.; van Gorp, P.J.; Busse, R.; Brandes, R.P. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free. Radic. Biol. Med. 2006, 41, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Sahlabadi, A.; Sadat, S.; Beigrezaei, S.; Pourmasomi, M.; Feizi, A.; Ghiasvand, R.; Hadi, A.; Clark, C.C.T.; Miraghajani, M. Dietary patterns and risk of non-alcoholic fatty liver disease. BMC Gastroenterol. 2021, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J.; on behalf of the International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Yagi, C.; Kusunoki, Y.; Tsunoda, T.; Murase, T.; Nakamura, T.; Osugi, K.; Ohigashi, M.; Morimoto, A.; Miyoshi, A.; Kakutani-Hatayama, M.; et al. Xanthine oxidoreductase activity is correlated with hepatic steatosis. Sci. Rep. 2022, 12, 12282. [Google Scholar] [CrossRef]
- Demaria, T.M.; Crepaldi, L.D.; Costa-Bartuli, E.; Branco, J.R.; Zancan, P.; Sola-Penna, M. Once a week consumption of Western diet over twelve weeks promotes sustained insulin resistance and non-alcoholic fat liver disease in C57BL/6 J mice. Sci. Rep. 2023, 13, 3058. [Google Scholar] [CrossRef]
- Kawachi, Y.; Fujishima, Y.; Nishizawa, H.; Nakamura, T.; Akari, S.; Murase, T.; Saito, T.; Miyazaki, Y.; Nagao, H.; Fukuda, S.; et al. Increased plasma XOR activity induced by NAFLD/NASH and its possible involvement in vascular neointimal proliferation. J. Clin. Investig. 2021, 6, e144762. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.-M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef]
- Johnson, R.J.; Perez-Pozo, S.E.; Lillo, J.L.; Grases, F.; Schold, J.D.; Kuwabara, M.; Sato, Y.; Hernando, A.A.; Garcia, G.; Jensen, T.; et al. Fructose increases risk for kidney stones: Potential role in metabolic syndrome and heat stress. BMC Nephrol. 2018, 19, 315. [Google Scholar] [CrossRef]
- Li, X.; Bhattacharya, D.; Yuan, Y.; Wei, C.; Zhong, F.; Ding, F.; D’agati, V.D.; Lee, K.; Friedman, S.L.; He, J.C. Chronic kidney disease in a murine model of non-alcoholic steatohepatitis (NASH). Kidney Int. 2024, 105, 540–561. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Johnson, R.J.; Andres-Hernando, A.; Roncal-Jimenez, C.; Sanchez-Lozada, L.G.; Tolan, D.R.; Lanaspa, M.A. Fructose Production and Metabolism in the Kidney. J. Am. Soc. Nephrol. 2020, 31, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Kosugi, T.; Gersch, M.; Connor, T.; Sanchez-Lozada, L.G.; Lanaspa, M.A.; Roncal, C.; Perez-Pozo, S.E.; Johnson, R.J.; Nakagawa, T.; et al. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. Am. J. Physiol. Renal Physiol. 2010, 298, F712–F720. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kang, D.-H. Fructose in the kidney: From physiology to pathology. Kidney Res. Clin. Pract. 2021, 40, 527–541. [Google Scholar] [CrossRef]
- Cirillo, P.; Gersch, M.S.; Mu, W.; Scherer, P.M.; Kim, K.M.; Gesualdo, L.; Henderson, G.N.; Johnson, R.J.; Sautin, Y.Y. Ketohexokinase-Dependent Metabolism of Fructose Induces Proinflammatory Mediators in Proximal Tubular Cells. J. Am. Soc. Nephrol. 2009, 20, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, H.E.; Abdelhady, M.A.; Aal, S.M.A.; Elrashidy, R.A. Dulaglutide mitigates high dietary fructose-induced renal fibrosis in rats through suppressing epithelial-mesenchymal transition mediated by GSK-3β/TGF-β1/Smad3 signaling pathways. Life Sci. 2022, 309, 120999. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-Y.; Yang, C.-T.; Lin, W.-H.; Yao, W.-Y.; Ou, H.-T.; Kuo, S. Chronic kidney outcomes associated with GLP-1 receptor agonists versus long-acting insulins among type 2 diabetes patients requiring intensive glycemic control: A nationwide cohort study. Cardiovasc. Diabetol. 2023, 22, 272. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol. Renal Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chang, H.; Gao, Y.; Wang, X.; Xu, W.; Liu, D.; Li, G.; Huang, G. Major Dietary Patterns and Risk of Asymptomatic Hyperuricemia in Chinese Adults. J. Nutr. Sci. Vitaminol. 2012, 58, 339–345. [Google Scholar] [CrossRef]
- Suzuki, H.; DeLano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schönbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef]
- Swei, A.; Lacy, F.; Delano, F.A.; Parks, D.A.; Schmid-Schönbein, G.W. A Mechanism of Oxygen Free Radical Production in the Dahl Hypertensive Rat. Microcirculation 1999, 6, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, C.; Aragón, A.; González, M.; Weiss, I.; Glaser, J.; Rivard, C.J.; Roncal-Jiménez, C.; Correa-Rotter, R.; Johnson, R.J. Heat stress, hydration and uric acid: A cross-sectional study in workers of three occupations in a hotspot of Mesoamerican nephropathy in Nicaragua. BMJ Open 2016, 6, e011034. [Google Scholar] [CrossRef]
- Ichida, K.; Amaya, Y.; Kamatani, N.; Nishino, T.; Hosoya, T.; Sakai, O. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical type I xanthinuria. J. Clin. Investig. 1997, 99, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, H.; Shi, W.; Lin, Y.; Ma, G.; Tao, G.; Gong, W.; Zhao, Q.; Du, M.; Wang, M.; et al. Genetic variants in XDH are associated with prognosis for gastric cancer in a Chinese population. Gene 2018, 663, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Steunou, A.-S.; Babot, M.; Durand, A.; Bourbon, M.-L.; Liotenberg, S.; Miotello, G.; Armengaud, J.; Ouchane, S. Discriminating Susceptibility of Xanthine Oxidoreductase Family to Metals. Microbiol. Spectr. 2023, 11, e0481422. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Sato, Y.; Milagres, T.; Hernando, A.A.; García, G.; Bjornstad, P.; Dawson, J.B.; Sorensen, C.; Newman, L.; Krisher, L.; et al. Experimental heat stress nephropathy and liver injury are improved by allopurinol. Am. J. Physiol. Renal Physiol. 2018, 315, F726–F733. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.K.; Naeher, T.M.; Broome, J.E.; Boedeker, E.C. Role of Host Xanthine Oxidase in Infection Due to Enteropathogenic and Shiga-Toxigenic Escherichia coli. Infect. Immun. 2013, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhu, X.; Hui, R.; Xing, Y.; Wang, J.; Shi, S.; Zhang, Y.; Zhu, L. Associations of metal exposure with hyperuricemia and gout in general adults. Front. Endocrinol. 2022, 13, 1052784. [Google Scholar] [CrossRef]
- Madigan, M.C.; McEnaney, R.M.; Shukla, A.J.; Hong, G.; Kelley, E.E.; Tarpey, M.M.; Gladwin, M.; Zuckerbraun, B.S.; Tzeng, E. Xanthine Oxidoreductase Function Contributes to Normal Wound Healing. Mol. Med. 2015, 21, 313–322. [Google Scholar] [CrossRef]
- Sorensen, C.; Garcia-Trabanino, R. A New Era of Climate Medicine—Addressing Heat-Triggered Renal Disease. New Engl. J. Med. 2019, 381, 693–696. [Google Scholar] [CrossRef]
- Hansson, E.; Glaser, J.; Jakobsson, K.; Weiss, I.; Wesseling, C.; Lucas, R.A.I.; Wei, J.L.K.; Ekström, U.; Wijkström, J.; Bodin, T.; et al. Pathophysiological Mechanisms by which Heat Stress Potentially Induces Kidney Inflammation and Chronic Kidney Disease in Sugarcane Workers. Nutrients 2020, 12, 1639. [Google Scholar] [CrossRef] [PubMed]
- Trabanino, R.G.; Aguilar, R.; Silva, C.R.; Mercado, M.O.; Merino, R.L. Nefropatía terminal en pacientes de un hospital de referencia en El Salvador. Rev. Panam. De Salud Publica-Pan Am. J. Public Health 2002, 12, 202–206. [Google Scholar] [CrossRef]
- John, O.; Gummudi, B.; Jha, A.; Gopalakrishnan, N.; Kalra, O.P.; Kaur, P.; Kher, V.; Kumar, V.; Machiraju, R.S.; Osborne, N.; et al. Chronic Kidney Disease of Unknown Etiology in India: What Do We Know and Where We Need to Go. Kidney Int. Rep. 2021, 6, 2743–2751. [Google Scholar] [CrossRef]
- Mani, M.K. Chronic renal failure in India. Nephrol. Dial. Transplant. 1993, 8, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, S.; Shivanthan, M.C.; Selvarajah, M. Chronic kidney disease of unknown etiology in Sri Lanka. Int. J. Occup. Environ. Health 2016, 22, 259–264. [Google Scholar] [CrossRef]
- Roncal-Jimenez, C.; García-Trabanino, R.; Barregard, L.; Lanaspa, M.A.; Wesseling, C.; Harra, T.; Aragón, A.; Grases, F.; Jarquin, E.R.; González, M.A.; et al. Heat Stress Nephropathy From Exercise-Induced Uric Acid Crystalluria: A Perspective on Mesoamerican Nephropathy. Am. J. Kidney Dis. 2016, 67, 20–30. [Google Scholar] [CrossRef]
- Laws, R.L.; Brooks, D.R.; Amador, J.J.; Weiner, D.E.; Kaufman, J.S.; Ramírez-Rubio, O.; Riefkohl, A.; Scammell, M.K.; López-Pilarte, D.; Sánchez, J.M.; et al. Changes in kidney function among Nicaraguan sugarcane workers. Int. J. Occup. Environ. Health 2015, 21, 241–250. [Google Scholar] [CrossRef]
- Wijkström, J.; Leiva, R.; Elinder, C.-G.; Leiva, S.; Trujillo, Z.; Trujillo, L.; Söderberg, M.; Hultenby, K.; Wernerson, A. Clinical and Pathological Characterization of Mesoamerican Nephropathy: A New Kidney Disease in Central America. Am. J. Kidney Dis. 2013, 62, 908–918. [Google Scholar] [CrossRef]
- Ryu, E.-S.; Kim, M.J.; Shin, H.-S.; Jang, Y.-H.; Choi, H.S.; Jo, I.; Johnson, R.J.; Kang, D.-H. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F471–F480. [Google Scholar] [CrossRef]
- Sorensen, C.J.; Butler-Dawson, J.; Dally, M.; Krisher, L.; Griffin, B.R.; Johnson, R.J.; Lemery, J.; Asensio, C.; Tenney, L.; Newman, L.S. Risk Factors and Mechanisms Underlying Cross-Shift Decline in Kidney Function in Guatemalan Sugarcane Workers. J. Occup. Environ. Med. 2019, 61, 239–250. [Google Scholar] [CrossRef]
- Wesseling, C.; Glaser, J.; Rodríguez-Guzmán, J.; Weiss, I.; Lucas, R.; Peraza, S.; da Silva, A.S.; Hansson, E.; Johnson, R.J.; Hogstedt, C.; et al. Chronic kidney disease of non-traditional origin in Mesoamerica: A disease primarily driven by occupational heat stress. Rev. Panam. Salud Publica—Pan Am. J. Public Health 2020, 44, e15. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.-Y.; Jia, P.-P.; Hu, H.; Liu, L.; Li, T.-Y.; Li, Y.-Z.; Pei, D.-S. Multi-omics reveal mechanisms underlying chronic kidney disease of unknown etiology (CKDu) pathogenesis using zebrafish. Environ. Pollut. 2023, 337, 122524. [Google Scholar] [CrossRef]
- Schaeffer, J.W.; Adgate, J.L.; Reynolds, S.J.; Butler-Dawson, J.; Krisher, L.; Dally, M.; Johnson, R.J.; James, K.A.; Jaramillo, D.; Newman, L.S. A Pilot Study to Assess Inhalation Exposures among Sugarcane Workers in Guatemala: Implications for Chronic Kidney Disease of Unknown Origin. Int. J. Environ. Res. Public Health 2020, 17, 5708. [Google Scholar] [CrossRef]
- Valcke, M.; Levasseur, M.-E.; da Silva, A.S.; Wesseling, C. Pesticide exposures and chronic kidney disease of unknown etiology: An epidemiologic review. Environ. Health 2017, 16, 49. [Google Scholar] [CrossRef]
- Butler-Dawson, J.; James, K.A.; Krisher, L.; Jaramillo, D.; Dally, M.; Neumann, N.; Pilloni, D.; Cruz, A.; Asensio, C.; Johnson, R.J.; et al. Environmental metal exposures and kidney function of Guatemalan sugarcane workers. J. Expo. Sci. Environ. Epidemiology 2021, 32, 461–471. [Google Scholar] [CrossRef]
- Rodrigues, P.; Furriol, J.; Bermejo, B.; Chaves, F.J.; Lluch, A.; Eroles, P. Identification of Candidate Polymorphisms on Stress Oxidative and DNA Damage Repair Genes Related with Clinical Outcome in Breast Cancer Patients. Int. J. Mol. Sci. 2012, 13, 16500–16513. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.; de Marco, G.; Furriol, J.; Mansego, M.L.; Pineda-Alonso, M.; Gonzalez-Neira, A.; Martin-Escudero, J.C.; Benitez, J.; Lluch, A.; Chaves, F.J.; et al. Oxidative stress in susceptibility to breast cancer: Study in Spanish population. BMC Cancer 2014, 14, 861. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; LaVallee, P.; Hoidal, J.R. Repressed Expression of the Human Xanthine Oxidoreductase Gene. E-Box and TATA-like Elements Restrict Ground State Transcriptional Activity. J. Biol. Chem. 2000, 275, 5918–5926. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Sasaki, T.; Ishikawa, M.; Hirasawa, N.; Hiratsuka, M. Functional Characterization of Genetic Polymorphisms Identified in the Promoter Region of the Xanthine Oxidase Gene. Drug Metab. Pharmacokinet. 2010, 25, 599–604. [Google Scholar] [CrossRef]
- Boban, M.; Kocic, G.; Radenkovic, S.; Pavlovic, R.; Cvetkovic, T.; Deljanin-Ilic, M.; Ilic, S.; Bobana, M.D.; Djindjic, B.; Stojanovic, D.; et al. Circulating purine compounds, uric acid, and xanthine oxidase/dehydrogenase relationship in essential hypertension and end stage renal disease. Ren. Fail. 2014, 36, 613–618. [Google Scholar] [CrossRef]
- Fujishiro, M.; Ishihara, H.; Ogawa, K.; Murase, T.; Nakamura, T.; Watanabe, K.; Sakoda, H.; Ono, H.; Yamamotoya, T.; Nakatsu, Y.; et al. Impact of Plasma Xanthine Oxidoreductase Activity on the Mechanisms of Distal Symmetric Polyneuropathy Development in Patients with Type 2 Diabetes. Biomedicines 2021, 9, 1052. [Google Scholar] [CrossRef]
- Okuyama, T.; Shirakawa, J.; Nakamura, T.; Murase, T.; Miyashita, D.; Inoue, R.; Kyohara, M.; Togashi, Y.; Terauchi, Y. Association of the plasma xanthine oxidoreductase activity with the metabolic parameters and vascular complications in patients with type 2 diabetes. Sci. Rep. 2021, 11, 3768. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.-D.; Wang, Y.-Z.; Zou, C.; She, X.-P.; Zheng, Z. The role of uric acid in the pathogenesis of diabetic retinopathy based on Notch pathway. Biochem. Biophys. Res. Commun. 2018, 503, 921–929. [Google Scholar] [CrossRef]
- Stamp, L.K.; Chapman, P.T. Allopurinol hypersensitivity: Pathogenesis and prevention. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101501. [Google Scholar] [CrossRef] [PubMed]
- FDA. FDA Adds Boxed Warning for Increased Risk of Death with Gout Medicine Uloric (Febuxostat). Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-adds-boxed-warning-increased-risk-death-gout-medicine-uloric-febuxostat (accessed on 2 June 2024).
- Rullo, R.; Cerchia, C.; Nasso, R.; Romanelli, V.; De Vendittis, E.; Masullo, M.; Lavecchia, A. Novel Reversible Inhibitors of Xanthine Oxidase Targeting the Active Site of the Enzyme. Antioxidants 2023, 12, 825. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, C.; Zhang, L.; Li, J. Identification of novel dual inhibitors targeting XOR and URAT1 via multiple virtual screening methods. J. Mol. Struct. 2022, 1256, 132567. [Google Scholar] [CrossRef]
- Singh, A.; Singh, K.; Sharma, A.; Kaur, K.; Chadha, R.; Bedi, P.M.S. Past, present and future of xanthine oxidase inhibitors: Design strategies, structural and pharmacological insights, patents and clinical trials. RSC Med. Chem. 2023, 14, 2155–2191. [Google Scholar] [CrossRef]
- Sundy, J.S.; Baraf, H.S.B.; Yood, R.A.; Edwards, N.L.; Gutierrez-Urena, S.R.; Treadwell, E.L.; Vázquez-Mellado, J.; White, W.B.; Lipsky, P.E.; Horowitz, Z.; et al. Efficacy and Tolerability of Pegloticase for the Treatment of Chronic Gout in Patients Refractory to Conventional Treatment: Two Randomized Controlled Trials. JAMA 2011, 306, 711–720. [Google Scholar] [CrossRef]
- Li, Z.; Liu, Q.; Mao, H.; Li, Z.; Dong, X.; Liu, Y.; Lin, J.; Chen, W.; Wang, H.; Johnson, R.J.; et al. Gender Difference in the Association of Hyperuricemia with Chronic Kidney Disease in Southern China. Kidney Blood Press. Res. 2012, 36, 98–106. [Google Scholar] [CrossRef]
- Bolognesi, A.; Bortolotti, M.; Battelli, M.G.; Polito, L. Gender Influence on XOR Activities and Related Pathologies: A Narrative Review. Antioxidants 2024, 13, 211. [Google Scholar] [CrossRef]
- Furuhashi, M.; Higashiura, Y.; Koyama, M.; Tanaka, M.; Murase, T.; Nakamura, T.; Akari, S.; Sakai, A.; Mori, K.; Ohnishi, H.; et al. Independent association of plasma xanthine oxidoreductase activity with hypertension in nondiabetic subjects not using medication. Hypertens. Res. 2021, 44, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Fariha, K.A.; Barman, Z.; Mou, A.D.; Miah, R.; Habib, A.; Tuba, H.R.; Ali, N. Assessment of the relationship between serum xanthine oxidase levels and type 2 diabetes: A cross-sectional study. Sci. Rep. 2022, 12, 20816. [Google Scholar] [CrossRef] [PubMed]


| Pre-Clinical Studies | ||||
| Disease | Model(s) | Study Design | Findings | Reference |
| DKD | Akita mouse (genetic model of type 1 diabetes mellitus) | Akita mice were treated with topiroxostat, a non-purine inhibitor of XOR for 4 weeks, and markers of XOR activity and diabetes-induced glomerular injury were measured. | Compared to non-diabetic C57BL/6J controls, vehicle-treated Akita mice developed progressive diabetic glomerular injury, which was associated with increased XOR activity and oxidative stress. Pharmacological inhibition of XOR activity with topiroxostat markedly decreased renal ROS and progressive DKD in Akita mice. | [7] |
| DKD | db/db mouse (model of obesity and type 2 diabetes mellitus) | db/db mice were treated with various doses of XOR inhibitors (topiroxostat and febuxostat) for 4 weeks, and markers of systemic and renal XOR activity and kidney injury were assessed. | Compared to control db/m littermates, control db/db mice exhibited increased systemic and intra-renal XOR activity, and this was associated with proteinuria. Treatment with topiroxostat significantly decreased XOR activity and oxidative stress in db/db mice. More importantly, topiroxostat rescued proteinuria in db/db mice in a dose-dependent fashion. While febuxostat significantly inhibited XOR activity in db/db mice, it did not significantly decrease proteinuria. | [122] |
| DKD | Streptozotocin-induced diabetic Sprague Dawley rat (chemically induced model of type 1 diabetes mellitus) | Diabetic Sprague Dawley rats were treated with febuxostat for 7 weeks, and markers of XOR activity and renal oxidative stress, renal macrophage infiltration, and proteinuria were measured. | In comparison to non-diabetic controls, diabetic Sprague Dawley rats exhibited increased XOR activity, renal inflammation, and renal injury, which were attenuated by treatment with febuxostat. | [113] |
| DKD | Akita, HFD-fed mice, STZ-induced diabetic B6-TG mice, and recombinant inbred BXD mice | This study employed multiomic and pharmacological approaches using the XOR inhibitors, allopurinol and febuxostat, to study the role of a XOR risk variant in the development of DKD | This study discovered that this identified variant is in the XOR promoter region and is a transcription factor binding site for C/EBPβ. Importantly, findings from this study demonstrated that the identified variants confer susceptibility to DKD. | [5] |
| CKD Non-DKD | 5/6th nephrectomy Wistar rats with oxonic acid-induced hyperuricemia | 5/6th nephrectomized Wistar rats were made hyperuricemic with oxonic acid and treated with febuxostat for 4 weeks. Plasma uric acid, renal hemodynamics, proteinuria, and histopathologic evaluation were carried out in response to treatment with febuxostat. | 5/6th nephrectomized rats treated with oxonic acid displayed hyperuricemia, impaired renal hemodynamics and proteinuria versus vehicle-treated 5/6 nephrectomy rats. The administration of febuxostat decreased hyperuricemia, improved renal function, and ameliorated proteinuria in 5/6th nephrectomized rats that received oxonic acid. | [123] |
| Obstructive nephropathy | Rat model of unilateral urethral obstructive (UUO) nephropathy | Sprague Dawley rats were pretreated with febuxostat before UUO surgery and then daily after surgery for up to 14 days. Rats were sacrificed on days 1, 4, and 14 after surgery. Markers of intrarenal and systemic XOR activity, oxidative stress, renal inflammation, and tubulointerstitial fibrosis were assessed in response to treatment with febuxostat. | Vehicle-treated UUO rats displayed increased XOR activity, renal inflammation, and tubulointerstitial fibrosis in comparison to sham rats. Pharmacological blockade of XOR with febuxostat rescued renal oxidative stress and inflammation and decreased tubulointerstitial fibrosis in UUO rats. | [124] |
| Hypertension-induced CKD | Dahl salt-sensitive rat (genetic model of salt-sensitive hypertension and renal injury) | Dahl salt-sensitive (SS) rats fed a high salt (8% NaCl) diet and treated with febuxostat daily for 4 or 8 weeks. XOR expression and XOR activity, oxidative stress, and progressive renal disease were measured in febuxostat-treated SS rats and their controls. | Compared to normal salt (0.6% NaCl) diet-fed SS rats, high salt-diet-fed SS rats exhibited increased renal XOR expression and XOR activity, which was associated with oxidative stress and the progression of renal disease in SS rats. Treatment with febuxostat decreased XOR expression and activity and attenuated the progression of renal disease in high salt diet-fed SS rats. | [125] |
| IgA Nephropathy | gddY mouse (rodent model of IgA nephropathy) | gddY mice either served as controls, or were treated with febuxostat for 9 weeks, during which XOR activity, inflammatory signaling, and the progression of renal disease were investigated. | Control gddY mice demonstrated increased XOR activity, renal inflammation and fibrosis and renal disease versus BALB/c controls. XOR blockade with febuxostat was associated with decreased renal inflammation and renal disease in gddY mice. | [126] |
| Hyperuricemic Nephropathy | Adenine + potassium oxonate-induced hyperuricemic Sprague Dawley rats. | Control or febuxostat-treated hyperuricemic rats were treated for 5 weeks, and markers of XOR activity, endoplasmic reticulum stress, apoptosis, and renal disease were assessed. | Treatment with febuxostat decreased XOR activity and renal urate deposition versus controls. Importantly, treatment with febuxostat ameliorated renal disease in hyperuricemic rats. | [127] |
| Clinical Studies | ||||
| Disease | Study Population | Study Design | Outcomes | Reference |
| DKD | Patients with diabetes, concomitant with hyperuricemia | Randomized, double-blinded, placebo-controlled, and parallel study of patients with DKD and hyperuricemia. Recruited patients received either placebo or topiroxostat for 28 weeks. uACR was primary endpoint, while eGFR and serum uric acid levels were secondary endpoints. | Topiroxostat decreased serum uric acid, while preventing renal function decline in the study population. However, topiroxostat did not have a significant effect on uACR in comparison to placebo. | [119] |
| DKD | DKD patients without gout | Cross sectional study of type 2 diabetes patients without a history of gout or allopurinol use. Serum creatinine and uric acid, HbA1C, and 24-hr urine protein excretion were measured. | Serum uric acid showed a positive correlation with proteinuria in diabetic patients. | [128] |
| DKD | Patients with diabetes and asymptomatic hyperuricemia | Randomized, parallel-controlled trial that investigated the effects of allopurinol on renal function in diabetic patients and hyperuricemia. | Effective control of serum uric acid with allopurinol was associated with decreased albumin excretion and the prevention on renal function decline in the study population. | [129] |
| Hyperuricemic CKD | Hyperuricemic patients with stage 3 CKD | Randomized, open label, parallel-group trial to investigate the renoprotective effects of febuxostat on hyperuricemic patients with stage 3 CKD. | Treatment with febuxostat decreased serum uric acid and urinary levels of albumin and β2-macroglobulin in hyperuricemic CKD patients versus the control group. However, febuxostat did not influence markers of renal function (serum creatinine and eGFR). | [130] |
| Hyperuricemic CKD | Hyperuricemic stage 3 CKD patients, with or without gout | This was a 22 week double-blind, multicentric, randomized trials that studied the safety and efficacy of topiroxostat in hyperuricemic patients with stage 3 CKD. | Topiroxostat decreased serum urate and uACR in hyperuricemic stage 3 CKD patients versus placebo group. The observed effect of topiroxostat on renal function (i.e., eGFR) was not statistically significant. Adverse events were generally comparable between topiroxostat and placebo groups. | [131] |
| Hyperuricemic CKD | Hyperuricemic patients with stage 3–4 CKD | Randomized controlled trial that examined the efficacy of febuxostat on markers of endothelial dysfunction and renal function in CKD patients with asymptomatic hyperuricemia. | Compared to the control group, febuxostat reduced serum uric acid and preserved renal function. However, febuxostat did not significantly decrease albuminuria or markers of endothelial dysfunction versus controls. | [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korsmo, H.W.; Ekperikpe, U.S.; Daehn, I.S. Emerging Roles of Xanthine Oxidoreductase in Chronic Kidney Disease. Antioxidants 2024, 13, 712. https://doi.org/10.3390/antiox13060712
Korsmo HW, Ekperikpe US, Daehn IS. Emerging Roles of Xanthine Oxidoreductase in Chronic Kidney Disease. Antioxidants. 2024; 13(6):712. https://doi.org/10.3390/antiox13060712
Chicago/Turabian StyleKorsmo, Hunter W., Ubong S. Ekperikpe, and Ilse S. Daehn. 2024. "Emerging Roles of Xanthine Oxidoreductase in Chronic Kidney Disease" Antioxidants 13, no. 6: 712. https://doi.org/10.3390/antiox13060712
APA StyleKorsmo, H. W., Ekperikpe, U. S., & Daehn, I. S. (2024). Emerging Roles of Xanthine Oxidoreductase in Chronic Kidney Disease. Antioxidants, 13(6), 712. https://doi.org/10.3390/antiox13060712