
The Impairment of Endothelial Autophagy Accelerates Renal Senescence by Ferroptosis and NLRP3 Inflammasome Signaling Pathways with the Disruption of Endothelial Barrier

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Immunohistochemical Immunofluorescence and Analysis
2.3. Western Blot Analysis
2.4. Antibodies
2.5. Electron Microscopic Analysis
2.6. Urine Analysis
2.7. Kidney Organoid Differentiation and Transwell Co-Culture System of ECs and Kidney Organoids
2.8. Measurement of Transepithelial Electrical Resistance
2.9. Transwell Permeability Assay
2.10. Iron Assay
2.11. Measurement of Reactive Oxygen Species
2.12. Three-Dimensional Cell Viability Assay and Live/Dead Cell Staining
2.13. Real-Time Quantitative PCR Analysis
2.14. Statistics
3. Results
3.1. EC-Specific Atg7 Deletion Accelerated Aging-Related Structural Changes and Fibrosis in Kidneys
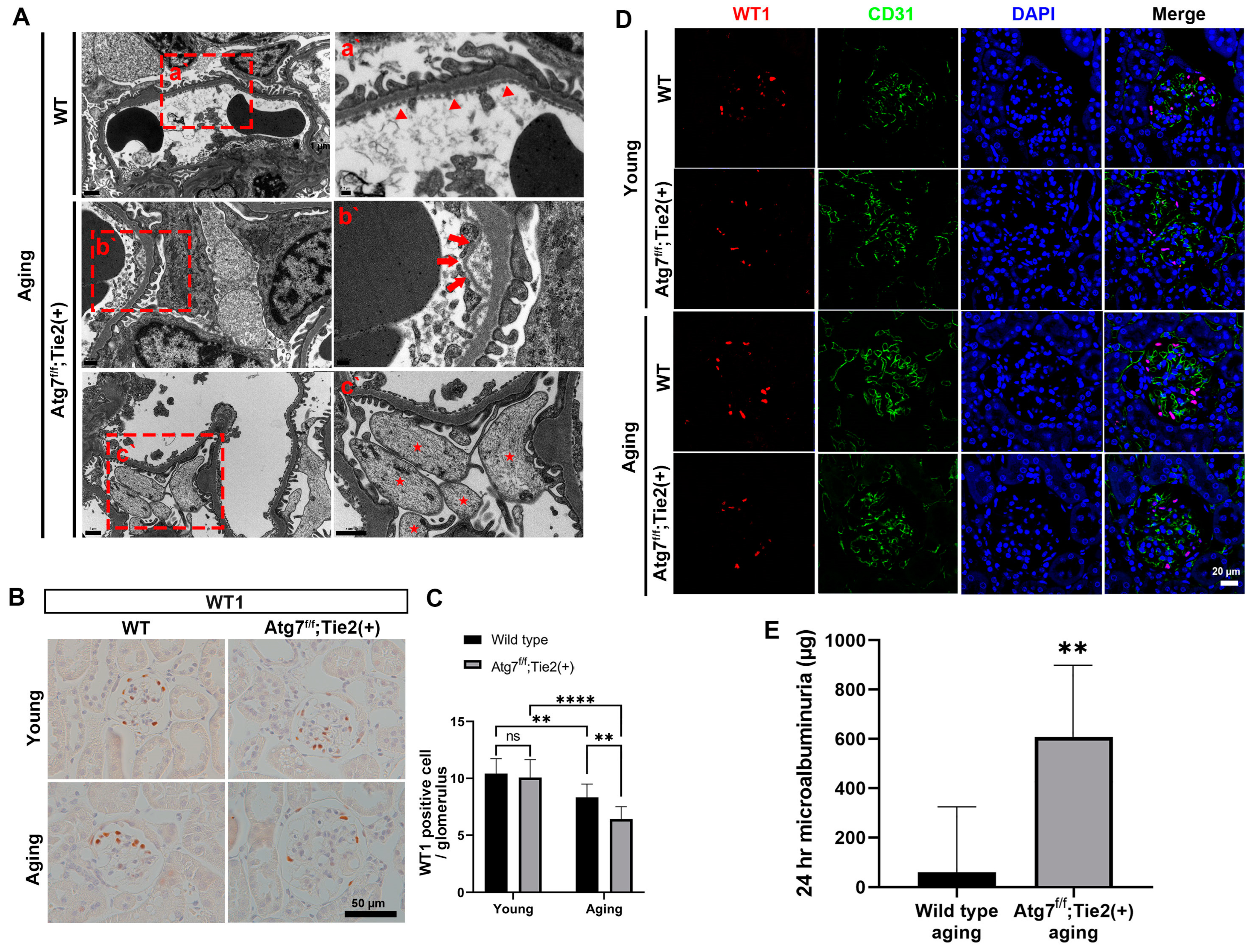
3.2. EC-Specific Atg7 Deletion in Aging Mice Induced the Detachment of EC with the Disruption of GBM Assembly and Increased Podocyte Loss Resulting in Microalbuminuria
3.3. EC-Specific Atg7 Deletion Increased Ferritin Accumulation in Renal Aging
3.4. Liproxstatin-1 Attenuated Renal Senescence from Endothelial Autophagy Deficiency by Regulating the Ferroptosis Pathway In Vivo
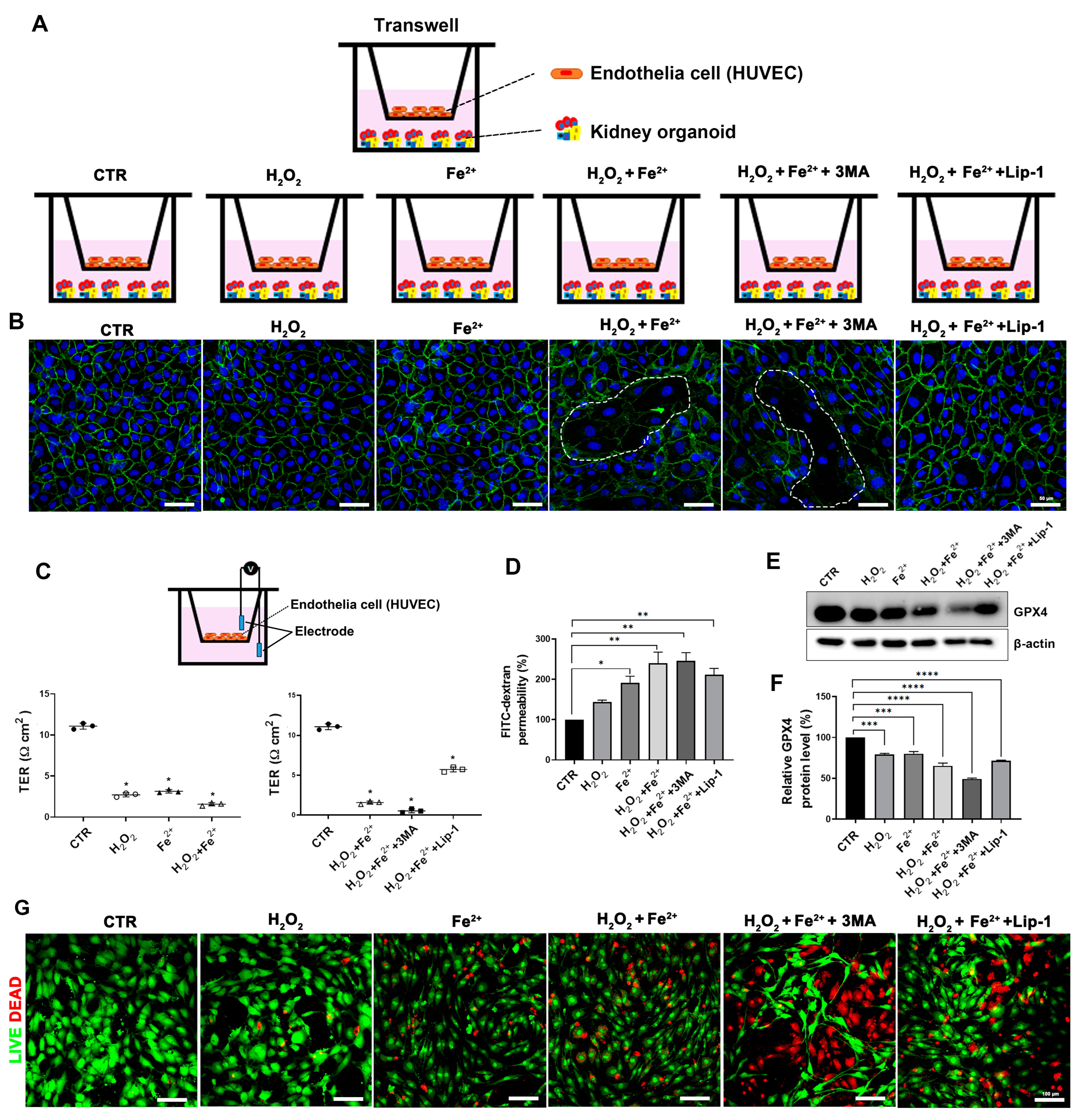
3.5. Disruption of the Endothelial Barrier by Autophagy Inhibition with Iron Loading and Oxidative Stress Accelerated the Ferroptotic Cell Death in the Human Kidney Organoid In Vitro Model
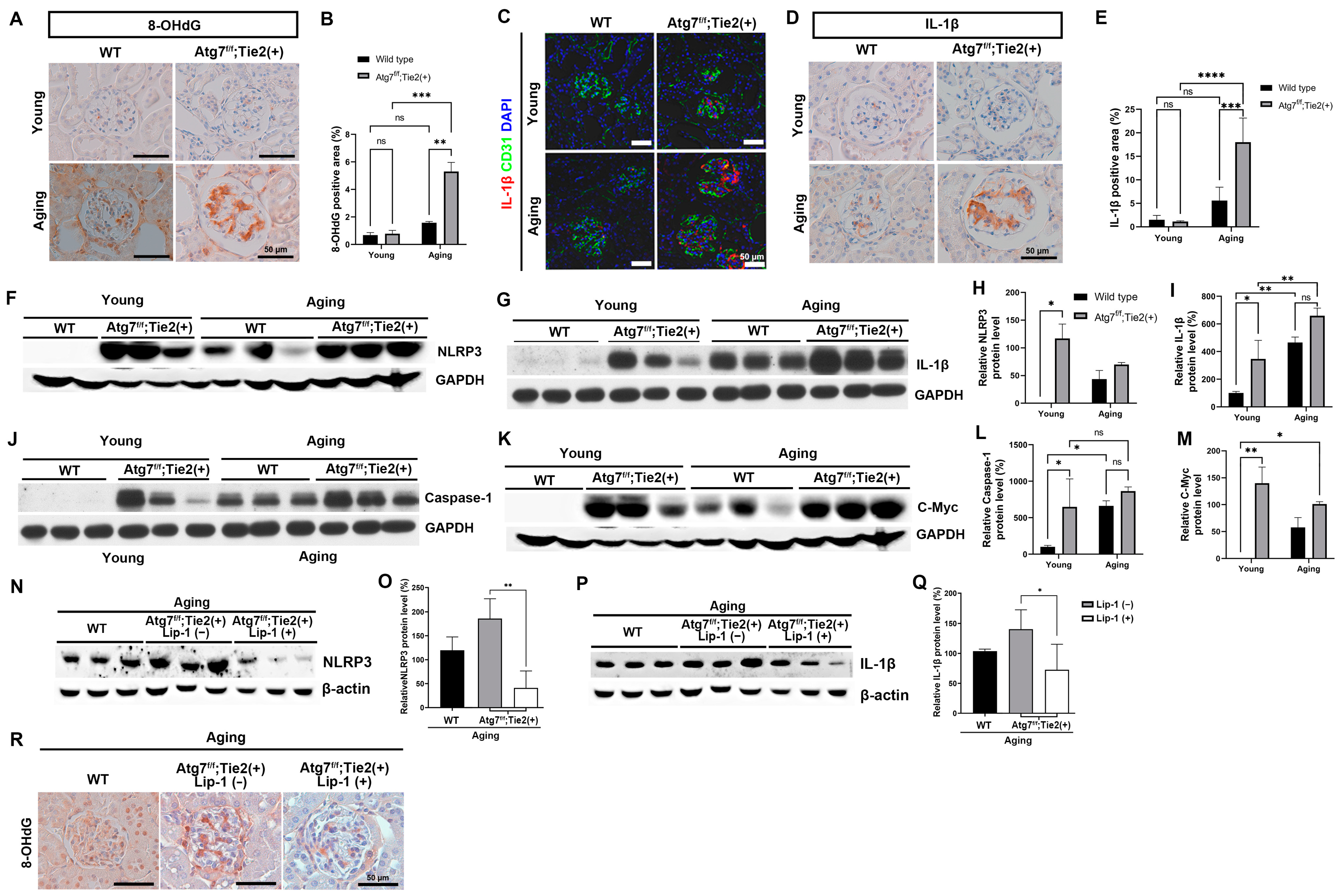
3.6. EC-Specific Atg7 Deletion Increased Oxidative Stress and Upregulated the NLRP3 Inflammasome Signaling Pathway in Renal Senescence
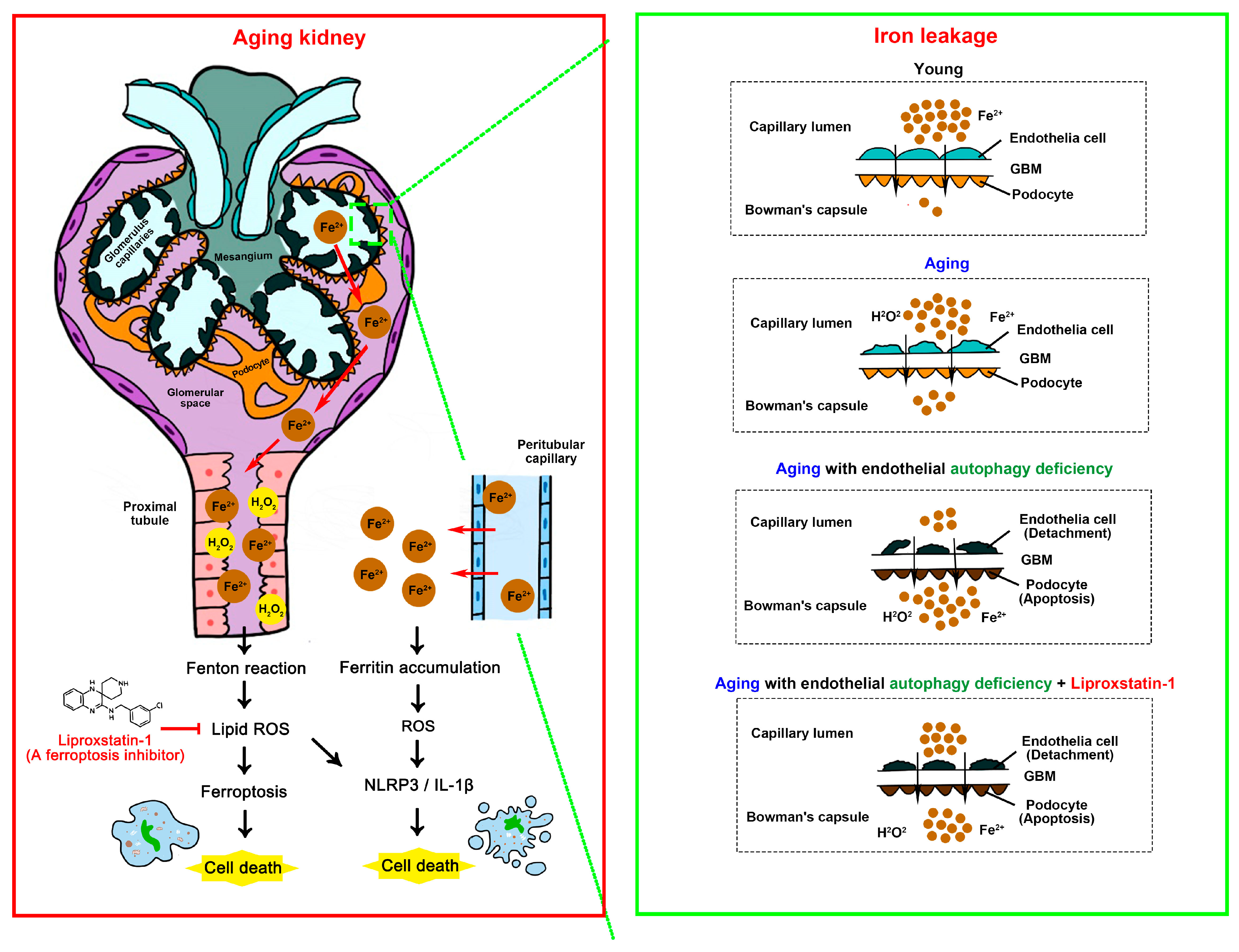
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolignano, D.; Mattace-Raso, F.; Sijbrands, E.J.; Zoccali, C. The aging kidney revisited: A systematic review. Ageing Res. Rev. 2014, 14, 65–80. [Google Scholar] [CrossRef]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef]
- Hong, Y.A.; Ban, T.H.; Kang, C.-Y.; Hwang, S.D.; Choi, S.R.; Lee, H.; Jung, H.-Y.; Kim, K.; Kwon, Y.E.; Kim, S.H. Trends in epidemiologic characteristics of end-stage renal disease from 2019 Korean Renal Data System (KORDS). Kidney Res. Clin. Pract. 2021, 40, 52. [Google Scholar] [CrossRef] [PubMed]
- Merchant, R.A.; Vathsala, A. Healthy aging and chronic kidney disease. Kidney Res. Clin. Pract. 2022, 41, 644. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jeong, S.A.; Kim, K.M.; Hwang, S.D.; Choi, S.R.; Lee, H.; Kim, J.H.; Kim, S.H.; Kim, T.H.; Koo, H.-S. Trends in clinical outcomes of older hemodialysis patients: Data from the 2023 Korean Renal Data System (KORDS). Kidney Res. Clin. Pract. 2024, 43, 263. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.-y.; Matsui, I.; Kitamura, H. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef]
- Minami, S.; Yamamoto, T.; Yamamoto-Imoto, H.; Isaka, Y.; Hamasaki, M. Autophagy and kidney aging. Prog. Biophys. Mol. Biol. 2023, 179, 10–15. [Google Scholar] [CrossRef]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Armelloni, S.; Mattinzoli, D.; Ikehata, M.; Chatziantoniou, C.; Alfieri, C.; Molinari, P.; Chadjichristos, C.E.; Malvica, S.; Castellano, G. Crosstalk mechanisms between glomerular endothelial cells and podocytes in renal diseases and kidney transplantation. Kidney Res. Clin. Pract. 2024, 43, 47. [Google Scholar] [CrossRef]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, J.; Namba, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Yamamoto, T.; Minami, S.; Sakai, S.; Fujimura, R.; Kaimori, J.-y. Antioxidant role of autophagy in maintaining the integrity of glomerular capillaries. Autophagy 2018, 14, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef]
- Schnaper, H.W.; Hayashida, T.; Hubchak, S.C.; Poncelet, A.-C. TGF-β signal transduction and mesangial cell fibrogenesis. Am. J. Physiol.-Ren. Physiol. 2003, 284, F243–F252. [Google Scholar] [CrossRef]
- Loeffler, I.; Wolf, G. Transforming growth factor-β and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i37–i45. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, J.Y.; Lee, S.Y.; Jeong, I.; Park, S.-Y.; Kim, J.W.; Nam, S.A.; Kim, H.W.; Kim, Y.K.; Lee, S. Deep learning predicts the differentiation of kidney organoids derived from human induced pluripotent stem cells. Kidney Res. Clin. Pract. 2023, 42, 75. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Denic, A.; Glassock, R.J.; Rule, A.D. Structural and functional changes with the aging kidney. Adv. Chronic Kidney Dis. 2016, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, N.; Shimazu, Y.; Kikuchi, K.; Nagata, M. Age-related renal microvascular changes: Evaluation by three-dimensional digital imaging of the human renal microcirculation using virtual microscopy. Int. J. Mol. Sci. 2016, 17, 1831. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.E.; Anderson, S.; Gordon, K.L.; Oyama, T.T.; Shankland, S.J.; Johnson, R.J. Tubulointerstitial disease in aging: Evidence for underlying peritubular capillary damage, a potential role for renal ischemia. J. Am. Soc. Nephrol. 1998, 9, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.J.; Saxena, R.; Liu, Z.; Vaziri, N.; Silva, F.G. Renal senescence in 2008: Progress and challenges. Int. Urol. Nephrol. 2008, 40, 823–839. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; ll Kim, S.; Lee, S.-Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846. [Google Scholar] [CrossRef]
- Dai, C.; Liu, Y. Hepatocyte growth factor antagonizes the profibrotic action of TGF-β1 in mesangial cells by stabilizing Smad transcriptional corepressor TGIF. J. Am. Soc. Nephrol. 2004, 15, 1402–1412. [Google Scholar] [CrossRef]
- Tsuchida, K.-I.; Zhu, Y.; Siva, S.; Dunn, S.R.; Sharma, K. Role of Smad4 on TGF-β–induced extracellular matrix stimulation in mesangial cells. Kidney Int. 2003, 63, 2000–2009. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, Q.; Pi, L.; Huang, B. Role of angiotensin II and JAK2 signal pathway in transdifferentation of renal tubular cells in mice after acute ischemic followed by reperfusion. Zhonghua Bing Li Xue Za Zhi = Chin. J. Pathol. 2009, 38, 466–471. [Google Scholar]
- Dai, C.; Yang, J.; Bastacky, S.; Xia, J.; Li, Y.; Liu, Y. Intravenous administration of hepatocyte growth factor gene ameliorates diabetic nephropathy in mice. J. Am. Soc. Nephrol. 2004, 15, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, F.S.; Advani, A. Endothelial-podocyte crosstalk: The missing link between endothelial dysfunction and albuminuria in diabetes. Diabetes 2013, 62, 3647–3655. [Google Scholar] [CrossRef]
- Glassock, R.J. Is the presence of microalbuminuria a relevant marker of kidney disease? Curr. Hypertens. Rep. 2010, 12, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Schmidt, I.M.; Claudel, S.; Palsson, R.; Waikar, S.S.; Srivastava, A. Association of Albuminuria with Chronic Kidney Disease Progression in persons with chronic kidney disease and Normoalbuminuria: A cohort study. Ann. Intern. Med. 2024, 177, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of ferroptosis in aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife 2020, 9, e56580. [Google Scholar] [CrossRef]
- Harris, J.; Lang, T.; Thomas, J.P.; Sukkar, M.B.; Nabar, N.R.; Kehrl, J.H. Autophagy and inflammasomes. Mol. Immunol. 2017, 86, 10–15. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732. [Google Scholar] [CrossRef]
- Tóth, M.L.; Sigmond, T.; Borsos, É.; Barna, J.; Erdélyi, P.; Takács-Vellai, K.; Orosz, L.; Kovács, A.L.; Csikós, G.; Sass, M. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef]
- Csiszar, A.; Toth, J.; Peti-Peterdi, J.; Ungvari, Z. The aging kidney: Role of endothelial oxidative stress and inflammation. Acta Physiol. Hung. 2007, 94, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Y.; Cao, L.; Lu, S.; Zhang, S.; Yang, R.; Wang, Y.; Zhang, N.; Yu, Y.; Wang, X. Insights on E1-like enzyme ATG7: Functional regulation and relationships with aging-related diseases. Commun. Biol. 2024, 7, 382. [Google Scholar] [CrossRef]
- Collier, J.J.; Suomi, F.; Oláhová, M.; McWilliams, T.G.; Taylor, R.W. Emerging roles of ATG7 in human health and disease. EMBO Mol. Med. 2021, 13, e14824. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.R.; Nomellini, V.; Faunce, D.E.; Kovacs, E.J. Innate immunity and aging. Exp. Gerontol. 2008, 43, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Guissart, C.; Oláhová, M.; Sasorith, S.; Piron-Prunier, F.; Suomi, F.; Zhang, D.; Martinez-Lopez, N.; Leboucq, N.; Bahr, A. Developmental consequences of defective ATG7-mediated autophagy in humans. N. Engl. J. Med. 2021, 384, 2406–2417. [Google Scholar] [CrossRef]
- Xiong, J. Atg7 in development and disease: Panacea or Pandora’s Box? Protein Cell 2015, 6, 722–734. [Google Scholar] [CrossRef]
- Kim, H.O.; Kim, H.-S.; Youn, J.-C.; Shin, E.-C.; Park, S. Serum cytokine profiles in healthy young and elderly population assessed using multiplexed bead-based immunoassays. J. Transl. Med. 2011, 9, 113. [Google Scholar] [CrossRef]
- Ryu, M.; Migliorini, A.; Miosge, N.; Gross, O.; Shankland, S.; Brinkkoetter, P.T.; Hagmann, H.; Romagnani, P.; Liapis, H.; Anders, H.J. Plasma leakage through glomerular basement membrane ruptures triggers the proliferation of parietal epithelial cells and crescent formation in non-inflammatory glomerular injury. J. Pathol. 2012, 228, 482–494. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.W.; Nam, S.A.; Koh, E.-S.; Kim, H.W.; Kim, S.; Woo, J.J.; Kim, Y.K. The Impairment of Endothelial Autophagy Accelerates Renal Senescence by Ferroptosis and NLRP3 Inflammasome Signaling Pathways with the Disruption of Endothelial Barrier. Antioxidants 2024, 13, 886. https://doi.org/10.3390/antiox13080886
Kim JW, Nam SA, Koh E-S, Kim HW, Kim S, Woo JJ, Kim YK. The Impairment of Endothelial Autophagy Accelerates Renal Senescence by Ferroptosis and NLRP3 Inflammasome Signaling Pathways with the Disruption of Endothelial Barrier. Antioxidants. 2024; 13(8):886. https://doi.org/10.3390/antiox13080886
Chicago/Turabian StyleKim, Jin Won, Sun Ah Nam, Eun-Sil Koh, Hyung Wook Kim, Sua Kim, Jin Ju Woo, and Yong Kyun Kim. 2024. "The Impairment of Endothelial Autophagy Accelerates Renal Senescence by Ferroptosis and NLRP3 Inflammasome Signaling Pathways with the Disruption of Endothelial Barrier" Antioxidants 13, no. 8: 886. https://doi.org/10.3390/antiox13080886