
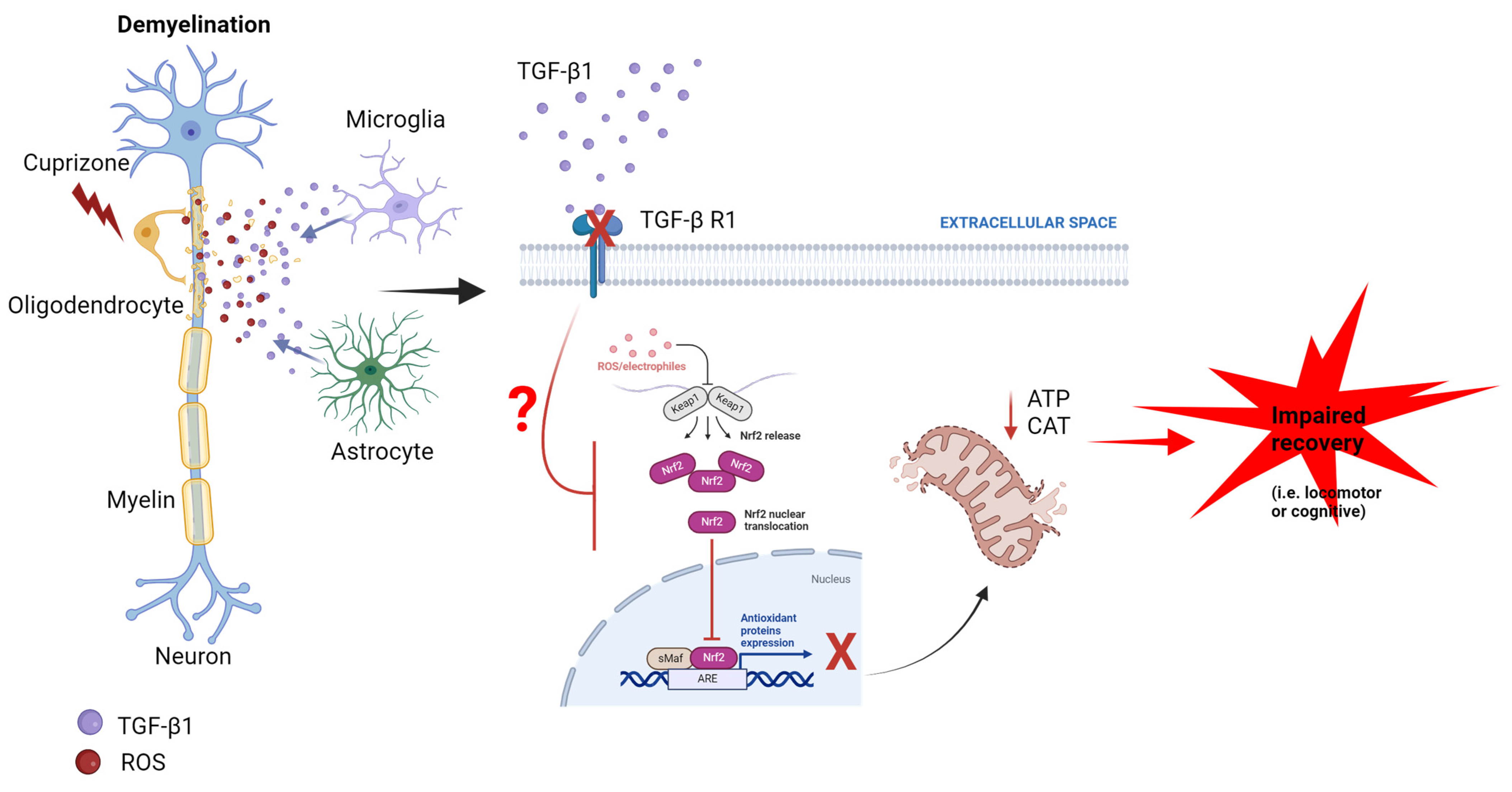
Evidence for TGF-β1/Nrf2 Signaling Crosstalk in a Cuprizone Model of Multiple Sclerosis

 , ,
, ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Reagents and Antibodies
2.2. Animals
2.3. Experimental Model
2.3.1. Cuprizone Administration
2.3.2. TGF-β1R-Blocker Administration
2.4. Immunostainings
2.5. Western Blot Analysis
2.6. ATP Bioluminescence Assay
2.7. Locomotor Rotarod Test
2.8. Evaluation of the Cognitive Function
2.9. Statistical Analysis
3. Results
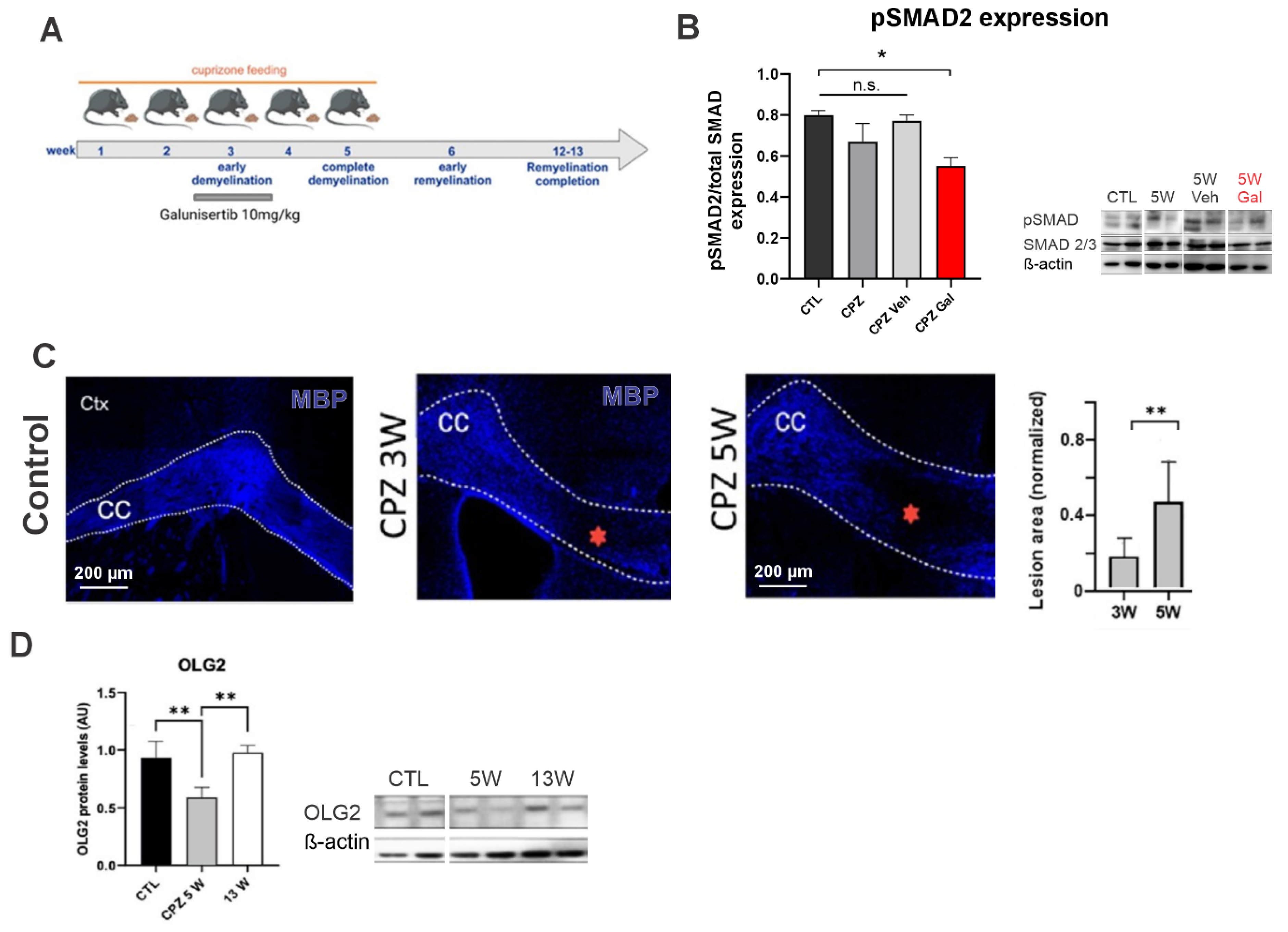
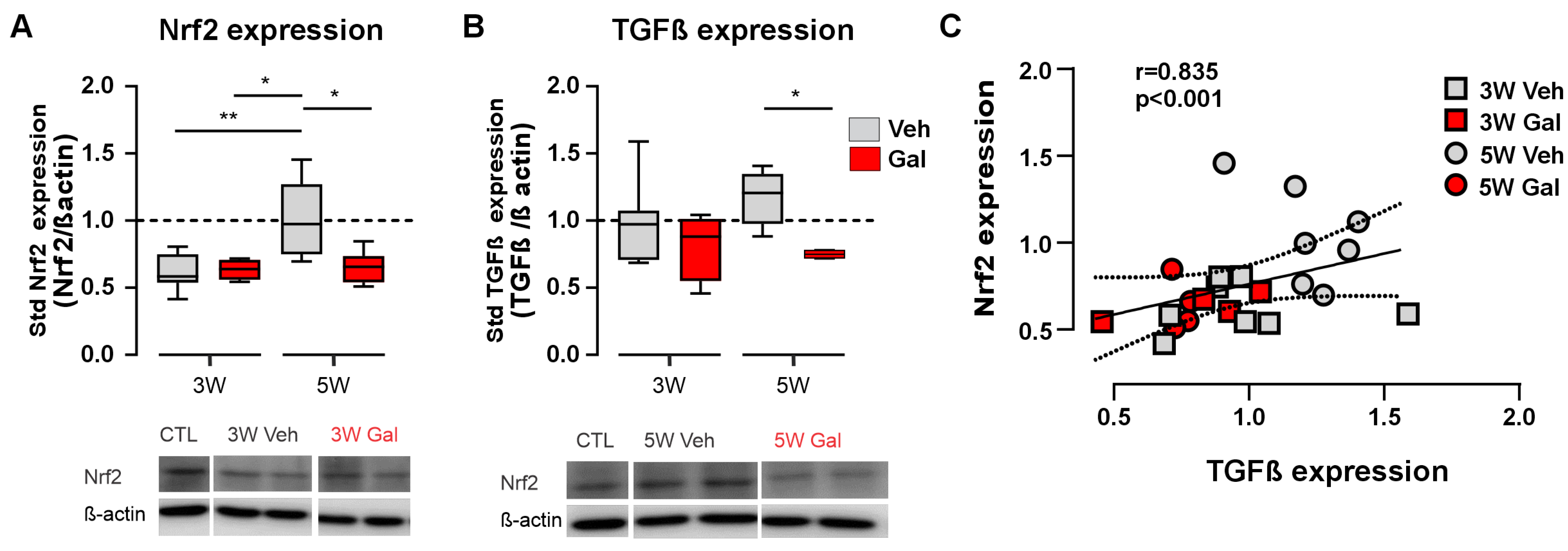
3.1. Blockage of the TGF-β1 Receptor Reduces Both Nrf2 and TGF-β1 Protein Levels in the CPZ Model of MS
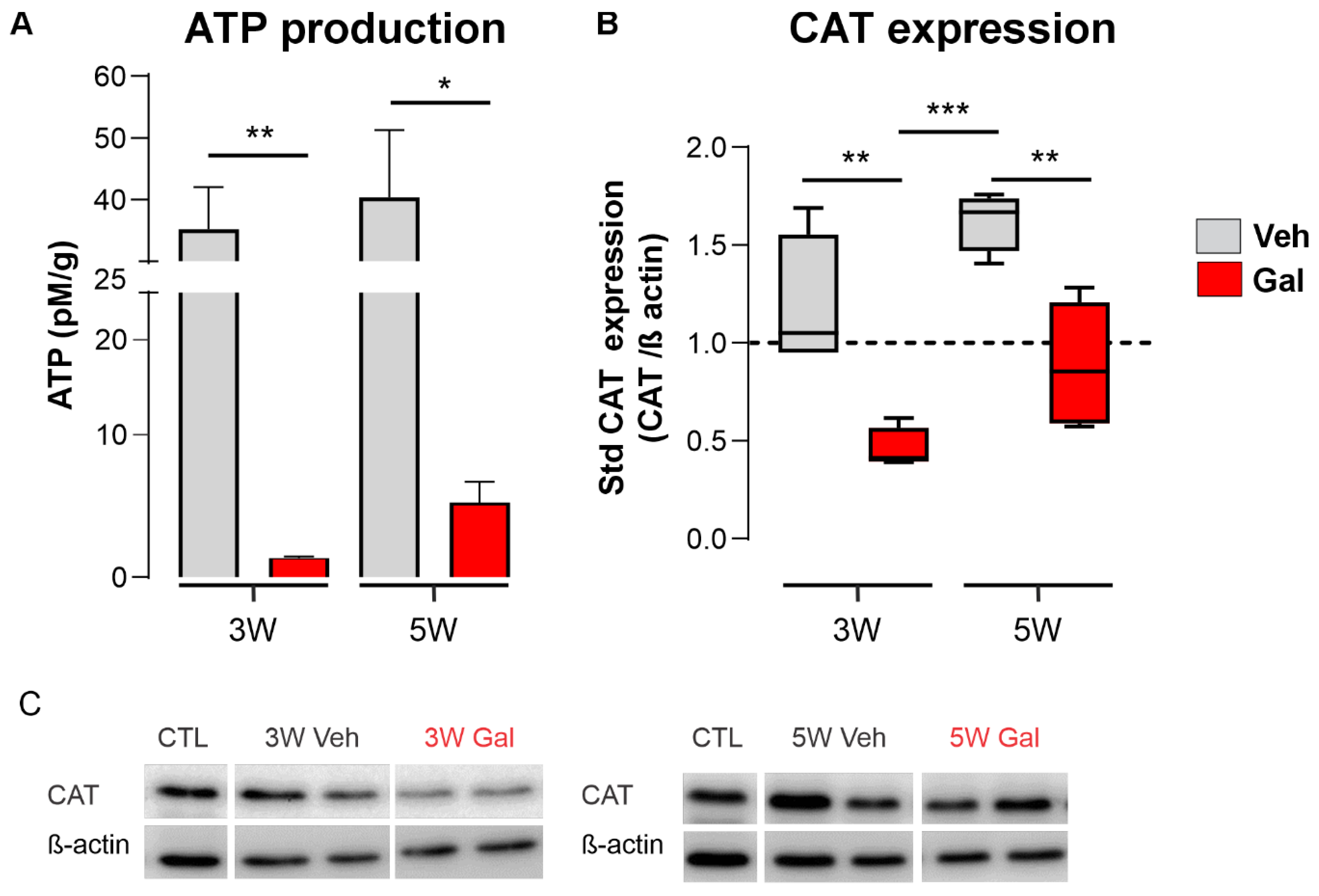
3.2. TGF-β1 Receptor Blocking Reduces ATP Production and Catalase Expression during Demyelination
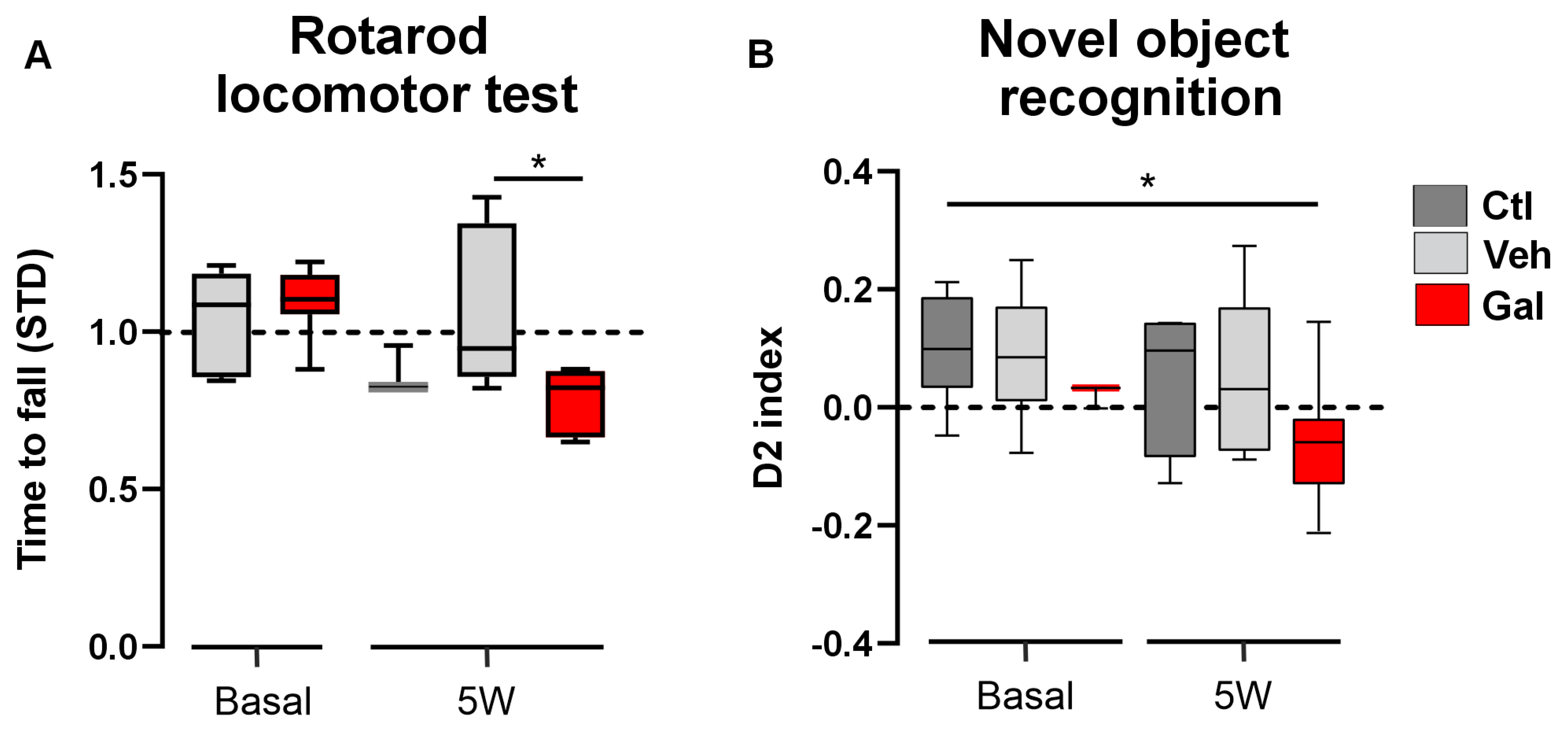
3.3. TGF-β1 Protects Animals from Locomotor and Cognition Impairment Induced by Cuprizone
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, R.; Giovannoni, G. Multiple Sclerosis—A Review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, G.; Thompson, A.; Berg, J.; Gannedahl, M.; Eriksson, J. New Insights into the Burden and Costs of Multiple Sclerosis in Europe. Mult. Scler. 2017, 23, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P.K. Multiple Sclerosis in Pregnancy. Continuum 2014, 20, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Velázquez, M.; Núñez, L.; Skromne, E.; Árcega, R.; Barroso, N.; Alberto, C.; Felipe, G.; Victor, G.; Rafael, J.; et al. Consenso Mexicano Para La Esclerosis Múltiple. Guía Diagnóstica y Terapéutica. Rev. Mex. Neuroci. 2007, 8, 155–162. [Google Scholar]
- Popescu, B.F.G.; Lucchinetti, C.F. Pathology of Demyelinating Diseases. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 185–217. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Stys, P.K. Virtual Hypoxia and Chronic Necrosis of Demyelinated Axons in Multiple Sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS Myelin—From Mechanisms to Experimental Medicines. Nat. Rev. Neurosci. 2017, 18, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Codarri, L.; Gyülvészi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L. RORgammat Drives Production of the Cytokine GM-CSF in Helper T Cells, Which Is Essential for the Effector Phase of Autoimmune Neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef]
- Hamaguchi, M.; Muramatsu, R.; Fujimura, H.; Mochizuki, H.; Kataoka, H.; Yamashita, T. Circulating Transforming Growth Factor-Β1 Facilitates Remyelination in the Adult Central Nervous System. Elife 2019, 8, e41869. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Dietrich, H.K.; Axtell, R.C.; Williams, A.M.; Egusquiza, R.; Wai, K.M.; Koshy, A.A.; Buckwalter, M.S. Astrocytic TGF-β Signaling Limits Inflammation and Reduces Neuronal Damage during Central Nervous System Toxoplasma Infection. J. Immunol. 2014, 193, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-Β1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. Neurobiol. 2019, 56, 4653–4679. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The Immunology of Stroke: From Mechanisms to Translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Tgf-Beta Pathway as a Potential Target in Neurodegeneration and Alzheimer’s. Curr. Alzheimer Res. 2006, 3, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, E.N. Role of Macrophages/Microglia in Multiple Sclerosis and Experimental Allergic Encephalomyelitis. J. Mol. Med. 1997, 75, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Flauzino, T.; Alfieri, D.F.; Pereira, W.L.d.C.J.; Oliveira, S.R.; Kallaur, A.P.; Lozovoy, M.A.B.; Kaimen-Maciel, D.R.; de Oliveira, K.B.; Simão, A.N.C.; Reiche, E.M.V. The Rs3761548 FOXP3 Variant Is Associated with Multiple Sclerosis and Transforming Growth Factor Β1 Levels in Female Patients. Inflamm. Res. 2019, 68, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Nataf, S.; Barritault, M.; Pays, L. A Unique TGFB1-Driven Genomic Program Links Astrocytosis, Low-Grade Inflammation and Partial Demyelination in Spinal Cord Periplaques from Progressive Multiple Sclerosis Patients. Int. J. Mol. Sci. 2017, 18, 2097. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. TGF-β as a Key Modulator of Astrocyte Reactivity: Disease Relevance and Therapeutic Implications. Biomedicines 2022, 10, 1206. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadóttir, K.; Benkhoucha, M.; Merkler, D.; Weber, M.S.; Payne, N.L.; Bernard, C.C.A.; Molnarfi, N.; Lalive, P.H. B Cell-Derived Transforming Growth Factor-Β1 Expression Limits the Induction Phase of Autoimmune Neuroinflammation. Sci. Rep. 2016, 6, 34594. [Google Scholar] [CrossRef]
- Kim, G.; Kim, J.; Rhie, S.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef]
- Branca, C.; Ferreira, E.; Nguyen, T.V.; Doyle, K.; Caccamo, A.; Oddo, S. Genetic Reduction of Nrf2 Exacerbates Cognitive Deficits in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2017, 26, 4823–4835. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Michaličková, D.; Šíma, M.; Slanař, O. New Insights in the Mechanisms of Impaired Redox Signaling and Its Interplay with Inflammation and Immunity in Multiple Sclerosis. Physiol. Res. 2020, 69, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-Mediated Neuroprotection in the MPTP Mouse Model of Parkinson’s Disease: Critical Role for the Astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 Suppresses Macrophage Inflammatory Response by Blocking Proinflammatory Cytokine Transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.; Yamamoto, M. Nrf2-MafG Heterodimers Contribute Globally to Antioxidant and Metabolic Networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFκB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weißflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the Astrocytic Nrf2/ARE System Ameliorates the Formation of Demyelinating Lesions in a Multiple Sclerosis Animal Model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Rosito, M.; Testi, C.; Parisi, G.; Cortese, B.; Baiocco, P.; Di Angelantonio, S. Exploring the Use of Dimethyl Fumarate as Microglia Modulator for Neurodegenerative Diseases Treatment. Antioxidants 2020, 9, 700. [Google Scholar] [CrossRef] [PubMed]
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl Fumarate Treatment in Multiple Sclerosis: Recent Advances in Clinical and Immunological Studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Xu, Z.; Qu, C.; Zhang, J.J. Dimethyl Fumarate Improves Cognitive Deficits in Chronic Cerebral Hypoperfusion Rats by Alleviating Inflammation, Oxidative Stress, and Ferroptosis via NRF2/ARE/NF-ΚB Signal Pathway. Int. Immunopharmacol. 2021, 98, 107844. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yamamoto, M. The KEAP1-NRF2 System and Neurodegenerative Diseases. Antioxid. Redox Signal. 2023, 38, 974–988. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, P.P.; Guevara, C.; Olesen, M.A.; Orellana, J.A.; Quintanilla, R.A.; Ortiz, F.C. Neurodegeneration in Multiple Sclerosis: The Role of Nrf2-Dependent Pathways. Antioxidants 2022, 11, 1146. [Google Scholar] [CrossRef] [PubMed]
- Abdulaal, W.H.; Asfour, H.Z.; Helmi, N.; Al Sadoun, H.; Eldakhakhny, B.; Alhakamy, N.A.; Alqarni, H.M.; Alzahrani, S.A.M.; El-Moselhy, M.A.; Sharkawi, S.S.; et al. Capsaicin Ameliorate Pulmonary Fibrosis via Antioxidant Nrf-2/PPAR-γ Pathway Activation and Inflammatory TGF-Β1/ NF-ΚB/COX II Pathway Inhibition. Front. Pharmacol. 2024, 15, 1333715. [Google Scholar] [CrossRef]
- Sueblinvong, V.; Tseng, V.; Smith, T.; Saghafi, R.; Mills, S.T.; Neujahr, D.C.; Guidot, D.M. TGFβ1 Mediates Alcohol-Induced Nrf2 Suppression in Lung Fibroblasts. Alcohol. Clin. Exp. Res. 2014, 38, 2731–2742. [Google Scholar] [CrossRef] [PubMed]
- Dreymueller, D.; Pruessmeyer, J.; Groth, E.; Ludwig, A. The Role of ADAM-Mediated Shedding in Vascular Biology. Eur. J. Cell Biol. 2012, 91, 472–485. [Google Scholar] [CrossRef]
- Churchman, A.T.; Anwar, A.A.; Li, F.Y.L.; Sato, H.; Ishii, T.; Mann, G.E.; Siow, R.C.M. Transforming Growth Factor-Beta1 Elicits Nrf2-Mediated Antioxidant Responses in Aortic Smooth Muscle Cells. J. Cell. Mol. Med. 2009, 13, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.K.; Mahns, D.A.; Coorssen, J.R.; Shortland, P.J. Behavioural Phenotypes in the Cuprizone Model of Central Nervous System Demyelination. Neurosci. Biobehav. Rev. 2019, 107, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beißel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-Induced Graded Oligodendrocyte Vulnerability Is Regulated by the Transcription Factor DNA Damage-Inducible Transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Gudi, V.; Gingele, S.; Skripuletz, T.; Stangel, M. Glial Response during Cuprizone- Induced de- and Remyelination in the CNS: Lessons Learned. Front. Cell. Neurosci. 2014, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical Development of Galunisertib (Ly2157299 Monohydrate), a Small Molecule Inhibitor of Transforming Growth Factor-Beta Signaling Pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Masuda, A.; Nakamura, T.; Abe, M.; Iwamoto, H.; Sakaue, T.; Tanaka, T.; Suzuki, H.; Koga, H.; Torimura, T. Promotion of Liver Regeneration and Anti-Fibrotic Effects of the TGF-β Receptor Kinase Inhibitor Galunisertib in CCl4-Treated Mice. Int. J. Mol. Med. 2020, 46, 427–438. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Schaer, D.A.; Li, Y.; Castaneda, S.P.; Murphy, M.Y.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.R.; Iversen, P.W.; et al. Targeting the TGFβ Pathway with Galunisertib, a TGFβRI Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.M.; Maurer, M.A.; Schwab, M.E. Four-Parameter Analysis in Modified Rotarod Test for Detecting Minor Motor Deficits in Mice. BMC Biol. 2023, 21, 177. [Google Scholar] [CrossRef] [PubMed]
- Bustos, F.; Ampuero, E.; Jury, N.; Aguilar, R.; Falahi, F.; Toledo, J.; Ahumada, J.; Lata, J.; Cubillos, P.; Henríquez, B.; et al. Epigenetic Editing of the Dlg4/PSD95 Gene Improves Cognition in Aged and Alzheimer’s Disease Mice. Brain A J. Neurol. 2017, 140, 3252–3268. [Google Scholar] [CrossRef] [PubMed]
- Ampuero, E.; Stehberg, J.; Gonzalez, D.; Besser, N.; Ferrero, M.; Diaz-Veliz, G.; Wyneken, U.; Rubio, F. Repetitive Fluoxetine Treatment Affects Long-Term Memories but Not Learning. Behav. Brain Res. 2013, 247, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.E.; Selesniemi, K.; Natale, D.R.C.; Brown, T.L. TGF-β Induces Smad2 Phosphorylation, ARE Induction, and Trophoblast Differentiation. Int. J. Stem Cells 2018, 11, 111–120. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Angel, P.; Lafyatis, R.; Hattori, K.; Kim, K.Y.; Sporn, M.B.; Karin, M.; Roberts, A.B. Autoinduction of Transforming Growth Factor Β1 Is Mediated by the AP-1 Complex. Mol. Cell. Biol. 1990, 10, 1492–1497. [Google Scholar] [CrossRef]
- Cattaneo, D.; Lamers, I.; Bertoni, R.; Feys, P.; Jonsdottir, J. Participation Restriction in People With Multiple Sclerosis: Prevalence and Correlations With Cognitive, Walking, Balance, and Upper Limb Impairments. Arch. Phys. Med. Rehabil. 2017, 98, 1308–1315. [Google Scholar] [CrossRef]
- Denissen, S.; De Cock, A.; Meurrens, T.; Vleugels, L.; Van Remoortel, A.; Gebara, B.; D’Haeseleer, M.; D’Hooghe, M.B.; Van Schependom, J.; Nagels, G. The Impact of Cognitive Dysfunction on Locomotor Rehabilitation Potential in Multiple Sclerosis. J. Cent. Nerv. Syst. Dis. 2019, 11, 1179573519884041. [Google Scholar] [CrossRef] [PubMed]
- Mistri, D.; Tedone, N.; Biondi, D.; Vizzino, C.; Pagani, E.; Rocca, M.A.; Filippi, M. Cognitive Phenotypes in Multiple Sclerosis: Mapping the Spectrum of Impairment. J. Neurol. 2024, 271, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- De Meo, E.; Portaccio, E.; Giorgio, A.; Ruano, L.; Goretti, B.; Niccolai, C.; Patti, F.; Chisari, C.G.; Gallo, P.; Grossi, P.; et al. Identifying the Distinct Cognitive Phenotypes in Multiple Sclerosis. JAMA Neurol. 2021, 78, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.W.; Severin, M.E.; Lovett-Racke, A.E. TGF-β Regulation of Encephalitogenic and Regulatory T Cells in Multiple Sclerosis. Eur. J. Immunol. 2017, 47, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Esmaeilzadeh, A.; Mohammadi, V.; Elahi, R. Transforming Growth Factor β (TGF-β) Pathway in the Immunopathogenesis of Multiple Sclerosis (MS); Molecular Approaches. Mol. Biol. Rep. 2023, 50, 6121–6131. [Google Scholar] [CrossRef] [PubMed]
- Guevara, C.; Ortiz, F. Glial-Derived Transforming Growth Factor Β1 (TGF-Β1): A Key Factor in Multiple Sclerosis Neuroinflammation. Neural Regen. Res. 2021, 16, 510–511. [Google Scholar] [CrossRef] [PubMed]
- Okita, Y.; Kamoshida, A.; Suzuki, H.; Itoh, K.; Motohashi, H.; Igarashi, K.; Yamamoto, M.; Ogami, T.; Koinuma, D.; Kato, M. Transforming Growth Factor-Induces Transcription Factors MafK and Bach1 to Suppress Expression of the Heme Oxygenase-1 Gene. J. Biol. Chem. 2013, 288, 20658–20667. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.-K.; Pokharel, Y.R.; Lim, S.C.; Han, H.-K.; Ryu, C.S.; Kim, S.K.; Kwak, M.K.; Kang, K.W. Inhibition of Liver Fibrosis by Solubilized Coenzyme Q10: Role of Nrf2 Activation in Inhibiting Transforming Growth Factor-Β1 Expression. Toxicol. Appl. Pharmacol. 2009, 240, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.J.; Kim, J.-Y.; Choi, Y.-K.; Kim, H.-J.; Jeong, J.-Y. Dimethylfumarate Attenuates Renal Fibrosis via NF-E2-Related Factor 2-Mediated Inhibition of Transforming Growth Factor-b/Smad Signaling. PLoS ONE 2012, 7, e45870. [Google Scholar] [CrossRef]
- Chan, T.S.; Teng, S.; Wilson, J.X.; Galati, G.; Khan, S.; O’Brien, P.J. Coenzyme Q Cytoprotective Mechanisms for Mitochondrial Complex I Cytopathies Involves NAD(P)H: Quinone Oxidoreductase 1(NQO1). Free. Radic. Res. 2002, 36, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Arfmann-Knubel, S.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schafer, H. The Crosstalk between Nrf2 and TGF-Β1 in the Epithelial-Mesenchymal Transition of Pancreatic Duct Epithelial Cells. PLoS ONE 2015, 10, e0132978. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; D’mello, V.; Caruso, D.; Abdul-Muneer, P.M. Traumatic Brain Injury-Induced Downregulation of Nrf2 Activates Inflammatory Response and Apoptotic Cell Death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef] [PubMed]
- Bento-Pereira, C.; Dinkova-Kostova, A.T. Activation of Transcription Factor Nrf2 to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Med. Res. Rev. 2021, 41, 785–802. [Google Scholar] [CrossRef] [PubMed]
- Lubrich, C.; Giesler, P.; Kipp, M. Motor Behavioral Deficits in the Cuprizone Model: Validity of the Rotarod Test Paradigm. Int. J. Mol. Sci. 2022, 23, 11342. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guevara, C.; Vicencio, S.C.; Pizarro, I.S.; Villavicencio-Tejo, F.; Quintanilla, R.A.; Astudillo, P.; Ampuero, E.; Varas, R.; Orellana, J.A.; Ortiz, F.C. Evidence for TGF-β1/Nrf2 Signaling Crosstalk in a Cuprizone Model of Multiple Sclerosis. Antioxidants 2024, 13, 914. https://doi.org/10.3390/antiox13080914
Guevara C, Vicencio SC, Pizarro IS, Villavicencio-Tejo F, Quintanilla RA, Astudillo P, Ampuero E, Varas R, Orellana JA, Ortiz FC. Evidence for TGF-β1/Nrf2 Signaling Crosstalk in a Cuprizone Model of Multiple Sclerosis. Antioxidants. 2024; 13(8):914. https://doi.org/10.3390/antiox13080914
Chicago/Turabian StyleGuevara, Coram, Sinay C. Vicencio, Ignacio S. Pizarro, Francisca Villavicencio-Tejo, Rodrigo A. Quintanilla, Pablo Astudillo, Estibaliz Ampuero, Rodrigo Varas, Juan A. Orellana, and Fernando C. Ortiz. 2024. "Evidence for TGF-β1/Nrf2 Signaling Crosstalk in a Cuprizone Model of Multiple Sclerosis" Antioxidants 13, no. 8: 914. https://doi.org/10.3390/antiox13080914