
Heavy Metal Exposure-Mediated Dysregulation of Sphingolipid Metabolism
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Toxicity Assay (MTT Assay)
2.3. Western Blot
2.4. Real-Time PCR
2.5. Cell Viability and Proliferation Assay
2.6. Human Lung (3D)-Perfusion Bioreactor
2.7. Sample Preparation and Extraction of Lipids for Lipidomics Analyses
2.8. LC-MS/MS of Sphingolipids
2.9. Immunofluorescent Staining
2.10. Measurement of Cellular Oxidative Stress
2.11. GeoMx Digital Spatial Profiling
2.12. Statistical Analysis
3. Results
3.1. Exposure to a High Dose of HM Induces Viability Changes in a Dose-Dependent Manner in Lung Epithelial Cells
3.2. Exposure to HM Alters Sphingolipid Metabolic Pathways in a Dose-Dependent Manner in Lung Epithelial Cells
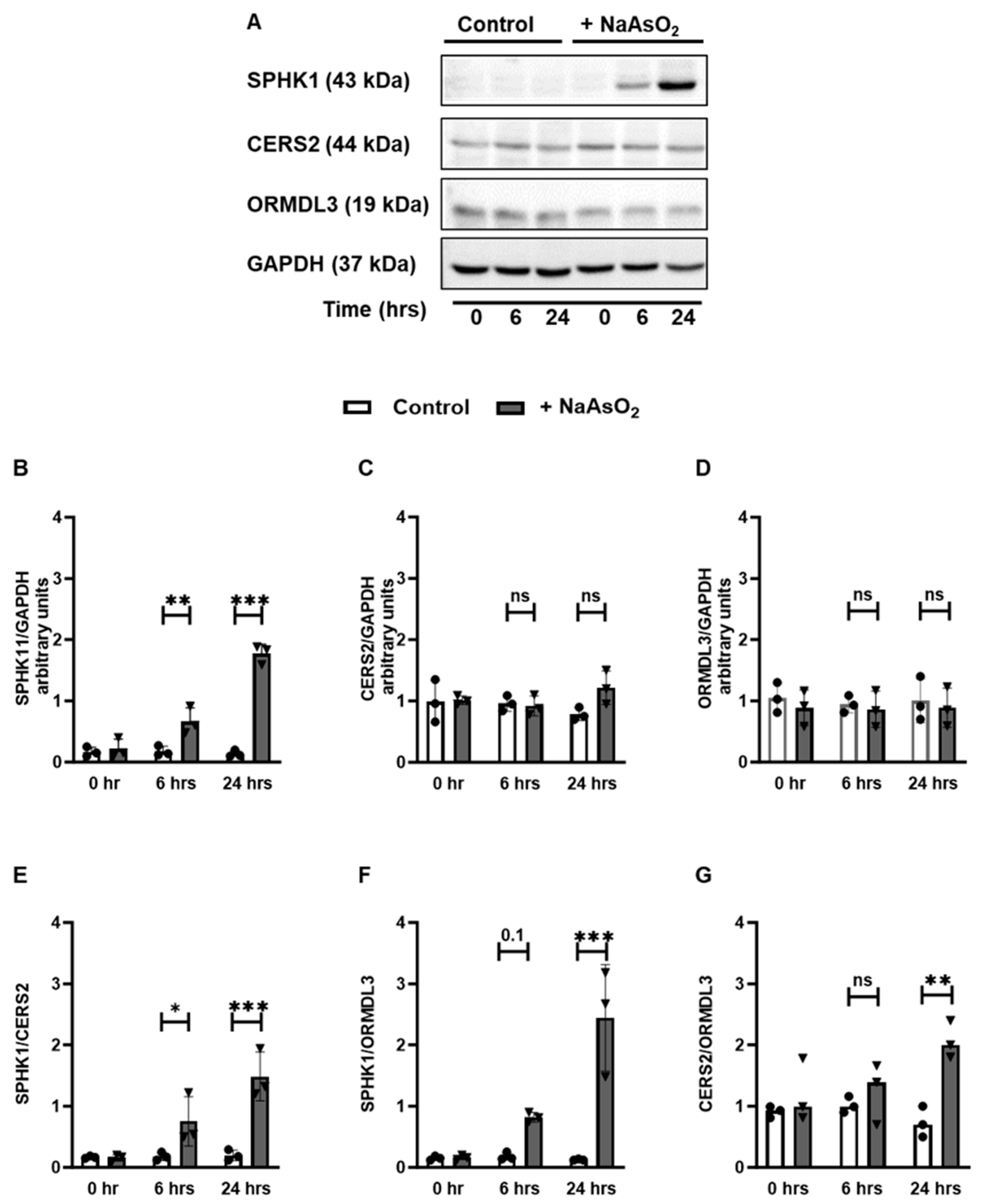
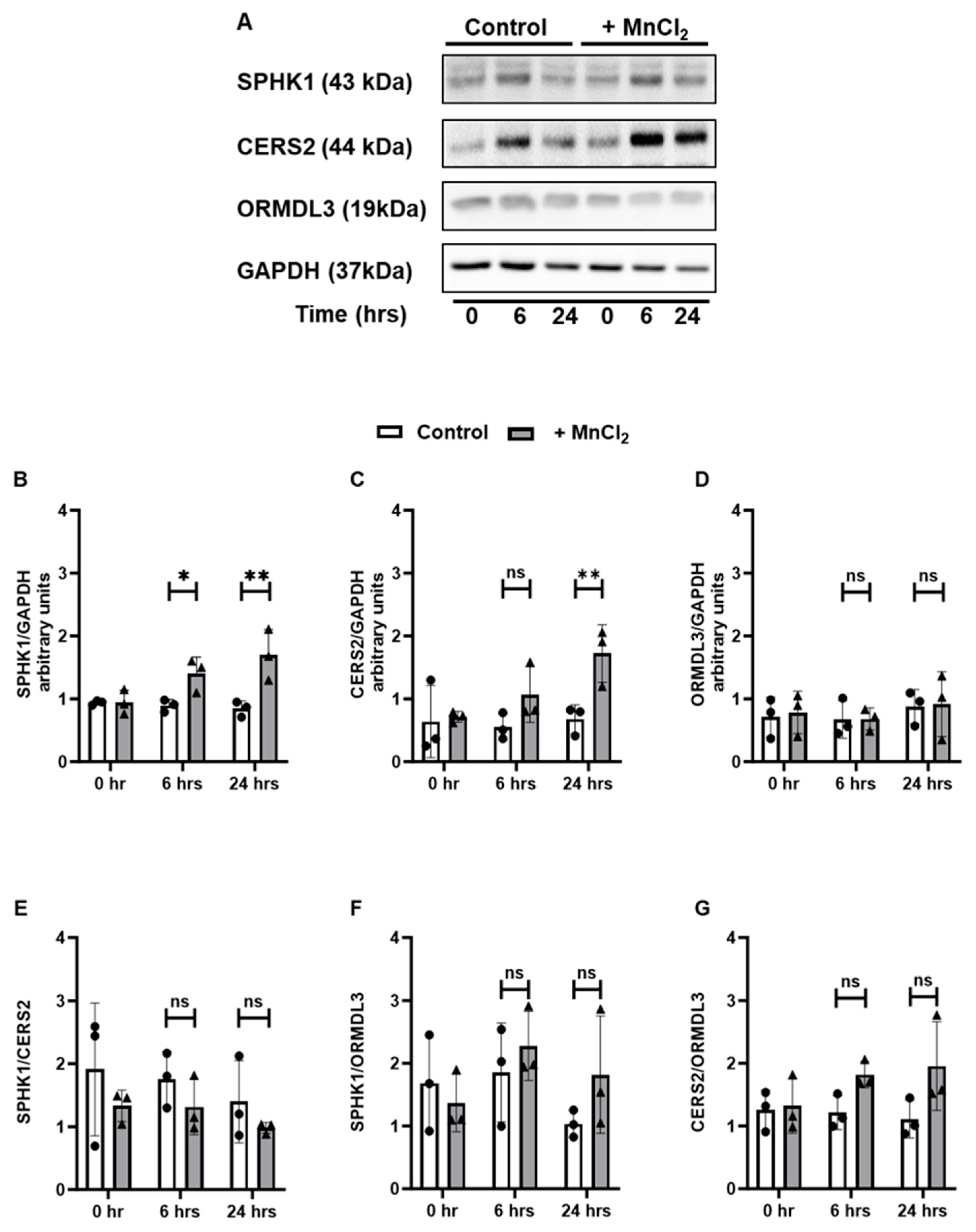
3.3. Exposure to HM Induces Time-Dependent Changes in the Sphingolipid Metabolic Pathways in Lung Epithelial Cells
3.4. Exposure to HM Alters the Expression of Sphingolipid Metabolizing Enzymes at the Transcript Level
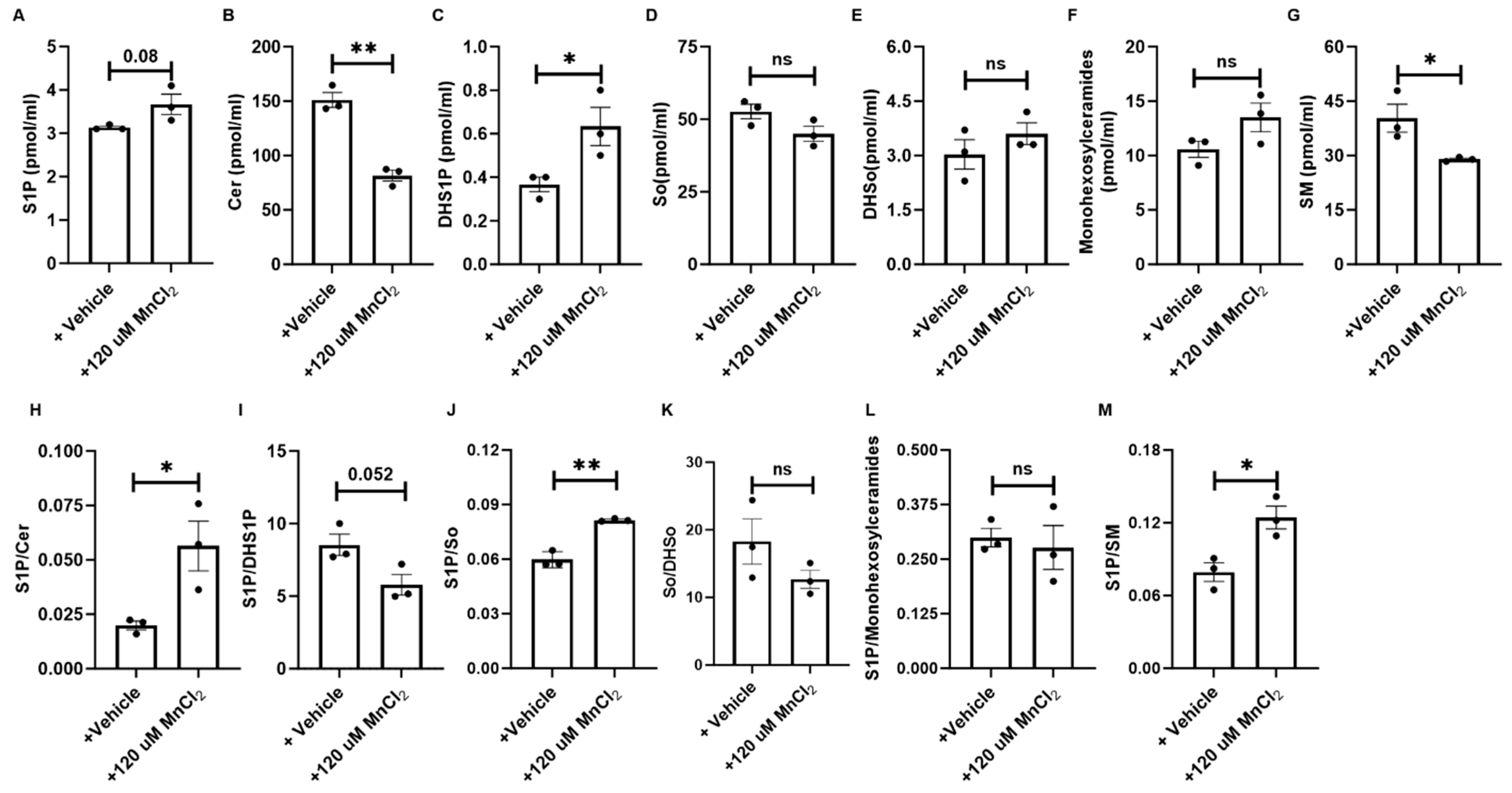
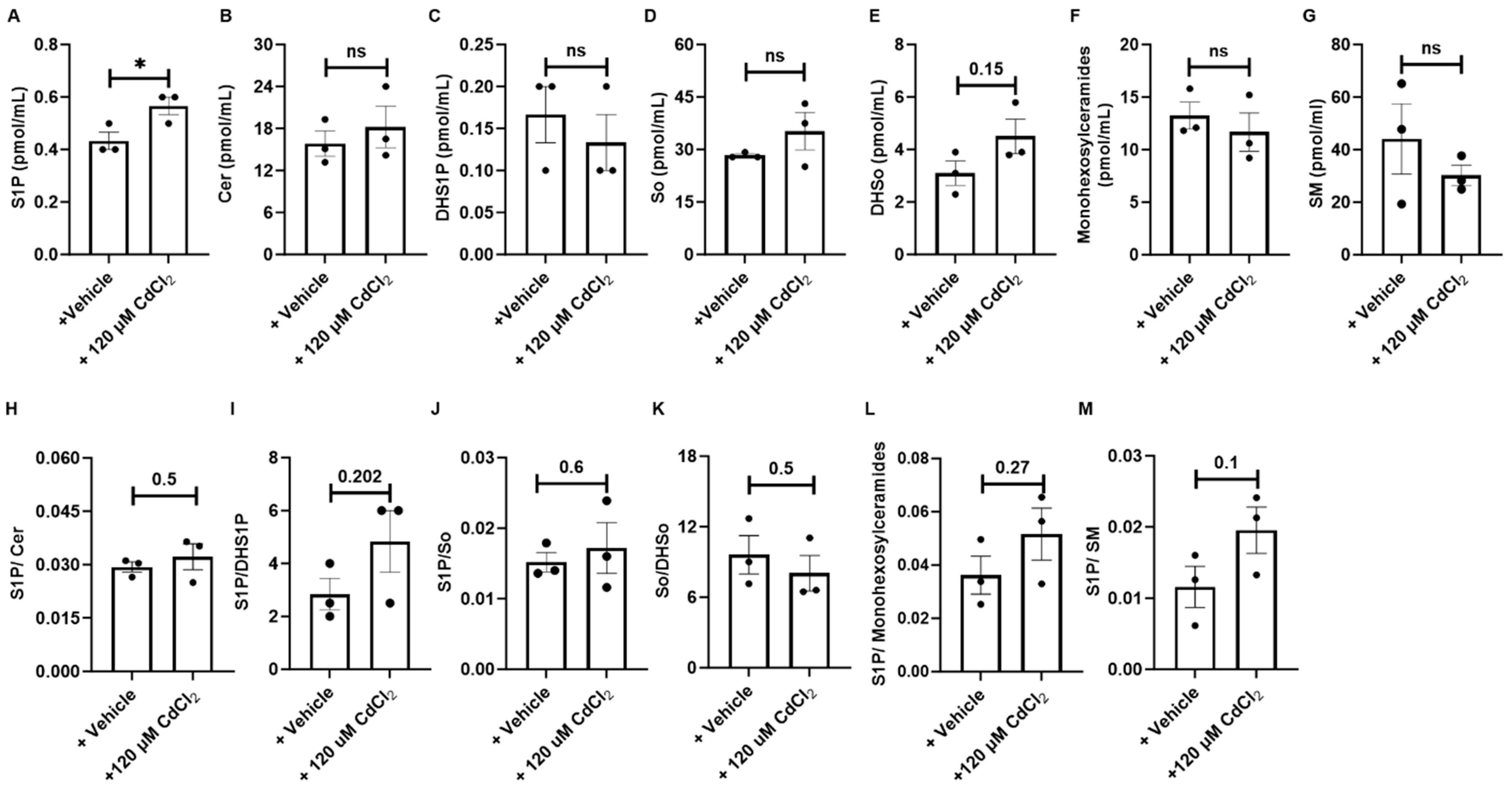
3.5. HM Exposure Induces Epithelial Cell-Derived Anti-Apoptotic Sphingolipid Metabolites via the Salvage and De Novo Synthesis Pathways
3.6. Exposure to HM Leads to a Limited Induction of Apoptosis in Epithelial Cells
3.7. HM Exposure in Human Lung Tissues Alters Expression of the Sphingolipid Enzymatic Pathway
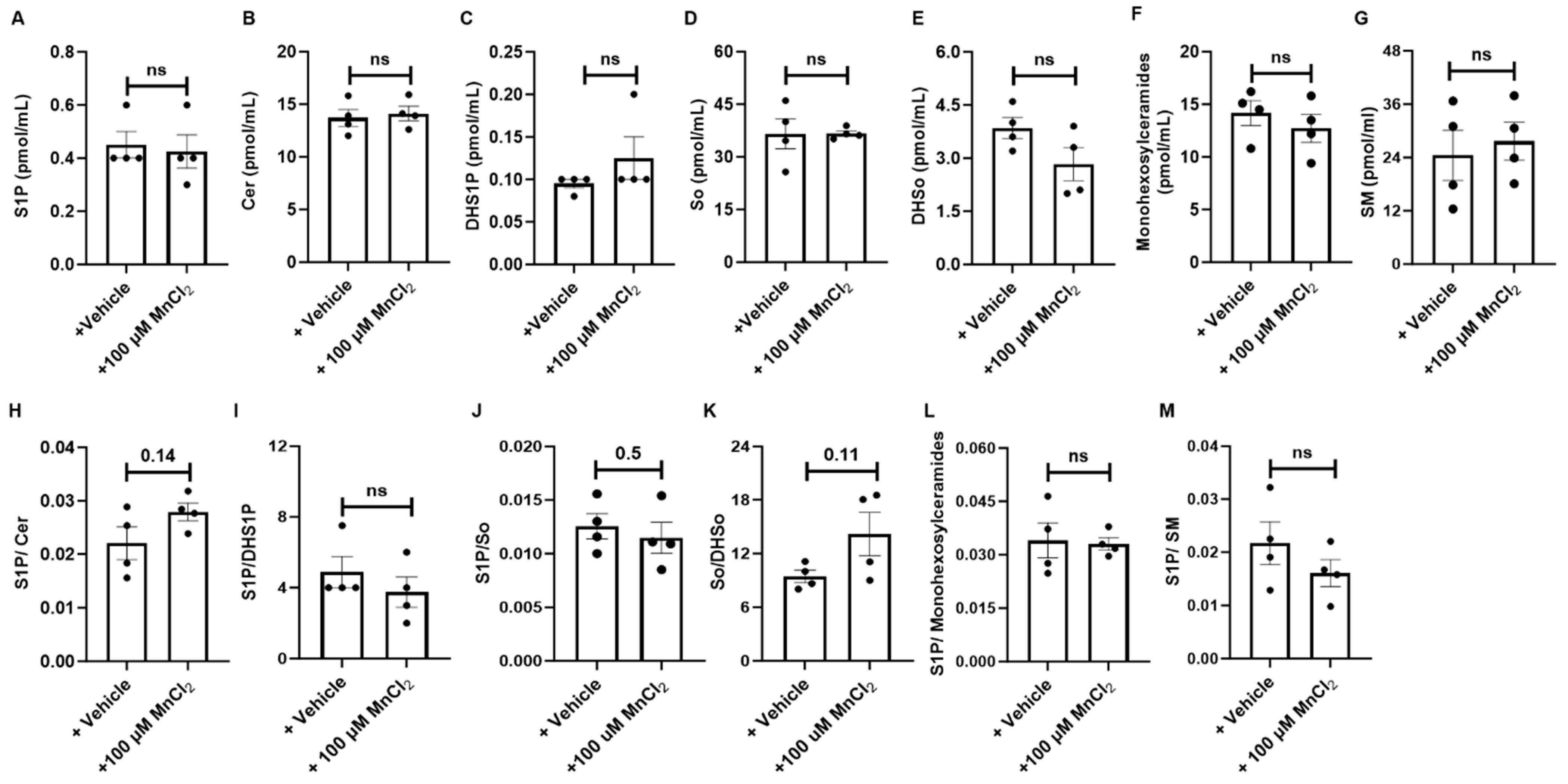
3.8. Inhibition of De Novo Synthesis and Induction of Salvage Pathway-Mediated Regulation of Sphingolipid Metabolism in HM-Exposed Lung Tissues
3.9. Exposure to HM Leads to Increased Expression of SPHK1 in Epithelial Cells and Other Structural Cells in Human Lung Tissues
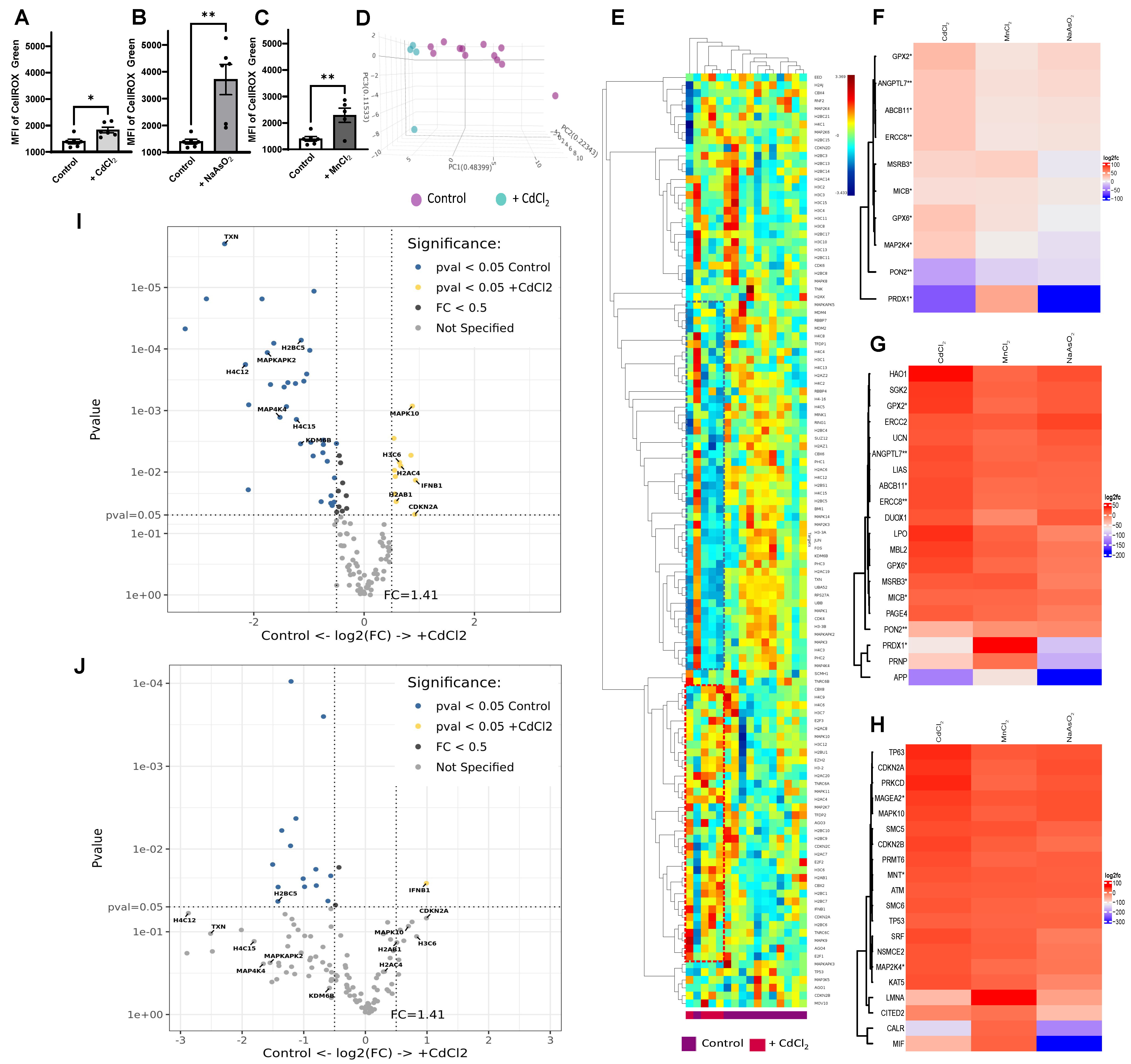
3.10. Upregulation of Oxidative Stress with HM Exposure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gehring, U.; Beelen, R.; Eeftens, M.; Hoek, G.; de Hoogh, K.; de Jongste, J.C.; Keuken, M.; Koppelman, G.H.; Meliefste, K.; Oldenwening, M.; et al. Particulate matter composition and respiratory health: The PIAMA Birth Cohort study. Epidemiology 2015, 26, 300–309. [Google Scholar] [CrossRef]
- Dunea, D.; Iordache, S.; Liu, H.Y.; Bohler, T.; Pohoata, A.; Radulescu, C. Quantifying the impact of PM2.5 and associated heavy metals on respiratory health of children near metallurgical facilities. Environ. Sci. Pollut. Res. Int. 2016, 23, 15395–15406. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, X.; Qin, Q.; Ye, K.; Wu, W.; Huo, X. Heavy metal exposure has adverse effects on the growth and development of preschool children. Environ. Geochem. Health 2019, 41, 309–321. [Google Scholar] [CrossRef]
- Zeng, X.; Xu, X.; Zheng, X.; Reponen, T.; Chen, A.; Huo, X. Heavy metals in PM2.5 and in blood, and children’s respiratory symptoms and asthma from an e-waste recycling area. Environ. Pollut. 2016, 210, 346–353. [Google Scholar] [CrossRef]
- Skalny, A.V.; Lima, T.R.R.; Ke, T.; Zhou, J.C.; Bornhorst, J.; Alekseenko, S.I.; Aaseth, J.; Anesti, O.; Sarigiannis, D.A.; Tsatsakis, A.; et al. Toxic metal exposure as a possible risk factor for COVID-19 and other respiratory infectious diseases. Food Chem. Toxicol. 2020, 146, 111809. [Google Scholar] [CrossRef]
- Cachon, B.F.; Firmin, S.; Verdin, A.; Ayi-Fanou, L.; Billet, S.; Cazier, F.; Martin, P.J.; Aissi, F.; Courcot, D.; Sanni, A.; et al. Proinflammatory effects and oxidative stress within human bronchial epithelial cells exposed to atmospheric particulate matter (PM2.5 and PM>2.5) collected from Cotonou, Benin. Environ. Pollut. 2014, 185, 340–351. [Google Scholar] [CrossRef]
- Wang, X.; Wang, B.; Zhou, M.; Xiao, L.; Xu, T.; Yang, S.; Nie, X.; Xie, L.; Yu, L.; Mu, G.; et al. Systemic inflammation mediates the association of heavy metal exposures with liver injury: A study in general Chinese urban adults. J. Hazard. Mater. 2021, 419, 126497. [Google Scholar] [CrossRef]
- Li, B.; Ma, Y.; Zhou, Y.; Chai, E. Research progress of different components of PM2.5 and ischemic stroke. Sci. Rep. 2023, 13, 15965. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972. [Google Scholar] [CrossRef]
- Huang, X.; Xie, J.; Cui, X.; Zhou, Y.; Wu, X.; Lu, W.; Shen, Y.; Yuan, J.; Chen, W. Association between Concentrations of Metals in Urine and Adult Asthma: A Case-Control Study in Wuhan, China. PLoS ONE 2016, 11, e0155818. [Google Scholar] [CrossRef]
- Yang, G.; Sun, T.; Han, Y.Y.; Rosser, F.; Forno, E.; Chen, W.; Celedon, J.C. Serum Cadmium and Lead, Current Wheeze, and Lung Function in a Nationwide Study of Adults in the United States. J. Allergy Clin. Immunol. Pract. 2019, 7, 2653–2660.e2653. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Wongtrakool, C.; Banton, S.A.; Li, S.; Orr, M.L.; Barr, D.B.; Neujahr, D.C.; Sutliff, R.L.; Go, Y.M.; Jones, D.P. Low-dose oral cadmium increases airway reactivity and lung neuronal gene expression in mice. Physiol. Rep. 2016, 4, e12821. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Surolia, R.; Li, H.; Wang, Z.; Liu, G.; Liu, R.M.; Mirov, S.B.; Athar, M.; Thannickal, V.J.; Antony, V.B. Low-dose cadmium exposure induces peribronchiolar fibrosis through site-specific phosphorylation of vimentin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L80–L91. [Google Scholar] [CrossRef] [PubMed]
- Dervan, P.A.; Hayes, J.A. Peribronchiolar fibrosis following acute experimental lung damage by cadmium aerosol. J. Pathol. 1979, 128, 143–149. [Google Scholar] [CrossRef]
- Park, S.; Lee, E.H.; Kho, Y. The association of asthma, total IgE, and blood lead and cadmium levels. J. Allergy Clin. Immunol. 2016, 138, 1701–1703.e1706. [Google Scholar] [CrossRef]
- Holgate, S.T. Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 2007, 120, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Davies, D.E.; Rorke, S.; Cakebread, J.; Murphy, G.; Powell, R.M.; Holloway, J.W. ADAM 33 and its association with airway remodeling and hyperresponsiveness in asthma. Clin. Rev. Allergy Immunol. 2004, 27, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.R.; Degheselle, O.; Smeets, K.; Van Kerkhove, E.; Cuypers, A. Cadmium-Induced Pathologies: Where Is the Oxidative Balance Lost (or Not)? Int. J. Mol. Sci. 2013, 14, 6116–6143. [Google Scholar] [CrossRef] [PubMed]
- Hormozi, M.; Mirzaei, R.; Nakhaee, A.; Izadi, S.; Dehghan Haghighi, J. The biochemical effects of occupational exposure to lead and cadmium on markers of oxidative stress and antioxidant enzymes activity in the blood of glazers in tile industry. Toxicol. Ind. Health 2018, 34, 459–467. [Google Scholar] [CrossRef]
- Odewabi, A.O.; Ekor, M. Levels of heavy and essential trace metals and their correlation with antioxidant and health status in individuals occupationally exposed to municipal solid wastes. Toxicol. Ind. Health 2017, 33, 431–442. [Google Scholar] [CrossRef]
- Briffa, J.; Sinagra, E.; Blundell, R. Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 2020, 6, e04691. [Google Scholar] [CrossRef]
- Fowler, B.A.; Whittaker, M.H.; Lipsky, M.; Wang, G.; Chen, X.Q. Oxidative stress induced by lead, cadmium and arsenic mixtures: 30-day, 90-day, and 180-day drinking water studies in rats: An overview. Biometals 2004, 17, 567–568. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, Y.; Shi, L.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 2022, 469, 153136. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Ma, J.; Zhou, M.; Wang, D.; Wang, B.; Nie, X.; Mu, G.; Zhang, X.; Chen, W. Heavy metals exposure, lipid peroxidation and heart rate variability alteration: Association and mediation analyses in urban adults. Ecotoxicol. Environ. Saf. 2020, 205, 111149. [Google Scholar] [CrossRef] [PubMed]
- Ijomone, O.M.; Ijomone, O.K.; Iroegbu, J.D.; Ifenatuoha, C.W.; Olung, N.F.; Aschner, M. Epigenetic influence of environmentally neurotoxic metals. Neurotoxicology 2020, 81, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Mabalirajan, U.; Rehman, R.; Ahmad, T.; Kumar, S.; Singh, S.; Leishangthem, G.D.; Aich, J.; Kumar, M.; Khanna, K.; Singh, V.P.; et al. Linoleic acid metabolite drives severe asthma by causing airway epithelial injury. Sci. Rep. 2013, 3, 1349. [Google Scholar] [CrossRef]
- Matsuoka, T.; Hirata, M.; Tanaka, H.; Takahashi, Y.; Murata, T.; Kabashima, K.; Sugimoto, Y.; Kobayashi, T.; Ushikubi, F.; Aze, Y.; et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000, 287, 2013–2017. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D.; Tager, A.M. T-cell trafficking in asthma: Lipid mediators grease the way. Nat. Rev. Immunol. 2004, 4, 711–724. [Google Scholar] [CrossRef]
- Nassan, F.L.; Wang, C.; Kelly, R.S.; Lasky-Su, J.A.; Vokonas, P.S.; Koutrakis, P.; Schwartz, J.D. Ambient PM2.5 species and ultrafine particle exposure and their differential metabolomic signatures. Environ. Int. 2021, 151, 106447. [Google Scholar] [CrossRef]
- Gruzieva, O.; Jeong, A.; He, S.; Yu, Z.; de Bont, J.; Pinho, M.G.M.; Eze, I.C.; Kress, S.; Wheelock, C.E.; Peters, A.; et al. Air pollution, metabolites and respiratory health across the life-course. Eur. Respir. Rev. 2022, 31, 220038. [Google Scholar] [CrossRef]
- Xu, Y.; Han, Y.; Wang, Y.; Gong, J.; Li, H.; Wang, T.; Chen, X.; Chen, W.; Fan, Y.; Qiu, X.; et al. Ambient Air Pollution and Atherosclerosis: A Potential Mediating Role of Sphingolipids. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Zhang, L.; Qin, L.; Ding, H.; Li, R.; Gu, W.; Chen, R.; Zhang, Y.; Rajagoplan, S.; Zhang, K.; et al. Airborne PM2.5 pollution: A double-edged sword modulating hepatic lipid metabolism in middle-aged male mice. Environ. Pollut. 2023, 324, 121347. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Sutliff, R.L.; Chandler, J.D.; Khalidur, R.; Kang, B.Y.; Anania, F.A.; Orr, M.; Hao, L.; Fowler, B.A.; Jones, D.P. Low-Dose Cadmium Causes Metabolic and Genetic Dysregulation Associated with Fatty Liver Disease in Mice. Toxicol. Sci. 2015, 147, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mu, X.; Zhang, J.; Huang, Q.; Alamdar, A.; Tian, M.; Liu, L.; Shen, H. Serum metabolomics reveals that arsenic exposure disrupted lipid and amino acid metabolism in rats: A step forward in understanding chronic arsenic toxicity. Metallomics 2015, 7, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, Y.; Ge, X.; Nie, D.; Wang, M.; Zhou, H.; Chen, M. In vitro toxicity evaluation of heavy metals in urban air particulate matter on human lung epithelial cells. Sci. Total Environ. 2019, 678, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Hough, K.P.; Wilson, L.S.; Trevor, J.L.; Strenkowski, J.G.; Maina, N.; Kim, Y.I.; Spell, M.L.; Wang, Y.; Chanda, D.; Dager, J.R.; et al. Unique Lipid Signatures of Extracellular Vesicles from the Airways of Asthmatics. Sci. Rep. 2018, 8, 10340. [Google Scholar] [CrossRef] [PubMed]
- Peuschel, H.; Sydlik, U.; Grether-Beck, S.; Felsner, I.; Stockmann, D.; Jakob, S.; Kroker, M.; Haendeler, J.; Gotic, M.; Bieschke, C.; et al. Carbon nanoparticles induce ceramide- and lipid raft-dependent signalling in lung epithelial cells: A target for a preventive strategy against environmentally-induced lung inflammation. Part Fibre Toxicol. 2012, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, H.M.; Onoda, A.; Shinkai, Y.; Nakamura, M.; El-Ghoneimy, A.A.; El-Sayed, Y.S.; Takeda, K.; Umezawa, M. The ceramide inhibitor fumonisin B1 mitigates the pulmonary effects of low-dose diesel exhaust inhalation in mice. Ecotoxicol. Environ. Saf. 2016, 132, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Ammit, A.J.; Hastie, A.T.; Edsall, L.C.; Hoffman, R.K.; Amrani, Y.; Krymskaya, V.P.; Kane, S.A.; Peters, S.P.; Penn, R.B.; Spiegel, S.; et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001, 15, 1212–1214. [Google Scholar] [CrossRef]
- Kume, H.; Takeda, N.; Oguma, T.; Ito, S.; Kondo, M.; Ito, Y.; Shimokata, K. Sphingosine 1-phosphate causes airway hyper-reactivity by rho-mediated myosin phosphatase inactivation. J. Pharmacol. Exp. Ther. 2007, 320, 766–773. [Google Scholar] [CrossRef]
- Olivera, A.; Mizugishi, K.; Tikhonova, A.; Ciaccia, L.; Odom, S.; Proia, R.L.; Rivera, J. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 2007, 26, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate in allergic responses, asthma and anaphylaxis. Pharmacol. Ther. 2007, 115, 390–399. [Google Scholar] [CrossRef]
- Ono, J.G.; Worgall, T.S.; Worgall, S. Airway reactivity and sphingolipids-implications for childhood asthma. Mol. Cell. Pediatr. 2015, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Kowal, K.; Zebrowska, E.; Chabowski, A. Altered Sphingolipid Metabolism Is Associated with Asthma Phenotype in House Dust Mite-Allergic Patients. Allergy Asthma Immunol. Res. 2019, 11, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Riemma, M.A.; Cerqua, I.; Romano, B.; Irollo, E.; Bertolino, A.; Camerlingo, R.; Granato, E.; Rea, G.; Scala, S.; Terlizzi, M.; et al. Sphingosine-1-phosphate/TGF-beta axis drives epithelial mesenchymal transition in asthma-like disease. Br. J. Pharmacol. 2022, 179, 1753–1768. [Google Scholar] [CrossRef] [PubMed]
- Roviezzo, F.; Di Lorenzo, A.; Bucci, M.; Brancaleone, V.; Vellecco, V.; De Nardo, M.; Orlotti, D.; De Palma, R.; Rossi, F.; D’Agostino, B.; et al. Sphingosine-1-phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 2007, 36, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef]
- van Echten-Deckert, G.; Zschoche, A.; Bar, T.; Schmidt, R.R.; Raths, A.; Heinemann, T.; Sandhoff, K. cis-4-Methylsphingosine decreases sphingolipid biosynthesis by specifically interfering with serine palmitoyltransferase activity in primary cultured neurons. J. Biol. Chem. 1997, 272, 15825–15833. [Google Scholar] [CrossRef]
- Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate signalling in mammalian cells. Biochem. J. 2000, 349, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Giannini, L.; Nistri, S.; Cinci, L.; Mastroianni, R.; Xu, W.; Comhair, S.A.; Li, D.; Cuzzocrea, S.; Matuschak, G.M.; et al. Ceramide: A key signaling molecule in a Guinea pig model of allergic asthmatic response and airway inflammation. J. Pharmacol. Exp. Ther. 2008, 324, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Gillard, B.K.; Clement, R.G.; Marcus, D.M. Variations among cell lines in the synthesis of sphingolipids in de novo and recycling pathways. Glycobiology 1998, 8, 885–890. [Google Scholar] [CrossRef]
- Becker, K.P.; Kitatani, K.; Idkowiak-Baldys, J.; Bielawski, J.; Hannun, Y.A. Selective inhibition of juxtanuclear translocation of protein kinase C betaII by a negative feedback mechanism involving ceramide formed from the salvage pathway. J. Biol. Chem. 2005, 280, 2606–2612. [Google Scholar] [CrossRef]
- Takeda, S.; Mitsutake, S.; Tsuji, K.; Igarashi, Y. Apoptosis occurs via the ceramide recycling pathway in human HaCaT keratinocytes. J. Biochem. 2006, 139, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.V.; Gorshkova, I.A.; Usatyuk, P.; Zhao, Y.; Saatian, B.; Hubbard, W.; Natarajan, V. De novo biosynthesis of dihydrosphingosine-1-phosphate by sphingosine kinase 1 in mammalian cells. Cell. Signal. 2006, 18, 1779–1792. [Google Scholar] [CrossRef]
- Gomez del Pulgar, T.; Velasco, G.; Sanchez, C.; Haro, A.; Guzman, M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem. J. 2002, 363, 183–188. [Google Scholar] [CrossRef]
- Worgall, T.S.; Veerappan, A.; Sung, B.; Kim, B.I.; Weiner, E.; Bholah, R.; Silver, R.B.; Jiang, X.C.; Worgall, S. Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Sci. Transl. Med. 2013, 5, 186ra167. [Google Scholar] [CrossRef]
- Price, M.M.; Oskeritzian, C.A.; Falanga, Y.T.; Harikumar, K.B.; Allegood, J.C.; Alvarez, S.E.; Conrad, D.; Ryan, J.J.; Milstien, S.; Spiegel, S. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J. Allergy Clin. Immunol. 2013, 131, 501–511.e501. [Google Scholar] [CrossRef]
- Lai, W.Q.; Goh, H.H.; Bao, Z.; Wong, W.S.; Melendez, A.J.; Leung, B.P. The role of sphingosine kinase in a murine model of allergic asthma. J. Immunol. 2008, 180, 4323–4329. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Cho, K.A.; Yoon, H.S.; Kim, S.Y.; Kim, H.Y.; Pewzner-Jung, Y.; Jung, S.A.; Park, W.J.; Futerman, A.H.; Park, J.W. Ceramide Synthase 2 Null Mice Are Protected from Ovalbumin-Induced Asthma with Higher T Cell Receptor Signal Strength in CD4+ T Cells. Int. J. Mol. Sci. 2021, 22, 2713. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Collins, S.R.; Bodenmiller, B.; Aebersold, R.; Simons, K.; Shevchenko, A.; Ejsing, C.S.; Weissman, J.S. Orm family proteins mediate sphingolipid homeostasis. Nature 2010, 463, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Schedel, M.; Michel, S.; Gaertner, V.D.; Toncheva, A.A.; Depner, M.; Binia, A.; Schieck, M.; Rieger, M.T.; Klopp, N.; von Berg, A.; et al. Polymorphisms related to ORMDL3 are associated with asthma susceptibility, alterations in transcriptional regulation of ORMDL3, and changes in TH2 cytokine levels. J. Allergy Clin. Immunol. 2015, 136, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.J.; Turvey, S.E. Functional analysis of the impact of ORMDL3 expression on inflammation and activation of the unfolded protein response in human airway epithelial cells. Allergy Asthma Clin. Immunol. 2013, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Goliwas, K.F.; Kashyap, M.P.; Khan, J.; Sinha, R.; Weng, Z.; Oak, A.S.W.; Jin, L.; Atigadda, V.; Lee, M.B.; Elmets, C.A.; et al. Ex Vivo Culture Models of Hidradenitis Suppurativa for Defining Molecular Pathogenesis and Treatment Efficacy of Novel Drugs. Inflammation 2022, 45, 1388–1401. [Google Scholar] [CrossRef] [PubMed]
- Goliwas, K.F.; Wood, A.M.; Simmons, C.S.; Khan, R.; Khan, S.A.; Wang, Y.; Berry, J.L.; Athar, M.; Mobley, J.A.; Kim, Y.I.; et al. Local SARS-CoV-2 Peptide-Specific Immune Responses in Lungs of Convalescent and Uninfected Human Subjects. medRxiv, 2022; preprint. [Google Scholar] [CrossRef]
- Singh, P.; Li, F.J.; Dsouza, K.; Stephens, C.T.; Zheng, H.; Kumar, A.; Dransfield, M.T.; Antony, V.B. Low dose cadmium exposure regulates miR-381-ANO1 interaction in airway epithelial cells. Sci. Rep. 2024, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, J.; Xue, C.; Wu, K.; Ouyang, W.; Zhang, D.; Yan, Y.; Huang, C. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in Beas-2B cells. J. Biol. Chem. 2006, 281, 24405–24413. [Google Scholar] [CrossRef]
- Liu, S.X.; Athar, M.; Lippai, I.; Waldren, C.; Hei, T.K. Induction of oxyradicals by arsenic: Implication for mechanism of genotoxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 1643–1648. [Google Scholar] [CrossRef]
- Yuan, Z.; Ying, X.P.; Zhong, W.J.; Tian, S.M.; Wang, Y.; Jia, Y.R.; Chen, W.; Fu, J.L.; Zhao, P.; Zhou, Z.C. Autophagy Attenuates MnCl2-induced Apoptosis in Human Bronchial Epithelial Cells. Biomed. Environ. Sci. 2016, 29, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sang, H.; Liu, Y.; Li, J. Manganese activates caspase-9-dependent apoptosis in human bronchial epithelial cells. Hum. Exp. Toxicol. 2013, 32, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Lin, H.; Muskhelishvili, L.; Latendresse, J.; Richter, P.; Heflich, R.H. Tight junction disruption by cadmium in an in vitro human airway tissue model. Respir. Res. 2015, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Bredow, S.; Falgout, M.M.; March, T.H.; Yingling, C.M.; Malkoski, S.P.; Aden, J.; Bedrick, E.J.; Lewis, J.L.; Divine, K.K. Subchronic inhalation of soluble manganese induces expression of hypoxia-associated angiogenic genes in adult mouse lungs. Toxicol. Appl. Pharmacol. 2007, 221, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Sudhadevi, T.; Basa, P.; Ha, A.W.; Natarajan, V.; Harijith, A. The Role of Sphingolipid Signaling in Oxidative Lung Injury and Pathogenesis of Bronchopulmonary Dysplasia. Int. J. Mol. Sci. 2022, 23, 1254. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, J.L. Sphingolipids and their enigmatic role in asthma. Adv. Biol. Regul. 2018, 70, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, H.M.; Amrani, Y.; Watterson, K.R.; Murthy, K.S.; Panettieri, R.A., Jr.; Spiegel, S. Sphingosine-1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB J. 2003, 17, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Ono, J.G.; Kim, B.I.; Zhao, Y.; Christos, P.J.; Tesfaigzi, Y.; Worgall, T.S.; Worgall, S. Decreased sphingolipid synthesis in children with 17q21 asthma-risk genotypes. J. Clin. Investig. 2020, 130, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Bouzigon, E.; Corda, E.; Aschard, H.; Dizier, M.H.; Boland, A.; Bousquet, J.; Chateigner, N.; Gormand, F.; Just, J.; Le Moual, N.; et al. Effect of 17q21 variants and smoking exposure in early-onset asthma. N. Engl. J. Med. 2008, 359, 1985–1994. [Google Scholar] [CrossRef]
- Miller, M.; Tam, A.B.; Cho, J.Y.; Doherty, T.A.; Pham, A.; Khorram, N.; Rosenthal, P.; Mueller, J.L.; Hoffman, H.M.; Suzukawa, M.; et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc. Natl. Acad. Sci. USA 2012, 109, 16648–16653. [Google Scholar] [CrossRef]
- Tripathi, P.; Deng, F.; Scruggs, A.M.; Chen, Y.; Huang, S.K. Variation in doses and duration of particulate matter exposure in bronchial epithelial cells results in upregulation of different genes associated with airway disorders. Toxicol. Vitr. 2018, 51, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Kops, G.J.; Dansen, T.B.; Polderman, P.E.; Saarloos, I.; Wirtz, K.W.; Coffer, P.J.; Huang, T.T.; Bos, J.L.; Medema, R.H.; Burgering, B.M. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002, 419, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, D.; Zhang, X.; Yalcin, S.; Luciano, J.P.; Brugnara, C.; Huber, T.; Ghaffari, S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J. Clin. Investig. 2007, 117, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Dijkers, P.F.; Kops, G.J.; Lens, S.M.; Coffer, P.J.; Burgering, B.M.; Medema, R.H. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J. Immunol. 2002, 168, 5024–5031. [Google Scholar] [CrossRef]
- Klotz, L.O.; Sanchez-Ramos, C.; Prieto-Arroyo, I.; Urbanek, P.; Steinbrenner, H.; Monsalve, M. Redox regulation of FoxO transcription factors. Redox Biol. 2015, 6, 51–72. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Single, S.; Liu, Y.; Hough, K.P.; Wang, Y.; Thannickal, V.J.; Athar, M.; Goliwas, K.F.; Deshane, J.S. Heavy Metal Exposure-Mediated Dysregulation of Sphingolipid Metabolism. Antioxidants 2024, 13, 978. https://doi.org/10.3390/antiox13080978
Ahmad S, Single S, Liu Y, Hough KP, Wang Y, Thannickal VJ, Athar M, Goliwas KF, Deshane JS. Heavy Metal Exposure-Mediated Dysregulation of Sphingolipid Metabolism. Antioxidants. 2024; 13(8):978. https://doi.org/10.3390/antiox13080978
Chicago/Turabian StyleAhmad, Shaheer, Sierra Single, Yuelong Liu, Kenneth P. Hough, Yong Wang, Victor J. Thannickal, Mohammad Athar, Kayla F. Goliwas, and Jessy S. Deshane. 2024. "Heavy Metal Exposure-Mediated Dysregulation of Sphingolipid Metabolism" Antioxidants 13, no. 8: 978. https://doi.org/10.3390/antiox13080978