
Pregnancy Disorders: A Potential Role for Mitochondrial Altered Homeostasis


 , , and
, , and
Abstract
:
1. Introduction
2. Mitochondria: Physiological Role and Homeostasis
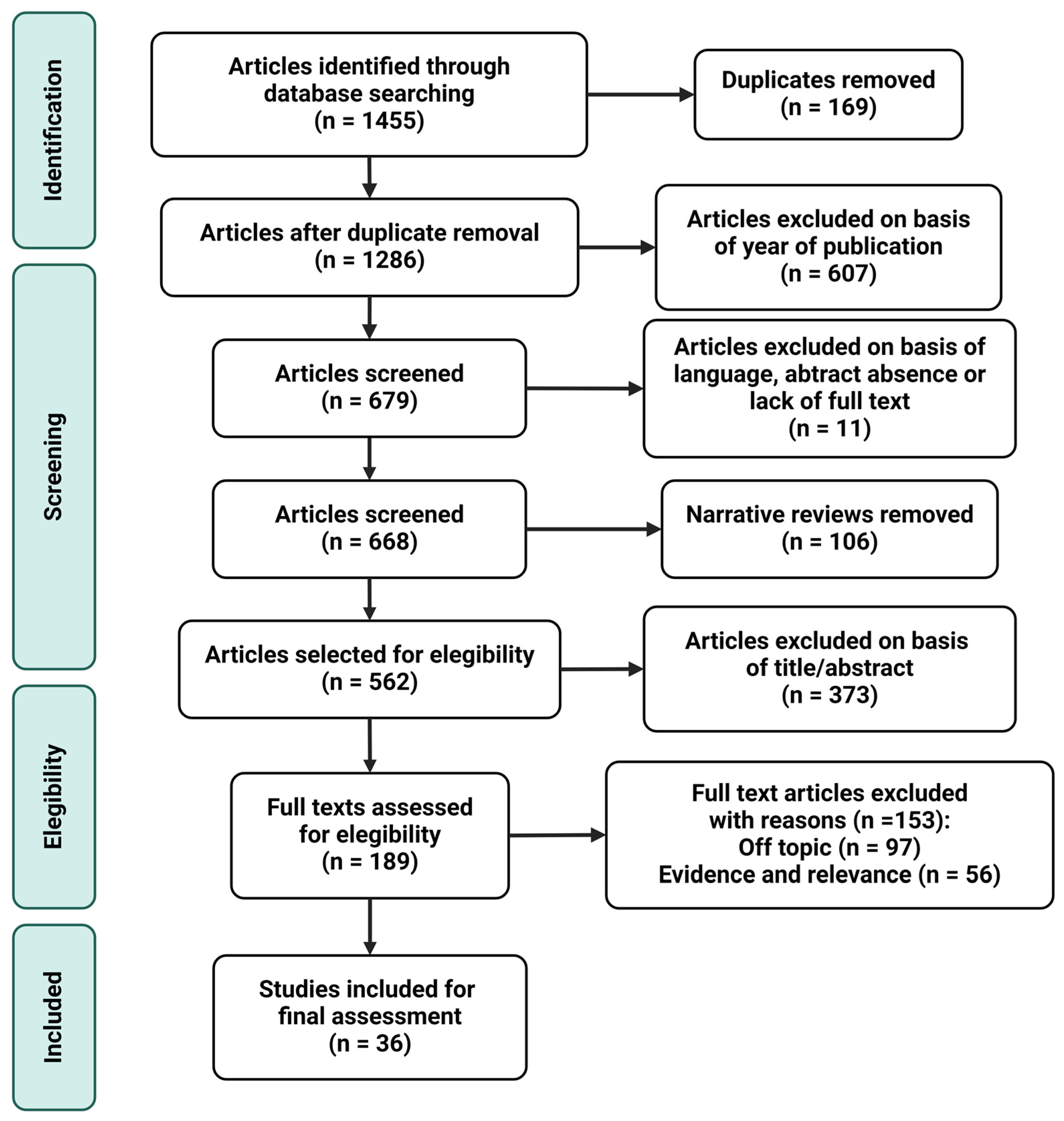
3. Materials and Methods
4. Results and Discussion
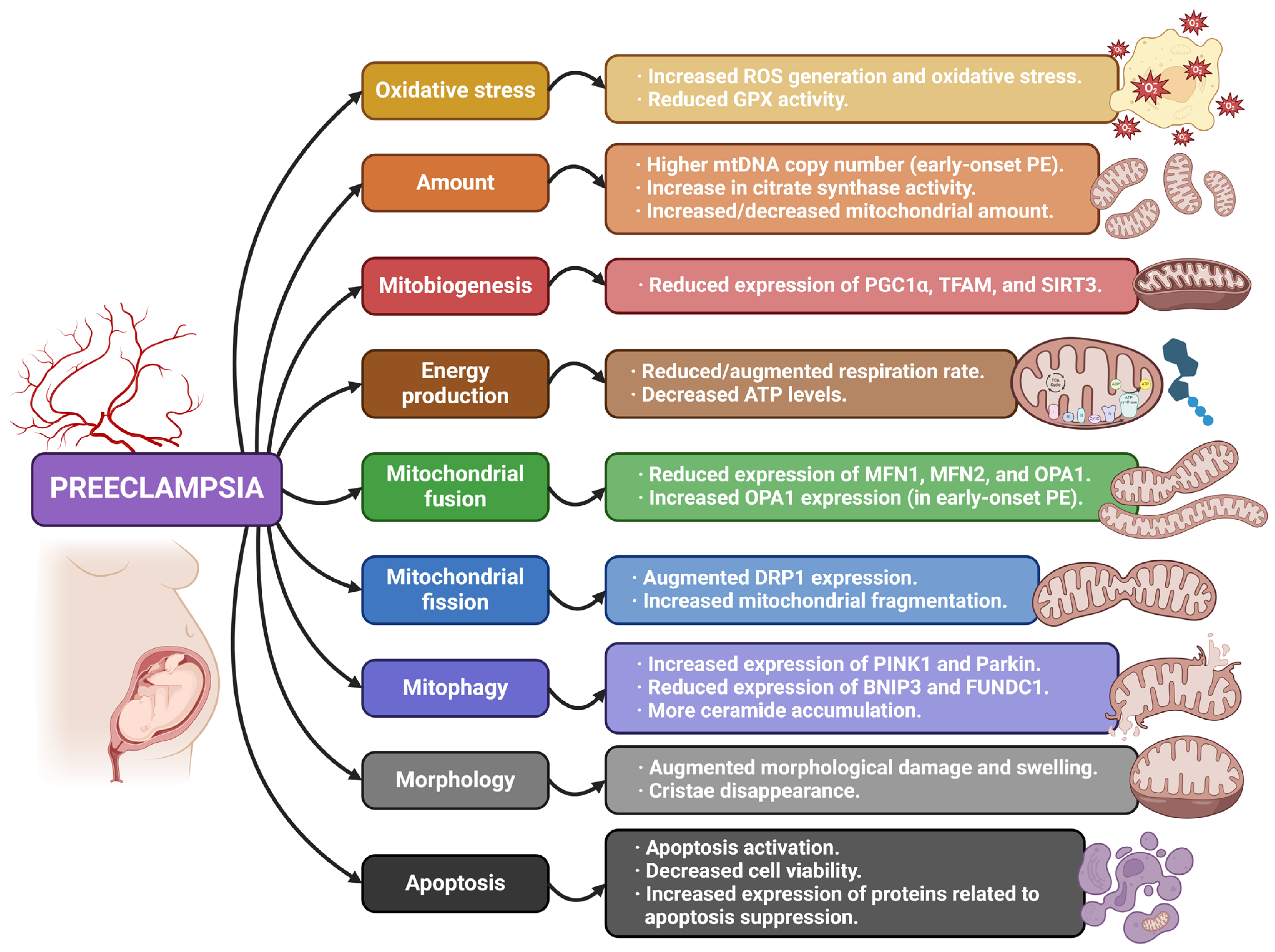
4.1. Mitochondria and Preeclampsia
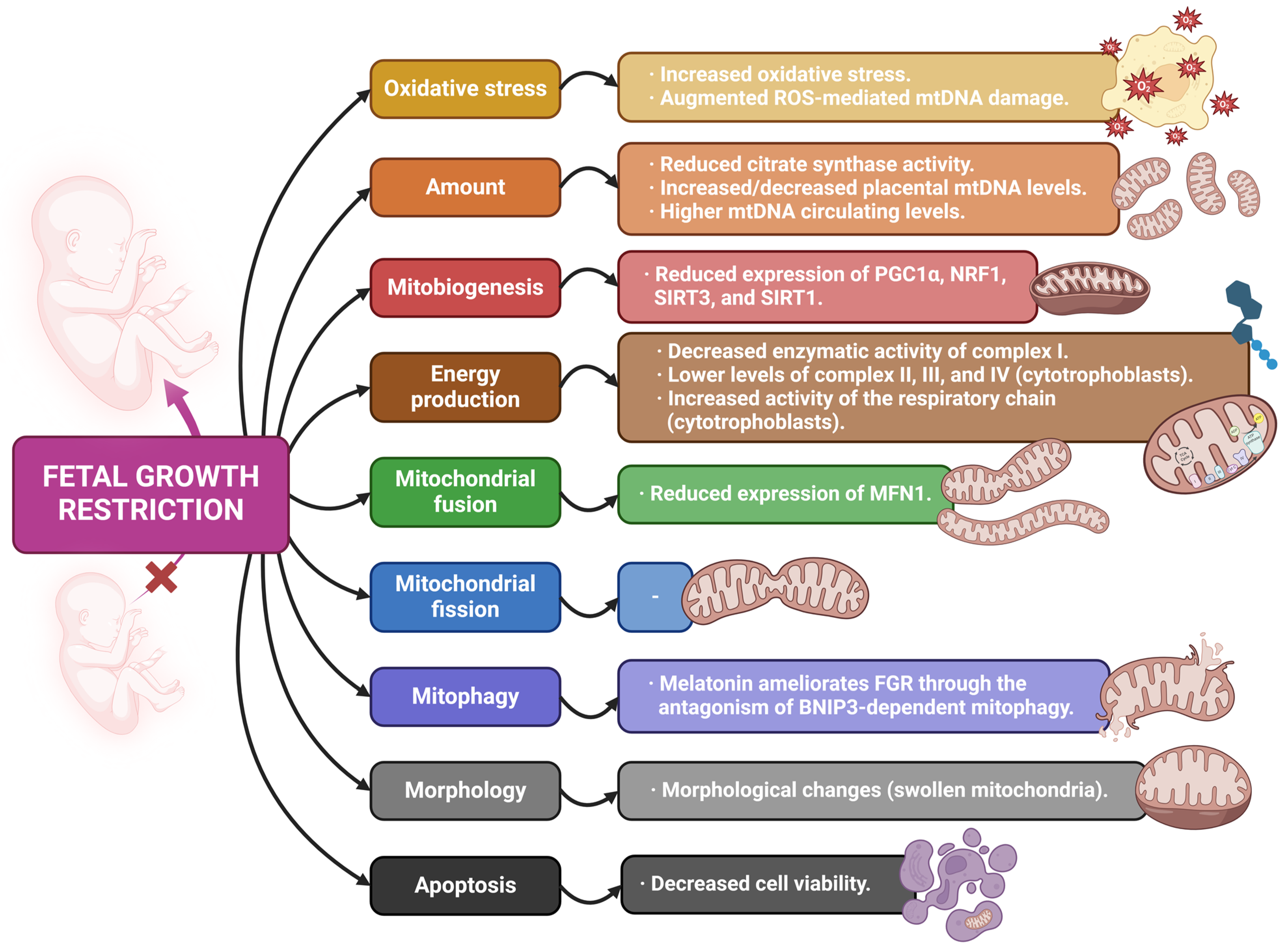
4.2. Mitochondria and Fetal Growth Restriction
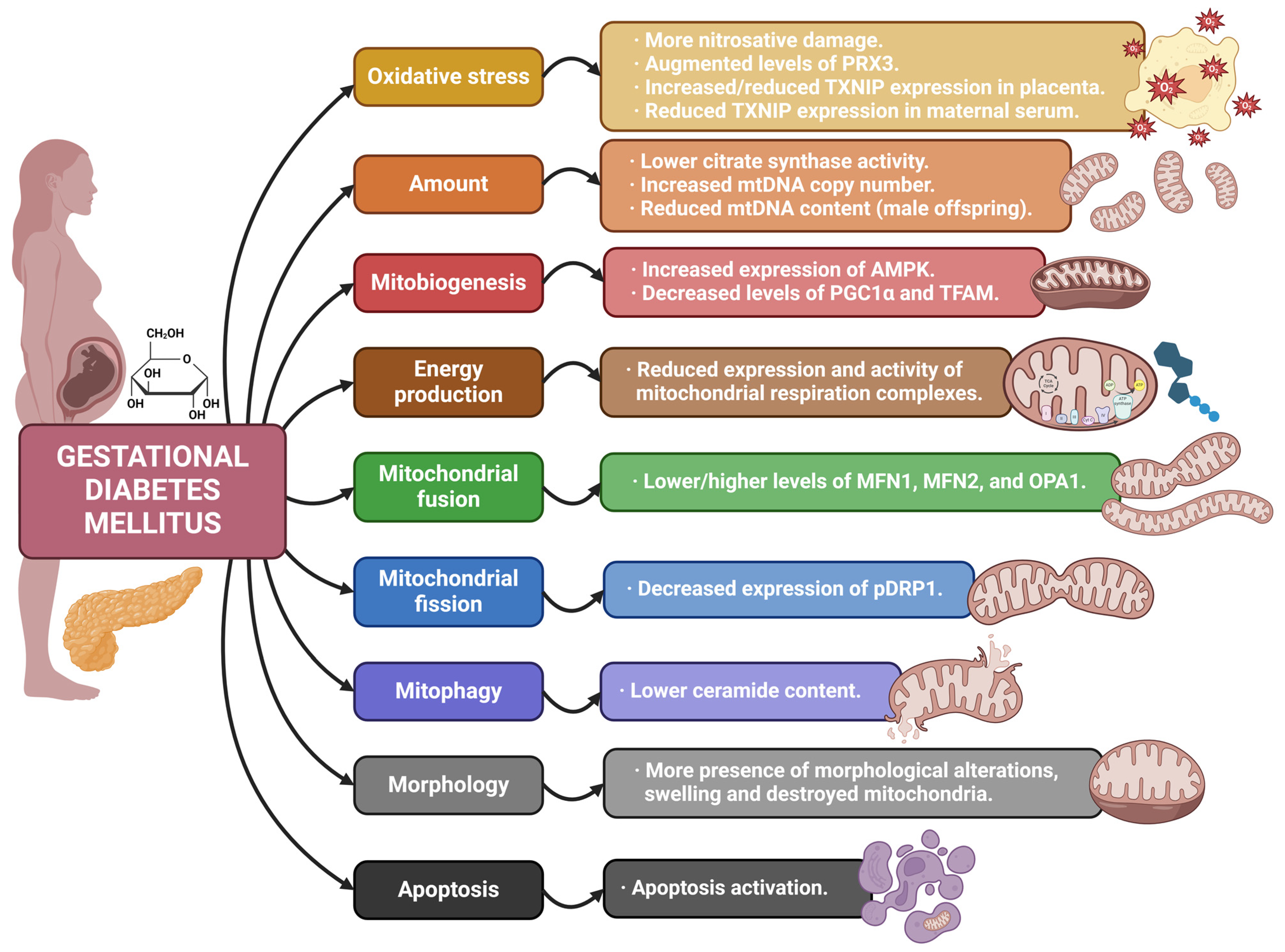
4.3. Mitochondria and Gestational Diabetes Mellitus
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Miller, W.L. Steroid hormone synthesis in mitochondria. Mol. Cell Endocrinol. 2013, 379, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.; Olvera-Sanchez, S.; Esparza-Perusquia, M.; Gomez-Chang, E.; Flores-Herrera, O. Multiple functions of syncytiotrophoblast mitochondria. Steroids 2015, 103, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Cui, X. Oxidative stress in the placenta. Histochem. Cell Biol. 2004, 122, 369–382. [Google Scholar] [CrossRef]
- Gupta, S.; Agarwal, A.; Sharma, R.K. The role of placental oxidative stress and lipid peroxidation in preeclampsia. Obstet. Gynecol. Surv. 2005, 60, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Elsharkawi, I.; Clapp, M.A.; Balcells, C. Effects of mitochondrial disease/dysfunction on pregnancy: A retrospective study. Mitochondrion 2019, 46, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J. Oxygen, the Janus gas; its effects on human placental development and function. J. Anat. 2009, 215, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Savitz, D.A.; Danilack, V.A.; Engel, S.M.; Elston, B.; Lipkind, H.S. Descriptive epidemiology of chronic hypertension, gestational hypertension, and preeclampsia in New York State, 1995–2004. Matern. Child. Health J. 2014, 18, 829–838. [Google Scholar] [CrossRef]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Colleoni, F.; Padmanabhan, N.; Yung, H.W.; Watson, E.D.; Cetin, I.; Tissot van Patot, M.C.; Burton, G.J.; Murray, A.J. Suppression of mitochondrial electron transport chain function in the hypoxic human placenta: A role for miRNA-210 and protein synthesis inhibition. PLoS ONE 2013, 8, e55194. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal 2012, 8, 343–357. [Google Scholar] [CrossRef]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Prieto, M.; Biarnés, X.; Vidossich, P.; Rovira, C. The Molecular Mechanism of the Catalase Reaction. J. Am. Chem. Soc. 2009, 131, 11751–11761. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Sesso, A.; Belizário, J.E.; Marques, M.M.; Higuchi, M.L.; Schumacher, R.I.; Colquhoun, A.; Ito, E.; Kawakami, J. Mitochondrial swelling and incipient outer membrane rupture in preapoptotic and apoptotic cells. Anat. Rec. 2012, 295, 1647–1659. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Pineda, F.J.; Hardwick, J.M. Mitochondrial fission and fusion dynamics: The long and short of it. Cell Death Differ. 2008, 15, 1147–1152. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, Q.; Gao, W.; Sehgal, S.A.; Wu, H. The multifaceted regulation of mitophagy by endogenous metabolites. Autophagy 2022, 18, 1216–1239. [Google Scholar] [CrossRef]
- Bassi, G.; Sidhu, S.K.; Mishra, S. The Expanding Role of Mitochondria, Autophagy and Lipophagy in Steroidogenesis. Cells 2021, 10, 1851. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021, 40, 101861. [Google Scholar] [CrossRef] [PubMed]
- Sultana, Z.; Maiti, K.; Aitken, J.; Morris, J.; Dedman, L.; Smith, R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am. J. Reprod. Immunol. 2017, 77, e12653. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Walsh, S.W.; Guo, J.D.; Zhang, J.Y. Maternal levels of prostacyclin, thromboxane, vitamin E, and lipid peroxides throughout normal pregnancy. Am. J. Obstet. Gynecol. 1991, 165, 1690–1694. [Google Scholar] [CrossRef] [PubMed]
- Gil-Acevedo, L.A.; Ceballos, G.; Torres-Ramos, Y.D. Foetal lipoprotein oxidation and preeclampsia. Lipids Health Dis. 2022, 21, 51. [Google Scholar] [CrossRef]
- Kaur, G.; Mishra, S.; Sehgal, A.; Prasad, R. Alterations in lipid peroxidation and antioxidant status in pregnancy with preeclampsia. Mol. Cell Biochem. 2008, 313, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sahay, A.S.; Sundrani, D.P.; Wagh, G.N.; Mehendale, S.S.; Joshi, S.R. Regional differences in the placental levels of oxidative stress markers in pre-eclampsia. Int. J. Gynaecol. Obstet. 2015, 129, 213–218. [Google Scholar] [CrossRef]
- Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Vázquez, C.M.; Mate, A.; Sobrevia, L.; Marín, R. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165354. [Google Scholar] [CrossRef]
- Butts, B.; Brown, J.A.; Denney, T.S., Jr.; Ballinger, S.; Lloyd, S.G.; Oparil, S.; Sanders, P.; Merriman, T.R.; Gaffo, A.; Singh, J.; et al. Racial Differences in XO (Xanthine Oxidase) and Mitochondrial DNA Damage-Associated Molecular Patterns in Resistant Hypertension. Hypertension 2022, 79, 775–784. [Google Scholar] [CrossRef]
- McCarthy, C.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, P.; Zhu, F.; Liao, J.; Wu, Y.; Hu, M.; Fu, H.; Qiao, J.; Lin, L.; Huang, B.; et al. The Potent Antioxidant MitoQ Protects Against Preeclampsia During Late Gestation but Increases the Risk of Preeclampsia When Administered in Early Pregnancy. Antioxid. Redox Signal 2021, 34, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Vishnyakova, P.A.; Volodina, M.A.; Tarasova, N.V.; Marey, M.V.; Tsvirkun, D.V.; Vavina, O.V.; Khodzhaeva, Z.S.; Kan, N.E.; Menon, R.; Vysokikh, M.Y.; et al. Mitochondrial role in adaptive response to stress conditions in preeclampsia. Sci. Rep. 2016, 6, 32410. [Google Scholar] [CrossRef]
- Shi, Z.; Long, W.; Zhao, C.; Guo, X.; Shen, R.; Ding, H. Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. PLoS ONE 2013, 8, e64351. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Mele, J.; Guo, C.; Myatt, L.G.; Myatt, L. MIR-210 modulates mitochondrial respiration in placenta with preeclampsia. Placenta 2012, 33, 816–823. [Google Scholar] [CrossRef]
- Yung, H.W.; Colleoni, F.; Dommett, E.; Cindrova-Davies, T.; Kingdom, J.; Murray, A.J.; Burton, G.J. Noncanonical mitochondrial unfolded protein response impairs placental oxidative phosphorylation in early-onset preeclampsia. Proc. Natl. Acad. Sci. USA 2019, 116, 18109–18118. [Google Scholar] [CrossRef]
- Holland, O.J.; Cuffe, J.S.M.; Dekker Nitert, M.; Callaway, L.; Kwan Cheung, K.A.; Radenkovic, F.; Perkins, A.V. Placental mitochondrial adaptations in preeclampsia associated with progression to term delivery. Cell Death Dis. 2018, 9, 1150. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L.; Muralimanoharan, S.; Maloyan, A. Effect of preeclampsia on placental function: Influence of sexual dimorphism, microRNA’s and mitochondria. Adv. Exp. Med. Biol. 2014, 814, 133–146. [Google Scholar] [CrossRef]
- Zhou, X.; Han, T.L.; Chen, H.; Baker, P.N.; Qi, H.; Zhang, H. Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 2017, 359, 195–204. [Google Scholar] [CrossRef]
- Udagawa, O.; Ishihara, T.; Maeda, M.; Matsunaga, Y.; Tsukamoto, S.; Kawano, N.; Miyado, K.; Shitara, H.; Yokota, S.; Nomura, M.; et al. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 2014, 24, 2451–2458. [Google Scholar] [CrossRef] [PubMed]
- Cushen, S.C.; Ricci, C.A.; Bradshaw, J.L.; Silzer, T.; Blessing, A.; Sun, J.; Zhou, Z.; Scroggins, S.M.; Santillan, M.K.; Santillan, D.A.; et al. Reduced Maternal Circulating Cell-Free Mitochondrial DNA Is Associated With the Development of Preeclampsia. J. Am. Heart Assoc. 2022, 11, e021726. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Enquobahrie, D.A.; Williams, M.A. A case-control study of maternal blood mitochondrial DNA copy number and preeclampsia risk. Int. J. Mol. Epidemiol. Genet. 2012, 3, 237–244. [Google Scholar] [PubMed]
- Williams, M.A.; Sanchez, S.E.; Ananth, C.V.; Hevner, K.; Qiu, C.; Enquobahrie, D.A. Maternal blood mitochondrial DNA copy number and placental abruption risk: Results from a preliminary study. Int. J. Mol. Epidemiol. Genet. 2013, 4, 120–127. [Google Scholar] [PubMed]
- Vangrieken, P.; Al-Nasiry, S.; Bast, A.; Leermakers, P.A.; Tulen, C.B.M.; Schiffers, P.M.H.; van Schooten, F.J.; Remels, A.H.V. Placental Mitochondrial Abnormalities in Preeclampsia. Reprod. Sci. 2021, 28, 2186–2199. [Google Scholar] [CrossRef]
- Bartho, L.A.; McKeating, D.R.; Hannan, N.J.; Kaitu’u-Lino, T.J.; Perkins, A.V. Transcriptional profiles of genes related to mitochondrial aging in placental pathologies. Mol. Hum. Reprod. 2022, 29, gaac026. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Guo, X.; Chen, R.; Feng, L. Downregulation of Mitofusin 2 in Placenta Is Related to Preeclampsia. Biomed. Res. Int. 2016, 2016, 6323086. [Google Scholar] [CrossRef]
- Ausman, J.; Abbade, J.; Ermini, L.; Farrell, A.; Tagliaferro, A.; Post, M.; Caniggia, I. Ceramide-induced BOK promotes mitochondrial fission in preeclampsia. Cell Death Dis. 2018, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhao, X.; Zhou, W.; Qi, H.; Zhang, H.; Han, T.L.; Baker, P. Impaired placental mitophagy and oxidative stress are associated with dysregulated BNIP3 in preeclampsia. Sci. Rep. 2021, 11, 20469. [Google Scholar] [CrossRef]
- Tong, J.; Zhao, W.; Lv, H.; Li, W.P.; Chen, Z.J.; Zhang, C. Transcriptomic Profiling in Human Decidua of Severe Preeclampsia Detected by RNA Sequencing. J. Cell Biochem. 2018, 119, 607–615. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Chen, G.; Chen, L.; Huang, Y.; Zhu, X.; Yu, Y. Increased FUN14 domain containing 1 (FUNDC1) ubiquitination level inhibits mitophagy and alleviates the injury in hypoxia-induced trophoblast cells. Bioengineered 2022, 13, 3620–3633. [Google Scholar] [CrossRef]
- Han, Y.W.; Yang, Z.; Ding, X.Y.; Yu, H. Differences in Liver Injury and Trophoblastic Mitochondrial Damage in Different Preeclampsia-like Mouse Models. Chin. Med. J. 2015, 128, 1627–1635. [Google Scholar] [CrossRef]
- Erlandsson, L.; Ducat, A.; Castille, J.; Zia, I.; Kalapotharakos, G.; Hedström, E.; Vilotte, J.L.; Vaiman, D.; Hansson, S.R. Alpha-1 microglobulin as a potential therapeutic candidate for treatment of hypertension and oxidative stress in the STOX1 preeclampsia mouse model. Sci. Rep. 2019, 9, 8561. [Google Scholar] [CrossRef] [PubMed]
- Cali, U.; Cavkaytar, S.; Sirvan, L.; Danisman, N. Placental apoptosis in preeclampsia, intrauterine growth retardation, and HELLP syndrome: An immunohistochemical study with caspase-3 and bcl-2. Clin. Exp. Obstet. Gynecol. 2013, 40, 45–48. [Google Scholar] [PubMed]
- Haché, S.; Takser, L.; LeBellego, F.; Weiler, H.; Leduc, L.; Forest, J.C.; Giguère, Y.; Masse, A.; Barbeau, B.; Lafond, J. Alteration of calcium homeostasis in primary preeclamptic syncytiotrophoblasts: Effect on calcium exchange in placenta. J. Cell Mol. Med. 2011, 15, 654–667. [Google Scholar] [CrossRef]
- Formanowicz, D.; Malińska, A.; Nowicki, M.; Kowalska, K.; Gruca-Stryjak, K.; Bręborowicz, G.; Korybalska, K. Preeclampsia with Intrauterine Growth Restriction Generates Morphological Changes in Endothelial Cells Associated with Mitochondrial Swelling-An In Vitro Study. J. Clin. Med. 2019, 8, 1994. [Google Scholar] [CrossRef]
- Berkane, N.; Liere, P.; Lefevre, G.; Alfaidy, N.; Nahed, R.A.; Vincent, J.; Oudinet, J.P.; Pianos, A.; Cambourg, A.; Rozenberg, P.; et al. Abnormal steroidogenesis and aromatase activity in preeclampsia. Placenta 2018, 69, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am. J. Pathol. 2008, 173, 451–462. [Google Scholar] [CrossRef]
- Krishna, U.; Bhalerao, S. Placental insufficiency and fetal growth restriction. J. Obstet. Gynaecol. India 2011, 61, 505–511. [Google Scholar] [CrossRef]
- Mathewson, K.J.; Chow, C.H.; Dobson, K.G.; Pope, E.I.; Schmidt, L.A.; Van Lieshout, R.J. Mental health of extremely low birth weight survivors: A systematic review and meta-analysis. Psychol. Bull. 2017, 143, 347–383. [Google Scholar] [CrossRef]
- Bujold, E.; Roberge, S.; Lacasse, Y.; Bureau, M.; Audibert, F.; Marcoux, S.; Forest, J.C.; Giguère, Y. Prevention of preeclampsia and intrauterine growth restriction with aspirin started in early pregnancy: A meta-analysis. Obstet. Gynecol. 2010, 116, 402–414. [Google Scholar] [CrossRef]
- Mandò, C.; De Palma, C.; Stampalija, T.; Anelli, G.M.; Figus, M.; Novielli, C.; Parisi, F.; Clementi, E.; Ferrazzi, E.; Cetin, I. Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E404–E413. [Google Scholar] [CrossRef]
- Biri, A.; Bozkurt, N.; Turp, A.; Kavutcu, M.; Himmetoglu, O.; Durak, I. Role of oxidative stress in intrauterine growth restriction. Gynecol. Obstet. Investig. 2007, 64, 187–192. [Google Scholar] [CrossRef]
- Naha, R.; Anees, A.; Chakrabarty, S.; Naik, P.S.; Pandove, M.; Pandey, D.; Satyamoorthy, K. Placental mitochondrial DNA mutations and copy numbers in intrauterine growth restricted (IUGR) pregnancy. Mitochondrion 2020, 55, 85–94. [Google Scholar] [CrossRef]
- Singh, A.; Jaiswar, S.P.; Priyadarshini, A.; Deo, S. Linking of oxidative stress and mitochondrial DNA damage to the pathophysiology of idiopathic intrauterine growth restriction. Int. J. Health Sci. 2023, 17, 15–22. [Google Scholar]
- Jones, R.; Peña, J.; Mystal, E.; Marsit, C.; Lee, M.J.; Stone, J.; Lambertini, L. Mitochondrial and glycolysis-regulatory gene expression profiles are associated with intrauterine growth restriction. J. Matern. Fetal Neonatal Med. 2020, 33, 1336–1345. [Google Scholar] [CrossRef]
- Guitart-Mampel, M.; Juarez-Flores, D.L.; Youssef, L.; Moren, C.; Garcia-Otero, L.; Roca-Agujetas, V.; Catalan-Garcia, M.; Gonzalez-Casacuberta, I.; Tobias, E.; Milisenda, J.C.; et al. Mitochondrial implications in human pregnancies with intrauterine growth restriction and associated cardiac remodelling. J. Cell Mol. Med. 2019, 23, 3962–3973. [Google Scholar] [CrossRef]
- Davy, P.; Nagata, M.; Bullard, P.; Fogelson, N.S.; Allsopp, R. Fetal growth restriction is associated with accelerated telomere shortening and increased expression of cell senescence markers in the placenta. Placenta 2009, 30, 539–542. [Google Scholar] [CrossRef]
- Biron-Shental, T.; Sukenik-Halevy, R.; Sharon, Y.; Goldberg-Bittman, L.; Kidron, D.; Fejgin, M.D.; Amiel, A. Short telomeres may play a role in placental dysfunction in preeclampsia and intrauterine growth restriction. Am. J. Obstet. Gynecol. 2010, 202, 381.e1–381.e7. [Google Scholar] [CrossRef]
- Lee, H.C.; Yin, P.H.; Lu, C.Y.; Chi, C.W.; Wei, Y.H. Increase of mitochondria and mitochondrial DNA in response to oxidative stress in human cells. Biochem. J. 2000, 348, 425–432. [Google Scholar] [CrossRef]
- Gutsaeva, D.R.; Carraway, M.S.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef]
- Poidatz, D.; Dos Santos, E.; Duval, F.; Moindjie, H.; Serazin, V.; Vialard, F.; De Mazancourt, P.; Dieudonné, M.N. Involvement of estrogen-related receptor-γ and mitochondrial content in intrauterine growth restriction and preeclampsia. Fertil. Steril. 2015, 104, 483–490. [Google Scholar] [CrossRef]
- Lattuada, D.; Colleoni, F.; Martinelli, A.; Garretto, A.; Magni, R.; Radaelli, T.; Cetin, I. Higher mitochondrial DNA content in human IUGR placenta. Placenta 2008, 29, 1029–1033. [Google Scholar] [CrossRef]
- Priliani, L.; Febinia, C.A.; Kamal, B.; Shankar, A.H.; Malik, S.G. Increased mitochondrial DNA copy number in maternal peripheral blood is associated with low birth weight in Lombok, Indonesia. Placenta 2018, 70, 1–3. [Google Scholar] [CrossRef]
- Peterside, I.E.; Selak, M.A.; Simmons, R.A. Impaired oxidative phosphorylation in hepatic mitochondria in growth-retarded rats. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1258–E1266. [Google Scholar] [CrossRef]
- Bartho, L.A.; O’Callaghan, J.L.; Fisher, J.J.; Cuffe, J.S.M.; Kaitu’u-Lino, T.J.; Hannan, N.J.; Clifton, V.L.; Perkins, A.V. Analysis of mitochondrial regulatory transcripts in publicly available datasets with validation in placentae from pre-term, post-term and fetal growth restriction pregnancies. Placenta 2021, 112, 162–171. [Google Scholar] [CrossRef]
- Zhu, H.L.; Shi, X.T.; Xu, X.F.; Zhou, G.X.; Xiong, Y.W.; Yi, S.J.; Liu, W.B.; Dai, L.M.; Cao, X.L.; Xu, D.X.; et al. Melatonin protects against environmental stress-induced fetal growth restriction via suppressing ROS-mediated GCN2/ATF4/BNIP3-dependent mitophagy in placental trophoblasts. Redox Biol. 2021, 40, 101854. [Google Scholar] [CrossRef]
- Zhu, H.L.; Shi, X.T.; Xu, X.F.; Xiong, Y.W.; Yi, S.J.; Zhou, G.X.; Liu, W.B.; Huang, M.M.; Gao, L.; Zhang, C.; et al. Environmental cadmium exposure induces fetal growth restriction via triggering PERK-regulated mitophagy in placental trophoblasts. Environ. Int. 2021, 147, 106319. [Google Scholar] [CrossRef]
- Parsons, A.M.; Bouma, G.J. A Potential Role and Contribution of Androgens in Placental Development and Pregnancy. Life 2021, 11, 644. [Google Scholar] [CrossRef]
- Shi, D.; Zhou, X.; Cai, L.; Wei, X.; Zhang, L.; Sun, Q.; Zhou, F.; Sun, L. Placental DNA methylation analysis of selective fetal growth restriction in monochorionic twins reveals aberrant methylated CYP11A1 gene for fetal growth restriction. FASEB J. 2023, 37, e23207. [Google Scholar] [CrossRef]
- Liu, Y.; Hou, W.; Meng, X.; Zhao, W.; Pan, J.; Tang, J.; Huang, Y.; Tao, M.; Liu, F. Heterogeneity of insulin resistance and beta cell dysfunction in gestational diabetes mellitus: A prospective cohort study of perinatal outcomes. J. Transl. Med. 2018, 16, 289. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Gesuita, R.; Di Renzo, G.C.; Meyyazhagan, A.; Tersigni, C.; Scambia, G.; Di Simone, N.; Marzioni, D. HtrA1 in Gestational Diabetes Mellitus: A Possible Biomarker? Diagnostics 2022, 12, 2705. [Google Scholar] [CrossRef]
- Alfadhli, E.M. Gestational diabetes mellitus. Saudi Med. J. 2015, 36, 399–406. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, I. Oxidant status in maternal and cord plasma and placental tissue in gestational diabetes. Placenta 2006, 27, 327–332. [Google Scholar] [CrossRef]
- Ramírez-Emiliano, J.; Fajardo-Araujo, M.E.; Zúñiga-Trujillo, I.; Pérez-Vázquez, V.; Sandoval-Salazar, C.; Órnelas-Vázquez, J.K. Mitochondrial content, oxidative, and nitrosative stress in human full-term placentas with gestational diabetes mellitus. Reprod. Biol. Endocrinol. 2017, 15, 26. [Google Scholar] [CrossRef]
- Lappas, M.; Hiden, U.; Desoye, G.; Froehlich, J.; Hauguel-de Mouzon, S.; Jawerbaum, A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid. Redox Signal 2011, 15, 3061–3100. [Google Scholar] [CrossRef]
- Sarina; Li, D. F.; Feng, Z.Q.; Du, J.; Zhao, W.H.; Huang, N.; Jia, J.C.; Wu, Z.Y.; Alamusi; Wang, Y.Y.; et al. Mechanism of Placenta Damage in Gestational Diabetes Mellitus by Investigating TXNIP of Patient Samples and Gene Functional Research in Cell Line. Diabetes Ther. 2019, 10, 2265–2288. [Google Scholar] [CrossRef]
- Qiu, C.; Hevner, K.; Abetew, D.; Sedensky, M.; Morgan, P.; Enquobahrie, D.A.; Williams, M.A. Mitochondrial DNA copy number and oxidative DNA damage in placental tissues from gestational diabetes and control pregnancies: A pilot study. Clin. Lab. 2013, 59, 655–660. [Google Scholar] [CrossRef]
- Pustovrh, M.C.; Jawerbaum, A.; Capobianco, E.; White, V.; Martínez, N.; López-Costa, J.J.; González, E. Oxidative stress promotes the increase of matrix metalloproteinases-2 and -9 activities in the feto-placental unit of diabetic rats. Free Radic. Res. 2005, 39, 1285–1293. [Google Scholar] [CrossRef]
- White, V.; Jawerbaum, A.; Sinner, D.; Pustovrh, C.; Capobianco, E.; González, E. Oxidative stress and altered prostanoid production in the placenta of streptozotocin-induced diabetic rats. Reprod. Fertil. Dev. 2002, 14, 117–123. [Google Scholar] [CrossRef]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Aston, C.E.; Lyons, T.; Chernausek, S.D. Effects of maternal diabetes and fetal sex on human placenta mitochondrial biogenesis. Placenta 2017, 57, 26–32. [Google Scholar] [CrossRef]
- Pasternak, Y.; Ohana, M.; Biron-Shental, T.; Cohen-Hagai, K.; Benchetrit, S.; Zitman-Gal, T. Thioredoxin, thioredoxin interacting protein and transducer and activator of transcription 3 in gestational diabetes. Mol. Biol. Rep. 2020, 47, 1199–1206. [Google Scholar] [CrossRef]
- Wang, L.; Hao, J.M.; Yu, A.Q.; Li, T.T.; Liu, R.R.; Li, L.; Li, J.; Li, X. The association of plasma peroxiredoxin 3 with insulin in pregnant women. Biochem. Biophys. Res. Commun. 2019, 508, 805–810. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Maloyan, A.; Myatt, L. Mitochondrial function and glucose metabolism in the placenta with gestational diabetes mellitus: Role of miR-143. Clin. Sci. 2016, 130, 931–941. [Google Scholar] [CrossRef]
- Kolac, U.K.; Kurek Eken, M.; Ünübol, M.; Donmez Yalcin, G.; Yalcin, A. The effect of gestational diabetes on the expression of mitochondrial fusion proteins in placental tissue. Placenta 2021, 115, 106–114. [Google Scholar] [CrossRef]
- Meng, Q.; Shao, L.; Luo, X.; Mu, Y.; Xu, W.; Gao, C.; Gao, L.; Liu, J.; Cui, Y. Ultrastructure of Placenta of Gravidas with Gestational Diabetes Mellitus. Obstet. Gynecol. Int. 2015, 2015, 283124. [Google Scholar] [CrossRef]
- Tiranti, V.; Rossi, E.; Ruiz-Carrillo, A.; Rossi, G.; Rocchi, M.; DiDonato, S.; Zuffardi, O.; Zeviani, M. Chromosomal localization of mitochondrial transcription factor A (TCF6), single-stranded DNA-binding protein (SSBP), and endonuclease G (ENDOG), three human housekeeping genes involved in mitochondrial biogenesis. Genomics 1995, 25, 559–564. [Google Scholar] [CrossRef]
- Fisher, J.J.; Vanderpeet, C.L.; Bartho, L.A.; McKeating, D.R.; Cuffe, J.S.M.; Holland, O.J.; Perkins, A.V. Mitochondrial dysfunction in placental trophoblast cells experiencing gestational diabetes mellitus. J. Physiol. 2021, 599, 1291–1305. [Google Scholar] [CrossRef]
- Mandò, C.; Anelli, G.M.; Novielli, C.; Panina-Bordignon, P.; Massari, M.; Mazzocco, M.I.; Cetin, I. Impact of Obesity and Hyperglycemia on Placental Mitochondria. Oxid. Med. Cell Longev. 2018, 2018, 2378189. [Google Scholar] [CrossRef]
- Duan, Y.; Sun, F.; Que, S.; Li, Y.; Yang, S.; Liu, G. Prepregnancy maternal diabetes combined with obesity impairs placental mitochondrial function involving Nrf2/ARE pathway and detrimentally alters metabolism of offspring. Obes. Res. Clin. Pract. 2018, 12, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Hastie, R.; Lappas, M. The effect of pre-existing maternal obesity and diabetes on placental mitochondrial content and electron transport chain activity. Placenta 2014, 35, 673–683. [Google Scholar] [CrossRef]
- Sobrevia, L.; Abarzúa, F.; Nien, J.K.; Salomón, C.; Westermeier, F.; Puebla, C.; Cifuentes, F.; Guzmán-Gutiérrez, E.; Leiva, A.; Casanello, P. Review: Differential placental macrovascular and microvascular endothelial dysfunction in gestational diabetes. Placenta 2011, 32 (Suppl. S2), S159–S164. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, G.; Valero, P.; Ramírez, M.A.; Sobrevia, L. Intracellular pH modulation in human umbilical vein endothelial cells requires sodium/proton exchangers activity in gestational diabesity but sodium/proton exchanger-1 activity in gestational diabetes with maternal pre-gestational normal weight or overweight. Physiol. Soc. 2019, 43, C039. [Google Scholar]
- Sultan, S.A.; Liu, W.; Peng, Y.; Roberts, W.; Whitelaw, D.; Graham, A.M. The Role of Maternal Gestational Diabetes in Inducing Fetal Endothelial Dysfunction. J. Cell Physiol. 2015, 230, 2695–2705. [Google Scholar] [CrossRef] [PubMed]
- Abbade, J.; Klemetti, M.M.; Farrell, A.; Ermini, L.; Gillmore, T.; Sallais, J.; Tagliaferro, A.; Post, M.; Caniggia, I. Increased placental mitochondrial fusion in gestational diabetes mellitus: An adaptive mechanism to optimize feto-placental metabolic homeostasis? BMJ Open Diabetes Res. Care 2020, 8, e000923. [Google Scholar] [CrossRef]
- Hill, M.; Pařízek, A.; Šimják, P.; Koucký, M.; Anderlová, K.; Krejčí, H.; Vejražková, D.; Ondřejíková, L.; Černý, A.; Kancheva, R. Steroids, steroid associated substances and gestational diabetes mellitus. Physiol. Res. 2021, 70, S617–S634. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Disease | Study Design | Major Findings |
|---|---|---|---|
| [32] | PE | In vitro | Human umbilical vein endothelial cells exposed to plasma from women with PE report a decrease in mitochondrial function, as well as augmented mitochondrial superoxide production. The pre-treatment of cells with the mitochondrial-targeted antioxidant MitoTempo protects them against cell death induced by peroxide, also normalizing mitochondrial metabolism and reducing mitochondrial superoxide production. |
| [33] | PE | Observational Animal (mouse) | Placentas from subjects with PE show significantly high oxidative stress, mitochondrial damage, and reduced GPX activity. MitoQ late administration ameliorates uterine perfusion pressure, while early administration proves to be detrimental. |
| [34] | PE | Observational | OPA1 gene and protein expression is increased in early-onset PE placentas. TFAM expression is downregulated in comparison to controls. mtDNA copy number in early-onset PE placentas is significantly higher compared to normal pregnancies. Increase in citrate synthase activity is found in both early- and late-onset PE. |
| [37] | PE | Observational | PE placentas show reduced OXPHOS capacity (with no changes in gene or protein expression). Mitochondrial unfolded protein response (mtUPR) may be associated with these results, possibly being a therapeutic target for placental function restoration. |
| [38] | PE | Observational | Expression of mitochondrial fission/fusion markers, apoptosis proteins, and OXPHOS complexes are altered in a different way in term and pre-term PE placental samples. PE term placentas report increased expression of fusion markers and proteins related to apoptosis suppression. Mitochondrial content and respiration rate are elevated in term PE placentas. |
| [40] | PE | Observational | There is an abnormal regulation of mitochondrial dynamics, autophagy, and biogenesis in PE placentas, with reduced protein expression of fusion markers (OPA1, MFN2, and MFN1), the mitophagy receptor BNIP3, as well as mitobiogenesis mediators (PGC1α and SIRT3). Compromised lipid metabolism in these samples may result from dysfunctional mitochondria. |
| [45] | PE | Observational | Mitochondrial content is lower in placentas from PE mothers, with an increase in glycolysis components. Gene and protein expression of mitochondrial biogenesis regulators is lower in PE placentas, whereas the abundance of mitophagy-related proteins like BNIP3 is higher. Apoptosis activation and inflammation are also reported in placentas from PE women. |
| [46] | PE FGR | Observational | Gene expression of mitochondrial dynamics markers experiment changes in those placentas affected by PE and FGR. The TOMM20/Parkin ratio arises as a potential marker to discern unhealthy placental tissue. |
| [47] | PE | Observational In vitro | MFN2 gene and protein expression, as well as ATP levels, are significantly decreased in placentas from PE pregnancies compared to regular ones. MFN2 expression and TEV-1 cells’ viability are reduced during hypoxia. TEV-1 cells’ viability and ATP levels are also reduced after MFN2 knockdown. Consequently, defects related to MFN2 could cause mitochondrial alterations and negatively affect trophoblastic cells’ viability in PE. |
| [48] | PE | Observational In vitro | Mitochondrial dynamics in PE placental tissue are tilted toward fission (augmented DRP1 and reduced OPA1 expression). Transmission electron microscopy shows increased fragmentation of the organelle in PE placentas. Ceramides are more accumulated in the mitochondria from PE placentas. Mitophagy markers PINK1 and Parkin report an increase in placentas from PE pregnancies. |
| [49] | PE | Observational | The mitophagy receptor BNIP3 shows reduced expression in PE placentas, together with altered autophagy and augmented mitochondrial damage. |
| [50] | PE | Observational | Decidual gene transcription is modified in severe PE. Several genes related to glycolysis/gluconeogenesis, HIF-1 signaling pathway, and mitophagy (BNIP3) are importantly downregulated in the decidua of PE placentas. |
| [52] | PE | In vitro Animal (mouse) | Reduced gene expression and low ubiquitination of FUNDC1 are found in hypoxic trophoblast cells of pregnant women with PE. |
| [53] | PE | Animal (mouse) | Mitochondria swelling and cristae disappearance are observed in the trophoblasts of experimental PE groups. |
| [57] | FGR PE | In vitro | Endothelial cells’ short exposition with serum from pregnant mothers with FGR and PE slightly reduces cell viability. Prolonged exposition leads to important morphological changes (swollen mitochondria), increased ROS generation, autophagy, decreased cell viability. |
| Reference | Disease | Study Design | Major Findings |
|---|---|---|---|
| [46] | PE FGR | Observational | Gene expression of mitochondrial dynamics markers changes in placentas affected by PE and FGR. The TOMM20/Parkin ratio arises as a potential marker to discern unhealthy placental tissue. |
| [57] | FGR PE | In vitro | Endothelial cells’ short exposition with serum from pregnant mothers with FGR and PE slightly reduces cell viability. Prolonged exposition leads to important morphological changes (swollen mitochondria), increased ROS generation, autophagy, decreased cell viability. |
| [63] | FGR | Observational | Lower mRNA levels of complex II, III, and IV are found in cytotrophoblast cells from FGR samples, without differences at the protein level. mtDNA is increased in FGR placentas, while mtDNA and NRF1 expression are significantly lower in isolated cytotrophoblast cells. The activity of cytotrophoblast respiratory chain is importantly augmented in placentas of FGR pregnancies. |
| [65] | FGR | Observational | mtDNA copy number is increased in FGR placental samples. Higher mutation rate is found in both coding and non-coding regions of mtDNA in several FGR placentas. SIRT3 expression is downregulated in FGR placentas. |
| [66] | FGR | Observational | mtDNA circulating levels are significantly higher in blood from mothers carrying FGR fetuses. SIRT3 expression is reduced in FGR placenta. Increased oxidative stress causes mtDNA damage in FGR. |
| [67] | FGR | Observational | The glycolysis-regulatory gene PDK1 is positively related to FGR. mtDNA content and oxidative stress are positively related to FGR. |
| [73] | FGR | Observational | FGR placentas show lower mtDNA and protein content (related to downregulation of ERRγ expression). Placental mtDNA content is directly correlated with fetal weight. PGC1α and SIRT1 gene expression is reduced in FGR+PE placentas. |
| [75] | FGR | Observational | mtDNA copy number in maternal venous blood is inversely associated with children’s birth weight, mostly in the third trimester. This might be a marker for identifying possible FGR. |
| [68] | FGR | Observational | FGR placentas report an important reduction in OXPHOS complex I enzymatic activity, together with complex I-stimulated oxygen consumption. They also show an increase in SIRT3 protein concentrations. Citrate synthase activity is significantly decreased in FGR newborns. |
| [77] | FGR | Observational | MFN1 protein expression is reduced in FGR placentas. TOMM20 gene and protein expression is increased in FGR placental tissue. |
| [78] | FGR | Animal (mouse) | Melatonin ameliorates Cd-caused FGR through the antagonism of BNIP3-dependent mitophagy in placental tissue, as well as the excessive release of ROS. |
| Reference | Disease | Study Design | Major Findings |
|---|---|---|---|
| [87] | GDM | Observational | Placentas with GDM are more susceptible to nitrosative damage as compared to normal placentas. GDM placentas report increased expression of AMPK, which may be associated with the maintenance of mitobiogenesis at a normal rate. |
| [89] | GDM | Observational In vitro | TXNIP expression in GDM placental tissue is increased compared to control. TXNIP expression in HTR-8/SVneo cells treated with high glucose is augmented, leading to ROS accumulation, mitochondrial defects, apoptosis, and migration inhibition. |
| [93] | GDM | Observational | Placental mitochondrial biogenesis is affected by GDM and offspring sex. GDM is associated with a reduction in PGC1α and TFAM levels in the placenta. Pregnancies with GDM and male offspring are related to reduced placental PGC1α and TFAM, as well as mtDNA content. Regarding female offspring, only decreased PGC1α is reported. |
| [94] | GDM | Observational | TXNIP gene expression is significantly elevated in serum of women with GDM. TXNIP gene expression is decreased in GDM placental tissue and cord blood. Pro-inflammatory alterations are related to low mRNA TXN/TXNIP ratio in maternal serum of GDM women but not in the placenta or in umbilical cord blood. |
| [95] | GDM | Observational | The H2O2 scavenger PRX3, with mitochondrial location, is increased in maternal plasma during pregnancies with GDM. It has an active role in the response to insulin release, suggesting it may be an indicator of high insulin resistance. |
| [96] | GDM | Observational | The expression of mitochondrial respiration complexes and PGC1α is significantly reduced in the placenta of women with GDM controlled by medication compared with women with GDM controlled by diet and controls. |
| [97] | GDM | Observational | Gene and protein expression fusion markers MFN1, MFN2, and OPA1 are lower in pre-DM and GDM placentas compared to healthy controls. PGC1α expression is reduced in these placentas as well. Proteins related to mitochondrial protein folding are also decreased in them. |
| [98] | GDM | Observational | GDM placentas show more swollen or destroyed mitochondria than those from regular pregnancies. |
| [101] | GDM | Observational | Obese women without GDM show increased mtDNA placental levels compared to normal weight women, suggesting increased biogenesis. The morphology of their mitochondria is similar. In obese women with GDM, mtDNA was not augmented compared to normal weight women, whereas morphological alterations are documented in their mitochondria. |
| [102] | GDM | Observational | Women with pre-existing maternal type 2 diabetes mellitus and obesity show elevated ROS production, decreased mtDNA content, reduced OXPHOS complexes I, II-III activities in placenta. |
| [100] | GDM | Observational | mtDNA copy number is higher in GDM placentas. Citrate synthase activity is shown to be lower in GDM placental samples. Placentas from GDM pregnancies report reduced respiration levels, mainly related to complexes I and II. Fusion proteins MFN1 and MFN2 report significant rises in GDM placentas. |
| [107] | GDM | Observational In vitro | Mitochondrial dynamics is tilted towards fusion in placentas of GDM women, supported by transmission electron microscopy and alterations in OPA1 (increases) and pDRP1 (decreased) expression. Placental ceramide content is lower in GDM. In vitro experiments show that increased insulin exposure promotes mitochondrial fusion. |
| Full Term | Abbreviation |
|---|---|
| Mitochondrial DNA | mtDNA |
| Reactive oxygen species | ROS |
| Preeclampsia | PE |
| Fetal growth restriction | FGR |
| Gestational diabetes mellitus | GDM |
| Oxidative phosphorylation | OXPHOS |
| Outer mitochondrial membrane | OMM |
| Inner mitochondrial membrane | IMM |
| Superoxide dismutase | SOD |
| Peroxisome proliferator-activated receptor-γ coactivator-1α | PGC1α |
| Nuclear respiratory factor 1 | NRF1 |
| Nuclear respiratory factor 2 | NRF2 |
| Mitochondrial transcription factor A | TFAM |
| Mitofusin 1 | MFN1 |
| Mitofusin 2 | MFN2 |
| Optic atrophy 1 protein | OPA1 |
| Dynamin-related protein 1 | DRP1 |
| Dynamin 2 | DNM2 |
| Mitochondrial fission 1 protein | FIS1 |
| PTEN-induced kinase 1 | PINK1 |
| E3 ubiquitin-protein ligase parkin | Parkin |
| Calcium binding and coiled-coil domain 2 | CALCOCO2/NDP52 |
| Optineurin | OPTN |
| FUN14 domain containing 1 | FUNDC1 |
| BCL2 interacting protein 3 | BNIP3 |
| BCL2-like 13 apoptosis facilitator | BCL2L13 |
| FKBP prolyl isomerase 8 | FKBP8 |
| Prohibitin 2 | PHB2 |
| Mitochondrial permeability transition pores | mPTP |
| Soluble Flt-1 | sFlt-1 |
| Soluble endoglin | sENG |
| Natural killer | NK |
| Catalase | CAT |
| Glutathione peroxidase | GPX |
| Mitochondrial unfolded protein response | mtUPR |
| Sirtuin 3 | SIRT3 |
| Mitochondrial import receptor subunit TOM20 homolog | TOMM20 |
| 20α-dihydroprogesterone | 20α-DHP |
| Intrauterine growth restriction IUGR | IUGR |
| Low birth weight | LBW |
| Estrogen-related receptor γ | ERRγ |
| Sirtuin 1 | SIRT1 |
| Type 2 diabetes mellitus | T2DM |
| Body mass index | BMI |
| Thioredoxin-interacting protein | TXNIP |
| Thioredoxin | TXN |
| Peroxiredoxin 3 | PRX3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toledano, J.M.; Puche-Juarez, M.; Galvez-Navas, J.M.; Moreno-Fernandez, J.; Diaz-Castro, J.; Ochoa, J.J. Pregnancy Disorders: A Potential Role for Mitochondrial Altered Homeostasis. Antioxidants 2024, 13, 979. https://doi.org/10.3390/antiox13080979
Toledano JM, Puche-Juarez M, Galvez-Navas JM, Moreno-Fernandez J, Diaz-Castro J, Ochoa JJ. Pregnancy Disorders: A Potential Role for Mitochondrial Altered Homeostasis. Antioxidants. 2024; 13(8):979. https://doi.org/10.3390/antiox13080979
Chicago/Turabian StyleToledano, Juan M., María Puche-Juarez, Jose Maria Galvez-Navas, Jorge Moreno-Fernandez, Javier Diaz-Castro, and Julio J. Ochoa. 2024. "Pregnancy Disorders: A Potential Role for Mitochondrial Altered Homeostasis" Antioxidants 13, no. 8: 979. https://doi.org/10.3390/antiox13080979