Abstract
Mutations in highly conserved genes encoding components of the electron transport chain (ETC) provide valuable insights into the mechanisms of oxidative stress and mitochondrial ROS (mtROS) in a wide range of diseases, including cancer, neurodegenerative disorders, and aging. This review explores the structure and function of the ETC in the context of its role in mtROS generation and regulation, emphasizing its dual roles in cellular damage and signaling. Using Caenorhabditis elegans as a model organism, we discuss how ETC mutations manifest as developmental abnormalities, lifespan alterations, and changes in mtROS levels. We highlight the utility of redox sensors in C. elegans for in vivo studies of reactive oxygen species, offering both quantitative and qualitative insights. Finally, we examine the potential of C. elegans as a platform for testing ETC-targeting drug candidates, including OXPHOS inhibitors, which represent promising avenues in cancer therapeutics. This review underscores the translational relevance of ETC research in C. elegans, bridging fundamental biology and therapeutic innovation.
1. Introduction
About 1 in 5000 adults suffer from primary mitochondrial diseases [1]. Most defects are due to either mutations in the nuclear genome or its mitochondrial equivalent (mtDNA), and most of these lie within the electron transport chain (ETC) [2]. Interestingly, mtDNA is prone to mutation—firstly, because it lacks wrapping histones and, secondly, because mtDNA repair is less efficient. This vulnerability is compounded by the proximity of mtDNA to the production of reactive oxygen species (ROS) from the ETC, leading to a 10- to 20-fold higher mtDNA mutation rate relative to nuclear DNA [3].
The components of the ETC are highly conserved from yeast to human [4], making the animal models of human genetic mutation tractable and attractive options to unravel the complex background of mitochondrial diseases, providing clues to possible therapeutic strategies [5,6].
Herein, we summarize knowledge about the structure and function of ETC complexes and give an overview of human diseases related to mutations of different complex subunits. Our focus is on homologous ETC mutations in the nematode Caenorhabditis elegans and their possible contributions to a better understanding of ETC functions. The ETC is a major source of ROS (mtROS) production in cells. By summarizing current views on redox balance and redox signaling, we demonstrate how C. elegans ETC mutant phenotypes are related to ROS. Our interest in this field was prompted by modelling complex II mutants by generating a clinically relevant SDHB mutant model in worms. As SDH germline mutations predispose to rare neuroendocrine tumors, we became interested in drug candidates targeting OXPHOS (thereby influencing ROS) as emerging therapeutic options in cancer treatment. Finally, we demonstrate how the nematode can be used as a test bed in evaluating effects of ETC-targeting agents.
2. Structure and Function of the Electron Transport Chain (ETC) Complexes and Human Diseases Related to ETC Subunit Mutations
The ETC, located in the inner mitochondrial membrane (IMM), ensures that electrons from reduced co-enzymes NADH and FADH2 generated during catabolic processes (also in the TCA cycle) are passed down an electrochemical gradient to elemental oxygen, the terminal electron acceptor, whilst pari passu drives protons outwardly into the intermembrane space. Proton re-entry across the IMM then drives ATP production [7] (Figure 1).
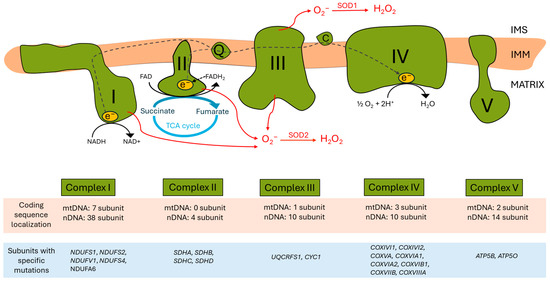
Figure 1.
Human mitochondrial electron transport chain (ETC) complexes and the oxidative phosphorylation system. In the upper part, the four complexes of the ETC and the ATP synthase within the mitochondrial inner membrane (IMM) are schematized, alongside the electron pathway (grey dashed arrows). The intermembrane space (IMS) is at the top, and the mitochondrial matrix is at the bottom. Complexes I and II are the two main entries of reducing equivalents in the ETC. Complex I transfers electrons from NADH to CoQ (ubiquinone) in the IMM, and during the process, 4H+ ions are translocated into the IMS. Complex II is the SDH complex, which transfers electrons from succinate, a tricarboxylic acid (TCA) cycle intermediate, to CoQ, as well as complex I. CoQ transfers its electrons derived from complexes I and II to complex III through the phospholipid bilayer. Complex III contributes to the electron transfer from CoQ to cytochrome C (C) in the IMS; during the process, another four H+ ions are pumped into the IMS. Cytochrome C is a hydrophilic electron transporter that can move in the intermembrane space and transfers electrons to complex IV. Complex IV, the terminal ETC component, allows for electron transfer from cytochrome C to oxygen as a terminal electron acceptor; reduced oxygen with two H+ ions results in H2O production on the matrix side. At the same time, the complex pumps two H+ ions to the IMS. Complex V/ATP synthase uses the proton gradient generated across the IMM for ATP synthesis. At some points of the ETC (complexes I, II, and III), electron leakage can occur, leading to the generation of reactive oxygen species (ROS) (red arrows). In the lower part of the figure, a table summarizes the number of subunits of each complex coded either in mitochondrial DNA (mtDNA) or in nuclear DNA (nDNA) (highlighted in light brown), as well as a brief list of corresponding human subunits carrying specific mutations (highlighted in light blue).
2.1. Complex I: NADH Oxidoreductase
Complex I of the electron transport chain is NADH oxidoreductase, which oxidizes NADH derived from the TCA cycle to NAD+ by directing the transfer of one electron pair from NADH to ubiquinone. The electron pair from NADH is first shuttled on a flavin-mononucleotide (FMN) to NADH dehydrogenase, and subsequently, inside complex I, electrons travel through Fe-S clusters to ubiquinone, which eventually becomes reduced to ubiquinol. As a consequence, four H+ ions are pumped into the intermembrane space.
Complex I, the largest complex of the respiratory chain, consists of 38 nuclear-encoded subunits and 7 mitochondrially encoded partners [8,9]. Complex I is L-shaped, with a hydrophilic peripheral arm protruding into the matrix, while the hydrophobic partner is embedded in the IMM. The peripheral arm contains the redox centers, the NADH-oxidizing N module and the Q module, which reduce ubiquinone. The proton-translocating machinery (P module) is localized in the membrane arm [10,11]. Fourteen conserved core subunits of complex I are necessary and sufficient for the catalysis of energy transduction: half of the core subunits belong to the peripheral arm, and the other half belong to the membrane arm. Thirty-one additional (supernumerary) subunits are distributed around the core; these subunits are functionally less well characterized but likely contribute towards the control of the assembly and the stability of the complex [12].
Most patients with primary mitochondrial diseases have complex I mutations [13]. Complex I deficiencies represent one-third of all early-onset mitochondrial disorders, with mutations in both nuclear and mitochondrial genes generating a wide range of clinical outcomes. The clinical phenotypes are classified into five groups as follows: Leigh syndrome, progressive leukoencephalopathy, neonatal cardiomyopathy, severe infantile lactic acidosis, and a group of unspecified encephalomyopathies [14].
Nuclear-encoded NDUFS1, NDUFS2, NDUFV1, and NDUFS4 genes are mutational hot spots for isolated complex I deficiency [8]. For example, numerous mutations (over 40) have been identified in N-module subunit NDUFV1, resulting in various phenotypes, such as hypotonia, lethargy, myopathy, or fatigue [15]. Substitutions of E104A, M292T, R118Q, M443K, E148K, and F84L in the NDUFS2 subunit in the Q module lead to Leigh syndrome [16,17], while R138Q, R333Q, and M292T mutations of NDUFS2 result in a Leigh-like syndrome [17]. Patients with R228Q, P229Q, and S413P mutations in NDUFS2 suffer from cardiomyopathy and encephalomyopathy [18]. N24A and R30A substitutions in supernumerary subunit NDUFB4 disrupt supercomplex assembly [19]. A recent report described neurological symptoms and/or elevated lactate levels in the cases of four unrelated children with various substitutions and/or deletions in the NDUFA6 gene, either in a homozygous or heterozygous state. Symptoms arose after 3 months and 2 years of age in the cases of two patients. The third patient died at the age of 13 weeks. NDUFA6 is a supernumerary subunit of the Q module, and investigations performed on fibroblasts of the above subjects showed complex I assembly defects [20].
Besides the structural subunits of electron transport, complex I assembly requires additional factors. The mutation of these factors can lead to structural defects of complex I. Patients carrying different mutations (deletions or substitutions of Val546Leu and Ala170Val) in complex I assembly factor ACAD9 suffer from an isolated oxidative phosphorylation complex I deficiency, affecting primarily muscles, the liver, nervous system, and heart [21,22,23,24].
2.2. Complex II: Succinate Dehydrogenase (SDH)
Complex II transfers electrons from FADH2 through Fe-S clusters to ubiquinone. The hydrophobic benzoquinone structure of ubiquinone/ubiquinol enables the transfer of electrons derived from complexes I and complex II to complex III through the phospholipid bilayer.
Mitochondrial complex II is also called the succinate dehydrogenase complex (SDH or succinate–ubiquinone oxidoreductase). A unique property of the SDH complex/complex II compared to other ETC components is that paralogous genes encoding the different subunits are located exclusively in the nuclear genome. SDH is the only complex of the respiratory chain that does not pump protons across the mitochondrial inner membrane during its active catalytic cycle. This complex plays a dual role in generating energy: in the TCA cycle, SDH oxidizes succinate to double-bonded fumarate; then, using electrons released by the succinate–fumarate conversion, it catalyzes the reduction of ubiquinone to ubiquinol in the mitochondrial respiratory chain.
The tetrameric SDH enzyme complex consists of four functionally distinct subunits, SDHA, SDHB, SDHC, and SDHD. All the four subunits are encoded by the nuclear genome. The 3D structure of the well-known porcine heart SDH shows that the enzyme complex has a hydrophilic head protruding into the mitochondrial matrix and a hydrophobic tail located within the mitochondrial inner membrane (IMM) [25,26].
Hydrophilic SDHA and SDHB subunits, together, form the catalytic site of the enzyme. SDHA is a flavoprotein containing the binding site for succinate and a covalently bound FAD prosthetic group. SDHB is a subunit displaying three strongly conserved iron–sulfur clusters ([2Fe-2S], [4Fe-4S], and [3Fe-4S]), which undergo oxidation-=–reduction processes as the electrons pass. Hydrophobic SDHC and SDHD subunits anchor the enzyme complex to the IMM and contain cytochrome-B [25,27].
The SDH enzymatic reaction starts with the binding of succinate to its binding site, subsequently leading to a conformational change in the SDHA subunit and oxidation of succinate with FAD, which, in turn, becomes reduced to FADH2. Next, electrons from FADH2 pass through the one electron-carrier Fe/S centers and, finally, convert/reduce ubiquinone to ubiquinol in two successive steps.
Mammalian SDH complexes display two ubiquinone binding sites: the proximal site (QP) is located closer to the matrix, at the interface of SDHB, SDHC, and SDHD subunits, and shows higher affinity for ubiquinone [25]. The other, distal ubiquinone binding site (QD) with lower affinity for ubiquinone is more distant from the matrix [27]. The importance of the QD site and the conserved haem moiety remains to be determined in eukaryotes [27].
The assembly of the SDH complex has recently been understood and requires the coordinated action of four assembly factors, which are necessary for the maturation of the soluble SDHA and SDHB subunits and subsequent assembly of the entire complex [28].
Germline mutations in the four SDH subunits of complex II predispose to various tumors: paragangliomas, pheochromocytomas, and gastrointestinal stromal tumors (GIST) [29]. Pheochromocytomas (PHEOs) and paragangliomas (PGLs) are rare neuroendocrine tumors arising from the adrenal medulla or from sympathetic and parasympathetic ganglia of the peripheral nerve system, respectively [30]. SDH-derived GISTs make up approximately 10% of these tumors [31], which are mainly formed in the stomach, and patients are usually younger than 30 years old [32]. SDH-derived paragangliomas can also present in dyads with GISTs (Carney-Stratakis syndrome) or in triads with GISTs and pulmonary adenoma (Carney triad). SDH-mutated PGLs/PHEOs have also been reported with pituitary adenomas. SDH-deficiency also occurs in 0.05–0.2% of renal cell carcinomas [33].
Many substitutions have been described in the SDHB subunit that are associated with GIST, renal cell carcinoma, multiple hamartomas, acute T-cell leukemia, PHEOs, and PGLs (reviewed in [34,35]). Some of them display regional localization, like the R46Q Maori mutation [36,37,38]. Germline mutations in the SDHC gene can also lead to PGL [39]. Some SDHA mutations, such as L511P, G233V, Arg31*, Arg512*, S445L, and UTRdel, cause PHEO and PGL [40], while other SDHA germline mutations have been reported in SDH-deficient GISTs [41].
CoQ (coenzyme Q/ubiquinone/ubiquinol) has a benzoquinone head group and a long isoprenoid side chain and resides in biological membranes [42]. In the ETC, CoQ acts as a transmitter of electrons from complexes I and II to complex III. CoQ7 mutations (for example, R54Q) result in a decrease in coenzyme Q10 production [43] and manifest in different clinical phenotypes. 1Met? mutation (c.3G > T (p.1Met?) mutation changes the canonical ATG start codon into an ATT sequence, blocking the initiation of translation, which leads to distal hereditary motor neuropathy [44], and Ala205HisfsTer48 and Met135Val cause cardiomyopathy and gastrointestinal obstruction [45], while Pro108Thr results in hereditary spastic paraplegia [46]. The increasing number of case reports describing different CoQ mutations [47,48] might provide clues to possible treatments.
2.3. Complex III: Cytochrome C Reductase
Complex III or cytochrome C reductase constitutes the central component of the ETC. The structure forms a symmetric dimer [49]; each of the monomers is composed of 11 different subunits [50]. Of the three proton-pumping complexes, complex III, has the fewest subunits: one encoded by mtDNA and 10 encoded on nDNA [50]. Complex III contains a series of catalytic subunits: cytochrome b, containing two CoQ binding sites and two heme b groups; UQCRFS1, the Rieske Fe–S protein; and CYC1, containing heme c as the prosthetic group. All of these contribute to electron transfer from ubiquinol (CoQ) through cytochrome C to complex IV whilst also contributing to the generation of the proton gradient across the mitochondrial membrane. An interesting step of complex III assembly is the post-translational cleavage of an N-terminal fragment of the UQCRFS1 protein known as ‘UQCRFS1N’. Cleavage is undertaken by the matrix processing peptidase activity of UQCRC1 and UQCRC2 [51]. UQCRFS1N is retained and bound in the interface between the UQCRC1 and UQCRC2 subunits [52].
UQCRFS1 mutants show a reduction in mitochondrial complex III activity. Homozygous Val72_Thr81del10, in combination with Val14Asp/p.Arg204*, leads to cardiomyopathy and alopecia totalis [53,54]. Trp96Cys and Leu215Phe mutations in human CYC1 manifest in insulin-responsive hyperglycemia [55].
CyCS (cytochrome C) shuttles electrons between complexes III and IV. Both Lys101del mutation in the α-helix of the C-terminal domain of CYCS and His27Tyr substitution result in autosomal dominant non-syndromic thrombocytopenia [56,57]. G41S, Y48H, and Tyr98His substitutions cause thrombocytopenia and affect cellular bioenergetics and apoptosis [58,59,60].
2.4. Complex IV: Cytochrome C Oxidase
Complex IV of the mitochondrial respiratory chain (also called cytochrome C oxidase; COX) (Figure 1) is composed of 13 subunits that can be divided into two groups: subunits I–III are the catalytic subunits of COX and are encoded in the mitochondrial genome, while subunits IV, VA, VB, VIA, VIB, VIC, VIIA, VIIB, and VIII are the so-called supernumerary subunits that are encoded in the nuclear genome [61].
Subunit II transfers electrons from cytochrome C to subunit I via its CuA site, composed of two copper atoms. The electrons are then transferred to the heme a group of subunit I and subsequently to the oxygen-binding site of subunit I, composed of heme a3, CuB, and a tyrosyl group. Four electrons are transferred simultaneously to the electron acceptor O2, which prevents the formation of ROS. Subunit III is responsible for the stabilization of the catalytic center and for proton pumping [62]. Less is known about the role of the supernumerary subunits, and it has been proposed that they have structural roles.
COX activity can be regulated through subunit IV. Depending on the ATP/ADP ratio of the mitochondrial matrix, subunit IV binds either ADP or ATP at its matrix domain. The conversion of bound ADP to ATP leads to the feedback inhibition of COX [63]. Other regulatory factors of COX include metabolite and protein binding, expression of supernumerary subunit isoforms, phosphorylation, and the formation of supercomplexes [62]. Mutations in complex IV structural subunits, assembly factors, or cofactor synthesis can lead to a variety of diseases, such as Leigh syndrome, Fanconi anemia, Charcot–Marie–Tooth syndrome, and neurological disorders (for a more comprehensive list, please see [29]). The K101N mutation in COXIVI1 leads to poor weight gain, short stature, and an increase in chromosomal breaks, resembling Fanconi anemia [64]. The P152T mutation of COXIVI1 causes clinical features resembling Leigh syndrome [65]. The E138K mutation in COXIVI2 causes calvarial hyperostosis, dyserythropoietic anemia, and exocrine pancreatic insufficiency [66]. The R107C mutation in COXVA presents with lactic acidemia, failure to thrive, and pulmonary arterial hypertension [67]. A 5 bp long deletional mutation of COXVIA1 causes the muscle wasting/neurological disease known as Charcot-Marie-Tooth disease [68], while the S39R substitution in COXVIA2 leads to muscle weakness [69]. The R19H mutation in COXVIB1 causes mitochondrial encephalomyopathy [70], while the R20C mutation in COXVIB1 not only causes encephalomyopathy but also cardiomyopathy and hydrocephalus [71]. Different COXVIIB mutations have been identified to cause microphthalmia with linear skin lesions [72]. Loss of COXVIIIA leads to Leigh-like syndrome and epilepsy [73].
2.5. Complex V: ATP Synthase
Complex V, also known as ATP synthase, plays a pivotal role in energy production within the cell via synthesis of ATP using the proton gradient across the inner mitochondrial membrane (IMM). This enzyme complex consists of multiple protein subunits divided into two main regions: the hydrophobic F0 and the hydrophilic F1 regions [74]. The F0 region, embedded within the IMM, includes a proton channel, which is crucial for the translocation of H+ ions. These ions are known to be essential for the mechanical rotation necessary for ATP synthesis. Key components of the F0 region include subunits a, b, and c, which form the proton channel [75]. The F1 portion is exposed to the mitochondrial matrix and houses the enzyme’s catalytic activity. It consists of five subunits (α3, β3, γ, δ, and ε) arranged in a complex structure. The gamma subunit acts as a central shaft surrounded by alternating alpha and beta subunits, which, together, form a hexameric ring. As protons move through the F0 region and enter the F1 region, they induce rotational motion in the gamma subunit, which, in turn, drives the cyclic interactions of ADP and inorganic phosphate (Pi) with the beta subunits, catalyzing ATP synthesis [76]. Under certain conditions, when mitochondrial electron transport is interrupted (after inhibition of the electron transport chain or exposure to uncouplers), ATP synthase can run in reverse by hydrolyzing ATP and transferring protons from the matrix to the intermembrane space, thereby building the necessary Δp (proton motive force) and maintaining membrane potential. This process consumes ATP, and there exists a defense mechanism against ATP depletion mediated by the mitochondrial ATPase. This mechanism requires endogenous inhibitor protein ATPIF1, which binds to the ATPase when the matrix pH falls (reviewed in [77,78]). Importantly, a polyphenolic compound, (+)-epicatechin (EPI), appears to selectively inhibit the ATPase activity of complex V without affecting ATP synthase activity [79].
ATP5B and ATP5O are significant genes associated with human ATP synthase. ATP5B encodes a subunit of the F1 region, specifically one of the beta subunits crucial for enzymatic activity. Mutations in ATP5B have been linked to various mitochondrial disorders, affecting ATP synthesis efficiency and influencing cellular energy metabolism [80,81]. ATP5O, encoding the O subunit (also known as OSCP: oligomycin sensitivity conferral protein) of ATP synthase, is part of the structural stator linking the F0 and F1 regions. It is crucial for stabilizing the interface between these enzyme sections. In humans, mutations in ATP5O have not been reported commonly but are thought to impact the assembly and stability of the ATP synthase complex, leading to diseases such as those of neuromuscular and cardiac systems.
All mutations in different ETC complex subunit genes discussed above are listed in Table 1.

Table 1.
Phenotypic outcomes of ETC dysfunction-related loss-of-function mutations in C. elegans, focusing on lifespan alterations. Regarding the human clinically relevant mutations, in this table, we presented some representative mutations.
3. Redox Signaling: Past and Present Research
The term reactive oxygen species (ROS) includes non-radical H2O2 (hydrogen peroxide) and its radical derivatives (i.e., containing an unpaired valence electron such as •HO2 (hydroperoxyl radical), also known as the hydrogen superoxide, .HO•: hydroxyl radical, OH−: hydroxyl anion, 1O2•: singlet oxygen, and O2−: superoxide) [82]. Based on their reactivity, ROS can be grouped into two categories: (1) low-reactivity ROS and (2) high-reactivity ROS. Superoxide and hydrogen peroxide, the most abundant and best studied ROS in vivo [83] are low-reactivity ROS. Free-radical superoxide and the non-radical hydrogen peroxide are produced when oxygen is reduced with one or two electrons, respectively. Low-reactivity ROS have a limited capacity to damage macromolecules [84], as specific enzymatic systems of antioxidants neutralize them in the cell. Superoxide is converted to hydrogen peroxide by superoxide dismutases [84]; hydrogen peroxide is detoxified by other types of enzymes, such as catalases, glutathione peroxidases, and peroxiredoxins. In contrast, high-reactivity ROS (for example, the hydroxyl radical) are harmful, as they directly attack macromolecules; in addition, cells lack specific antioxidant systems to eliminate them.
Mitochondrial ROS (mtROS), including superoxide, hydrogen peroxide, and the hydroxyl radical derived from complex I (CI), complex II (CII), and complex III (CIII) of the ETC [85,86], are the most abundant source of reactive oxygen species in the cell, but ROS are also produced in peroxisomes during fatty acid β-oxidation and in the endoplasmic reticulum (ER) during disulfide bond formation of protein folding [87]. The ER, mitochondria, and peroxisomes are often called ‘the redox triangle’; we note that they are in direct contact with each other through membrane contact sites and generate and transport oxidants into the cytoplasm. In addition, essentially all cellular organelles communicate via redox signaling [88].
Past research identified mtROS as toxic agents causing oxidative damage to biological molecules, thereby contributing to age-related diseases. In the last two decades, we have learned that mtROS produced in specific places at specific times and intensities act as signaling molecules and determine the downstream effects of mitochondrial redox signaling, which is essential to maintain cellular homeostasis [89].
Technologies to measure different ROS have undergone significant progress in the last decade [90]. While early measurements of mtROS were performed on isolated mitochondria in vitro, recently, fluorescent probes (either small dyes or genetically encoded fluorescent reporters) have been developed to allow for in vivo measurements [91]. C. elegans also contributes to measurement of some types of reactive oxygen species in vivo. Using H2O2 redox sensor HyPer under the control of a ubiquitous promoter, it is possible to determine the tissue/cell type and the developmental stage where hydrogen peroxide is produced [92]. Grx1-roGFP1 and Grx1-roGFP2 probes detect the GSSG/2GSH ratio, which monitors the glutathione redox potential in vivo [92]. These redox sensors directly or indirectly provide quantitative and qualitative (temporal and spatial) information about the production of given reactive oxygen species in C. elegans, adding to the tractability of this organism.
3.1. Past Research Focused on Investigation of Defense Against Oxidative Stress Mechanisms That Eliminate Excess ROS
As elevated levels of ROS have been implicated in the pathogenesis of cancer, neurodegenerative disorders, and cardiovascular diseases (age-related disorders) [93], investigations in the past focused on harmful effects of ROS, i.e., the oxidative stress caused by increased ROS levels. ROS and other free radicals were thought to be byproducts of cellular metabolism [94]. Due to their capacity to cause oxidative damage, mtROS were considered main drivers of aging [83]. The goal has been to seek out signaling pathways that drive homeostasis, protect cells against oxidative damage, and eliminate ROS. Research has identified three key players as ROS-regulated transcription factors: NRF2 (nuclear factor erythroid 2-related), NF-κB (nuclear factor kappa-light-chain enhancer of activated B cells), and p53. Classically, NRF2 is responsible for cell homeostasis by eliminating DNA-damaging agents, including ROS. NRF2 binds to antioxidant response elements (AREs) of its target genes, such as HMOX1 (heme oxygenase), GCL (glutamate-cysteine ligase), NQO1 (NAD(P)H quinone oxidoreductase 1), and GSTs (glutathione S transferases). Keap1 (Kelch ECH-associating protein 1) is the repressor of NRF2 that promotes its degradation [95]; however, elevated ROS lead to the oxidation of Keap1, which prevents the degradation of NRF2, thereby resulting in NRF2 activation [96]. The regulation of p53 by ROS differentially depends on ROS concentrations: low ROS prompt the upregulation of p53-activated antioxidant genes, while in the case of a higher ROS level, p53 activates pro-oxidant genes, inducing cell death [97]. Such data point to a new approach involving terms such as eustress and hormesis, as discussed below.
C. elegans also possesses an evolutionarily conserved protective system to deal with harmful increases in ROS [98]. Phase II detoxification enzymes are present in the nematode [98].
In the worm intestine, which is often the first tissue facing pathogens and xenobiotics, ROS-induced activation of detoxifying genes is mediated by NRF homolog SKN-1, which acts as a transcription factor through ARE binding [99]. Indeed, ROS-protecting enzymes in the gut may be sufficient for C. elegans stress resistance [100,101]. Upon oxidative stress, SKN-1 is activated via a conserved p38 MAP kinase cascade, which leads to PMK- 1/MAPK-mediated phosphorylation and nuclear localization of SKN-1, as well as subsequent induction of stress-responsive genes [102]. This implies a regulatory interface between ROS and protein phosphorylation.
Other transcriptional regulators are also involved in the activation of ROS-protective gene expression in worms. Among them, the FOXO homolog DAF-16 transcription factor plays an important role. Although DAF-16 has different target genes from SKIN-1, they share similar regulatory mechanisms; DAF-16 is also activated through the stress-activated p38 MAPK pathway [103]. Under normal, stress-free growth conditions, both DAF-16 and SKN-1 are phosphorylated on specific residues by kinases of the insulin/insulin-like signaling pathway, resulting in DAF-16 and SKN-1 retention in the cytoplasm, inhibiting their translocation to the nucleus [104,105]. Whether ROS levels interfere with this phosphorylation remains to be determined.
Importantly, the activity of master transcription factors DAF-16 and SKN-1 is essential in a wide range of stress responses and is required for longevity [98,106,107].
3.2. The Current View: Redox Flux—Old Lamps for New Flammation via Eustress to Hormesis
Although excess ROS cause oxidative damage to biomolecules, it has become clear that a lack of appropriate oxidant levels also impairs signaling events and damages cellular processes. Recent evidence suggests that ROS, if expressed with the correct concentration and sub-cellular localization, can be beneficial and act as signaling molecules, regulating different physiological processes (reviewed in [108]).
Cells maintain a delicate balance between the level of oxidants and regulatory antioxidants. If the redox balance is shifted towards oxidants, oxidative distress occurs that favors antioxidants, potentially leading to reductive distress. Thus, an emerging idea is that the levels oxidants and antioxidants are balanced by redox regulation (reviewed in [109]). Redox regulation involves redox sensing, redox signaling, and subsequent signal translation to cellular stress responses. Stress responses cause changes in the activity of redox-regulated proteins and influence gene expression by modifying the epigenetic landscape. For example, SIRT1/sirtuin is under redox regulation, and its oxidation results in inactivation of its deacetylase function [110]. Oxidation reactions are counterbalanced by reduction reactions using NADH and NADPH. Briefly, NADH/NADPH is linked to sulfur metabolism through thioredoxin and glutathione (GSH) systems, which are necessary to maintain the redox state of proteins and are the primary targets of redox modifications in cells. Redox protein modifications occur on sulfur- and selenium-containing constituents of proteins (e.g., cysteine, methionine, selenocysteine, and selenomethionine residues). C. elegans studies (below) demonstrate how redox modification on given cysteine residues of specific proteins can lead to different biological responses.
For example, ROS can directly sulfenylate a cysteine residue within the ER stress sensor IRE-1 kinase, which results in activation of the p38 MAPK/SKN-1 axis, thereby increasing stress resistance and lifespan in C. elegans [111]. A recent study showed that mitochondrial superoxide enhances an RAS-dependent ROS signaling pathway called RDRS that controls the expression of a large set of genes regulating a variety of cellular functions with enhanced RDRS signaling in response to mtROS that results in lifespan extension [112]. Superoxide is converted by superoxide dismutase SOD-1 into cytoplasmic hydrogen peroxide, yet oxidizes a cysteine residue (C118) of LET-60/RAS. Hydrogen peroxide generated by redox pathways in different cellular compartments serves as a second messenger and has a major role in maintaining redox homeostasis [88,109].
In summary, the maintenance of redox homeostasis requires constant monitoring and reprogramming of redox fluctuations. The dynamic turnover of oxidative metabolism is a continuous challenge, resulting in a low level of oxidative stress (eustress) operating within a physiological range. Low levels of the oxidant species lead to cell adaptation to stress that protects against higher levels of oxidative stress; this is the principle of hormesis [113,114].
3.3. Complexities of ROS Signaling in Normal Health Conditions and Disease
In this chapter, we highlight some examples of how ROS signaling contributes to normal cellular functions and how cellular ROS levels can be targeted for therapeutic use.
ROS are accumulated during reverse electron transport (RET) in a site-specific way in respiratory complex I. A significant amount of ROS generated by this process (also called RET-ROS) is necessary for several cellular functions, for example, macrophage activation in response to bacterial infection.
Targeting ROS levels is also a developing approach to treat diseases. For example, increasing ROS in cancer cells to toxic levels in order to kill them by apoptosis is the principle of pro-oxidant therapies, as we discuss below.
3.3.1. RET-ROS Signaling in Health and Disease
The process of reverse electron transport (RET) was first described in 1961 [115], when the addition of succinate into isolated mitochondria resulted in the redirection of electrons from ubiquinol to complex I and the reduction of NAD+ to NADH by the NADH dehydrogenase module of complex I [116]. RET is an energetically unfavorable reaction that only occurs when there is a high ubiquinol/ubiquinone ratio and high proton motive force (Δp) in the mitochondria. During RET, NAD+ is reduced at the expense of ATP, and protons are pumped from the matrix to the intermembrane space. RET is also a major source of ROS in cells [116]. ROS generated during RET (RET-ROS) has been shown to initiate an anti-inflammatory response of macrophages after bacterial infection. ROS are required to produce anti-inflammatory cytokines in macrophages. Note that in order to boost ROS levels, macrophages shift their metabolism from OXPHOS to glycolysis to generate ATP and increase succinate oxidation by complex II/SDH with an elevated mitochondrial membrane potential, which, in turn, leads to RET and RET-ROS production [117]. Interestingly, RET-ROS signaling was blocked by ectopic expression of an alternative oxidase (AOX) derived from Ciona intestinalis in mice. AOX prevents the over-reduction of CoQ [118] and inhibits the inflammatory phenotype of mouse macrophages [117]. Another beneficial role of RET-ROS signaling was reported in the adaptation of the carotid body to hypoxia [119]. In contrast, deregulated RET-ROS has been identified as a main factor causing stroke, and decreases in the NAD+/NADH ratio induced by RET have been linked to many age-related disorders, such as neurodegeneration and cancer (reviewed in [120]).
3.3.2. Targeting ROS in Cancer
Targeting ROS for therapeutic benefit involves either reducing excessive ROS to alleviate oxidative damage or increasing ROS to cytotoxic levels to kill cancer cells. Antioxidants such as N-acetylcysteine (NAC), vitamin C, and vitamin E are commonly investigated for their potential to scavenge ROS and protect against oxidative stress in various diseases. However, the clinical efficacy of antioxidants in cancer therapy has been mixed, with some studies suggesting that antioxidants might protect cancer cells from ROS-induced cytotoxicity [121].
On the other hand, pro-oxidant therapies aim to exploit the elevated basal ROS levels in cancer cells. Agents such as arsenic trioxide, menadione, and elesclomol selectively induce lethal levels of ROS in cancer cells. This therapeutic strategy is based on the concept that cancer cells, already under oxidative stress, are closer to the threshold of ROS-induced apoptosis, making them more vulnerable to further ROS insults [122].
4. Effects Exerted by Mutations in ETC Genes on Development and Lifespan of Nematodes
As humanity gained a high level of medical and technical knowledge, our research turned towards enhancing not only the quantity but also the quality of our extended life. This is a challenging task, as in most cases, advancing age (longer lifespan) is accompanied by reduced well-being (as known as healthspan) [123]. While lifespan is the total number of years of an individual’s life, healthspan is that period which is free from disabilities and/or chronic diseases. These two dimensions are closely bonded and cannot be discussed without the inspection of the phenomenon called aging, which is affected by various factors, like genetic, cellular, and environmental features. But one of the key drivers of aging is the decay of mitochondrial function, causing energetic decline at the cellular level [123]. Mitochondrial dysfunction has a crucial impact on lifespan, as well as on healthspan. Therefore, investigating the consequences of mitochondrial dysfunction in a variety of complex model organisms is vital in developing new strategies to improve quality of life in our aging society [123].
With its many advantages—e.g., short life cycle and 65–70% genome sequence homology to humans—Caenorhabditis elegans is a popular model in many aspects of developmental biology, especially in the field of aging [124]. This simple model organism is also capable of easily determining the difference between lifespan and healthspan, as standardized methods have been developed recently, like the measurement of movement, pharyngeal pumping, brood size, etc. [124,125,126]. Many characteristics of C. elegans and human mtDNA are similar, such as their structure and size (13.7 vs. 16.6 kb), maternal inheritance, polyploidy, and heteroplasmy [127,128]. Furthermore, most biochemical properties of worm mitochondria are similar to those of mammals, such as oxygen consumption profile; supercomplex formation; and, most importantly, TCA cycle and ETC activities [129,130]. Amongst 91 human genes encoding ETC components, 72 have orthologs in C. elegans [131]. The conservation rate of these subunits is significant; for instance, sequence similarity at the amino acid level between C. elegans and human homologous complex I subunits varies between 25% and 99.2%. These data, together, support an extensive evolutionary conservation between C. elegans and humans in terms of ETC structure and function [132].
4.1. Mutations of Complex I Subunits
Thirty-one nuclear-encoded mammalian complex I subunit genes have orthologs in C. elegans [132]. Homologs of NDUFV1, NDUFS2, NDUFB4, and NDUFA6 have been extensively characterized in the nematode. Grad and Lemire generated transgenic worms carrying clinically relevant point mutations in the highly conserved nuo-1 gene in C. elegans. A352V, T434M, and the A443F mutant versions of NUO-1 correspond to the disease-causing substitutions in the NDUFV1 protein, A341V, T423M, and A432F, respectively. These conserved residues are important for protein folding, and the mutant NUO-1 proteins seem more susceptible to degradation. All the mutants displayed decreased lifespan, low brood size, and hypersensitivity to hyperoxia and (pro-oxidant) paraquat [133]. Importantly, transgenic worms showed hallmarks of complex I dysfunction such as an increase in lactate and the lactate:pyruvate ratio (lactic acidosis) and decreased NADH-dependent mitochondrial respiration. nuo-1(RNAi) phenocopied lactic acidosis, reminiscent of that which is observed in some patients with mitochondrial disorders [134].
A deletional allele (ua1) of NDUFV1 homolog nuo-1 (NADH ubiquinone oxidoreductase 1) results in L3 larval arrest and increased lifespan [135]. L3-to-L4 transition has a high energy demand underpinned by increases in oxygen consumption [135] and mtDNA copy number. A lack of ETC subunits in nuo-1 mutants results in severely decreased energy production that does not allow for the transition to the L4 stage.
In contrast, the fc21 deletional allele of NDUSFS2 homolog gas-1 (general anesthetic sensitivity abnormal 1) shows slowed development and reduced lifespan [136]. Furthermore gas-1 loss-of-function mutant animals are hypersensitive to volatile anesthetics [137]. This ‘worm to cot side’ discovery originated in C. elegans to guide clinical recommendations in mitochondrial disorder patients for proper dosing of volatile anesthetics [138]. Similarly, experiments conducted on the nematode showed that riboflavin rescued complex I stability and partially rescued complex I and IV activities in nuo-1 point mutants [139]. Based on these data, riboflavin supplementation has been proposed as a possible therapeutic intervention to treat NDUFV1 and NDUFV2 defects, in addition to also possibly treating defects in the other flavoproteins, like NDUFV3 [140].
Another complex I mutant, NDUFB4 homolog nuo-6(qm200), showed decreased respiration and electron transport rates, despite increases in ATP concentrations. Nuo-6(qm200)-like gas-1(fc21) and nuo-1(ua1) display slower embryonic and postembryonic development. Nuo- 6(qm200) animals have an elevated lifespan [141].
Interestingly, ETC complex I dysfunction was rescued by hypoxia in a mouse neurological disease model [142]. Recently, another group used the nematode as a model to reveal the unknown mechanism behind this finding [143]. NDUFA6/nuo-3(G60D), which phenocopies hypoxia rescue or hypoxia, directly restored complex I activity and rescued ETC flux and, in some cases, complex I levels (nduf-7(et19) and gas-1(fc21)). In a screen, NDUFS2/GAS-1(V161) and NDUFS7/NDUF-7(M80)—critical residues in the ubiquinone binding pocket—were identified as suppressors of nuo-3(G60D) [143]. Meisel et al. showed that these mutants also blocked the rescue of nduf-7(et19) and gas-1(fc21) by hypoxia, pointing to a shared rescue mechanism between nuo-3(G60D) and hypoxia. Results suggest that the activity of complex I mutants is rescued either by altering the conformation of the ubiquinone binding pocket, or by influencing the local chemical environment. In summary, Meisel et al. showed that hypoxia and nuo-3(G60D) rescue complex I mutants through a shared mechanism via key residues in the ubiquinone binding site [143].
Deficiency of ACDH-12 (Acyl CoA dehydrogenase), the homolog of complex I assembly factor ACAD9, affects the function and formation of complex I. RNAi knockdown of acdh-12 shortens lifespan [144].
Knocking down any of the complex I subunits, for example, gas-1(fc21) [145], leads to increased complex II-dependent respiration [132].
4.2. Mutations of Complex II Subunits
Interestingly, SDHA and SDHB (but not C and D) subunits are evolutionarily highly conserved at the amino acid level [146]. As the SDH complex has been the focus of our work recently, we performed pairwise alignments of the relevant worm and human SDH subunit homologs, and the results of sequence alignments underpin this statement. Indeed, in C. elegans SDHA and SDHB subunits show higher similarity and identity to their human counterparts at the amino acid level (SDHA: 83% and 73%; SDHB: 84% and 60%, respectively) compared to SDHC and SDHD subunits, where these similarity and identity values are lower (SDHC: 54% and 30%; SDHD: 59% and 43%, respectively). Interestingly many agricultural pest control agents target the SDHx complex (complex II) [147], and some of them are being considered for use in human anti-fungal treatments [148]. However, it is important to note that many of these substances are nematicides as well; for example, fluopyram is described as a selective nematicide [149]. The SDHx complex is well characterized in the nematode; mev-1 encodes CYT-1, cytochrome b560, which is the large subunit of succinate–ubiquinone oxidoreductase in SDH (the SDHC subunit). It was the first discovered oxygen sensitivity mutant [150]: Ishii et al. originally screened for mutants sensitive to methyl viologen (paraquat), which is a redox-active herbicide, and identified mev-1(kn1)- (G71E) as a paraquat-sensitive mutant. This homozygous point mutation not only disturbs the biochemical process of succinate/fumarate conversion but also shows an effect on the respiratory electron transport chain by electron leakage. mev-1(kn1) animals display a short lifespan, reduced brood-size phenotype, and oxygen hypersensitivity, while the deletional allele of mev-1(tm1081) is lethal [151,152]. These observations have subsequently been recapitulated in a mouse model, showing that these SDHC-related processes are evolutionarily conserved [153]. Interestingly, Pujol et al. described an elevated gas-1(fc21) lifespan upon sdhc-1 and sdhd-1 knockdown [154]. Since then, based on these traits (accelerated aging and susceptibility to oxidative stress) the mev-1 mutants have been used frequently as a model of mitochondrial malfunction [155] and as test beds in the search for new anti-aging antioxidants [156].
The other well characterized SDH complex member in worms is B-subunit ortholog sdhb-1. Huang and Lemire discovered that different mutations in the sdhb-1 gene resulted in superoxide generation and premature aging [157]. They observed impaired SDH function and assembly, increased superoxide anion production, and perturbed mitochondrial respiration due to a mutation in Pro211, a conserved proline residue—which corresponds to Pro197 in the human counterpart—near the proximal quinone binding site (Qp) in SDHB-1. Furthermore, they showed that different missense Pro211 mutations (Pro211His, Pro211Leu, Pro211Phe, and Pro211Arg) cause embryonic lethality with incomplete penetrance and shortened lifespan in the surviving animals. Huang and Lemire also showed that sdhb-1 null mutants carrying the gk165 deletional allele arrest development in the L2/L3 larval stage [157]. Another group studied the effect of SDH deficiency on the hypoxia system in worms [158]. One consequence of HIF (hypoxia-inducible factor) activation in worms is a defect in egg laying. To study HIF activation, Braun et al. knocked down SDHB in a subset of neurons thought to be responsible for egg laying. They found that sdhb-1 depletion resulted in the expected excess succinate level, which inhibited prolyl-hydroxylase EGL-9. EGL-9 inhibition prevented HIF-1 hydroxylation and degradation, thereby activating HIF-1 signaling, which led, inter alia, to the retention of eggs [158].
Lately, our lab has built on these data to make a new disease model: the SDHB Arg244His mutation was introduced into the worm, which is a clinically relevant germline mutation, corresponding to Arg230His causing human paraganglioma [159]. We showed that this point mutation makes the SDH enzyme defective, with abnormal succinate accumulation and reduced oxygen consumption. It has been proposed that excess succinate levels are also characteristic of PHEO/PGL tumors, where SDH mutations cause mitochondrial succinate accumulation in the cytoplasm and HIF-1α (hypoxia-inducible factor alpha) activation, which favors pseudohypoxia-driven tumorigenesis [160] and can lead to inflammation [161]. Arg244His worms also develop abnormally, but unlike sdhb-1 null mutants—which arrest in the L2/L3 larval stage—these point mutants reach adulthood but remain sterile and show slowed development. Our data contrast with the larval arrest of the null mutants that occurs at the L2/L3 stage, likely because loss of SDH activity is compensated for by the glyoxylate cycle, which is most active during embryonic development and peaks at the L1 larval stage, declining thereafter by the end of the L2 stage [130]. However, both sdhb-1 deletion and Arg244His missense mutation shorten lifespan. In line with these data, silencing of sdhb-1 by RNAi also resulted in a shortened lifespan [154]. This shortened lifespan reflects the dual role of the SDH complex: if SDH function is decreased, not only is the mitochondrial electron transport function damaged, but succinate-to-fumarate conversion in the TCA cycle is also impaired. Finally, Arg244His mutant worms display a rewired metabolism—an aberrant glycolysis reminiscent of the Warburg effect—which could explain the developmental differences between Arg244His animals and sdhb-1 null mutants [159].
Lastly, sdha-1 is the third member of the SDH complex that has also been characterized in worms. sdha-1 loss-of-function mutant animals are viable but show slower movement and a reduced oxygen consumption rate and contain smaller and less networked mitochondria. Their development is slower from larval stages L2 to L3. Males cannot copulate due to an anal depressor muscle defect [162]. Goncalves et al. discovered metabolic changes in the absence of SDHA-1 function: the expression of phosphoenolpyruvate carboxykinase (PEPCK; pck-1 and pck-2, parts of the gluconeogenesis pathway) is increased. In contrast, sdha-1 overexpression has the opposite effect, suggesting that mitochondrial function and levels of anabolic processes are inversely correlated [162]. These the above observations illustrate multiple examples of metabolic adaptation—a hallmark displayed by tumors—in nematodes carrying different SDH deficiencies [163,164]. Altogether, C. elegans can reveal developmental abnormalities, metabolic changes, and oxidative effects caused by SDHx mutations.
4.3. Mutations Affecting CoQ Synthesis
It has been said that life depends on the stepwise transfer of electrons downhill through energy gradients. CoQ (Coenzyme Q/ubiquinone/ubiquinol) acts as transmitter of the electrons transferred from complexes I and II to complex III. The multistep synthesis of CoQ9 in C. elegans requires the activity of coq-1, coq-2, coq-3, coq-8, and clk-1 (also called coq-7) genes. Knockout mutations in coq-1, coq-2, coq-3, and coq-8 result in animals that can only survive for one generation on normal E. coli bacteria (E. coli strain OP50 produces CoQ8) but whose progeny become fertile [165,166,167,168]. Survival of the first homozygous generation is due to maternally deposited gene products and CoQ in the egg [169]. In contrast, clk-1 (stands for clock) mutants can reproduce indefinitely on normal E. coli strain OP50 but cannot grow on bacteria that do not produce CoQ8 [170]. clk-1 mutants accumulate biosynthetic intermediate demethylubiquinone-9 (DMQ9), which partially compensates for the absence of CoQ9 and allows these mutants to grow as long as dietary CoQ8 is available [170,171]. clk-1 is actually one of the very first identified longevity genes (reviewed by Wang et al. [42]).
The two most studied mutations of clk-1 are the qm30 null allele and the e2519 missense allele. All clk-1 phenotypes are present in both mutants but are weaker in e2519 mutants [172]: clk-1 mutants show lifespan extension, reduced fecundity, decreased pharynx pumping, defecation, and locomotion, alongside slow embryonic and postembryonic development [172,173]. All clk-1 mutant phenotypes (lifespan, behavioral phenotypes, and altered expression of mitochondrial quality control genes) were rescued by pharmacologically restoring CoQ biosynthesis, indicating that clk-1 mutant phenotypes are entirely due to defects in CoQ biosynthesis [174]. coq-3(ok506) mutants have a shorter lifespan and retain fertility, but only a handful of the second-generation mutants develop to adulthood [168,175].
4.4. Mutations of Complex III Subunits and Cytochrome C
The C. elegans genome possess eight complex III subunit homologs: [131] among the three well characterized subunits, two are encoded in the nucleus (isp-1, the homolog of the mammalian Rieske iron sulfur protein and cyc-1/cytochrome C1), and one is encoded in the mitochondrial genome ctb-1/(cytochrome b). isp-1(qm150) mutants show “slow phenotypes”: they lay reduced numbers of eggs; have slower embryonic and postembryonic development and lower fecundity; and, importantly, live longer than wild-type animals [176]. Jafari et al. identified intragenic suppressors (such as A149T/V and A151T) in the highly conserved six-amino-acid tether region (DQRALA) of ISP-1 [177]. Suppressor mutants restored isp-1(qm150) phenotypes, including all “slow phenotypes” and lifespan extension. Furthermore, mutations analogous to Rieske protein Rip1 in budding yeast show similar effects, proving the conservation of this structure–function relationship across highly divergent species. isp-1(qm150) carries a proline-to-serine amino acid change at position 225, which lies in the head unit of ISP. Prolines are important structurally, as they make the peptide backbone rigid [178]. The ISP head region is a key element in ISP function. Based on multiple studies, the tether region seems to be a flexible element responsible for positioning the head region of ISP between three main locations: either to the electron donor or to the acceptor site or into an intermediate position [50,179]. In conclusion, mutations in the tether region are capable of compensating for the steric alteration of the ISP head caused by isp-1(qm150). Regarding lifespan phenotypes, similar to isp-1(qm150) mutation, RNAi against another complex III subunit cyc-1 also extends lifespan [180]. This lifespan extension is suppressed by icl-1(ok531) due to the inactivation of the glyoxylate pathway [181]. It has been suggested that defective ISP function is compensated for by the expression of SOD3 (superoxide dismutase) [176] and ICL-1, the key enzyme of the glyoxylate shunt [182]. As expected, isp-1(qm150) mutants display a defective complex III function. The respiratory complexes can form higher-order assemblies called supercomplexes or respirasomes, consisting of monomeric CI and dimeric CIII and monomeric CIV [183]; it also was shown that complex I activity is not only impaired but that the I:III:IV supercomplex is also disrupted in these animals. It has been suggested that the assembly of supercomplexes may increase the efficiency of the electron transport chain, reducing the rate of ROS production [184]. Together, these data lead to the conclusion that ISP-1 may act as a stabilizer of the supercomplex through either protein–protein interactions or protein-conformational linkage [176].
Consistent with these data, RNAi specific for cytochrome C1 homolog cyc-1 also extended lifespan [180], which was suppressed in the icl-1(ok531) mutant background, as a consequence of inactivation of the glyoxylate pathway [181].
Interestingly, a mutant allele of cytochrome b homolog ctb-1(qm189) suppresses the slow phenotypes mentioned above but not the elevated lifespan of isp-1(qm150) [176]. Feng et al. concluded that ctb-1(qm189) mutation has a double effect: on one hand, it causes loss of function in complex III, and on the other hand, possess gain-of-function in stabilizing the I:III:IV supercomplex, thereby compensating for the effects of isp-1(qm150).
CyC (cytochrome C) in C. elegans is encoded by two genes: cyc-2.1. and cyc-2.2. To date, these genes have not been deeply investigated. However, C. elegans CyC possesses similar properties as its mammalian homolog [185]. The germline serves as key tissue in lifespan regulation. cyc-2.1 knockdown by RNAi in the germline extends lifespan by activating the intestinal mitochondrial unfolded protein response (UPRmt), AMPK (AMP-activated kinase), and mitochondrial fission [186].
4.5. Mutations of Complex IV and V Subunits
Complex IV (cytochrome C oxidase or COX) is regulated by signals of the intramitochondrial ATP/ADP ratio [187]. In electron transfer, complex IV involves two hemes (cytochrome a and cytochrome a3) and two copper centers (CuA and CuB). RNAi against COX-IV, COX-VB/cco-1, and COX-VA subunits decreases fecundity and slows development [188,189], while COX-VIIC (RNAi) causes arrest at the second larval stage [189]; in all cases, extended lifespan is observed [180,189].
Finally, complex V of the MRC is the ATP synthase, which is not directly involved in electron transport, unlike the other complexes. In C. elegans, the homologs of human ATP5B and ATP5O genes are integral to the known roles of mitochondrial dysfunction and aging. The C. elegans homolog of ATP5B, known as atp-2, is critical for the proper function of ATP synthase’s F1 subunit. atp-2 (ua2) loss-of-function mutants have been demonstrated to result in reduced ATP synthesis, leading to decreased mitochondrial function and altered metabolic states that can accelerate aging or increase stress resistance, arrest at the third larval stage, and decreased pharynx pumping [131,135,190]. Similarly, the ATP5O homolog, oscp-1, plays a vital role in maintaining the stability and efficiency of ATP synthase. Studies have reported that disruptions in oscp-1 (also known as atp-3) can lead to compromised energy production and are associated with phenotypes indicative of mitochondrial pathology. leading to effects such as reduced brood size, developmental delays, and increased sensitivity to mitochondrial toxins. Studies of C. elegans have also demonstrated that alterations in the homologous gene can disrupt the normal function and assembly of mitochondrial ATP synthase, reflecting phenotypic changes such as reduced fecundity, slower growth rates, and increased lifespan [180,191]. Retardation in gonadal development in response to atp-3 silencing is the consequence of decreased ATP production, and it is known that germline development is very sensitive to available energy sources [192]. Xu et al. silenced Y82E9BR.3, the C. elegans ORF corresponding to human ATP synthase C subunit, which resulted in slowed development and sterility [193]. The C. elegans genome encodes two inhibitor proteins of the F1Fo-ATPase, MAI-1 and MAI-2. MAI-2 is localized to mitochondria: worms lacking MAI-2 function have an enhanced mitochondrial membrane potential and decreased physiological germ cell apoptosis, suggesting that MAI-2 might play a role in apoptosis by regulating mitochondrial membrane potential [194].
These studies highlight the immense importance of ATP synthase components in energy metabolism and stress response within C. elegans, demonstrating that such effects are not only observed in higher mammalians but genetic model systems like C. elegans that also exhibit a similar phenotype, thereby making this model a valuable tool for understanding the genetic and biochemical bases of mitochondrial pathologies.
5. Caenorhabditis elegans Serves as Test Bed for Drugs Acting on ETC Components
C. elegans can serve as a model system to test drugs acting on ETC components. For example, drug candidates inhibiting OXPHOS have emerged as promising targets of cancer therapeutics that have been tested on nematodes, focusing on lifespan effects. Antidiabetic agent metformin slows cancer progression both in vitro and in vivo by inhibiting mitochondrial complex I and increases ROS production with subsequent activation of HIF-1α [195]. Metformin extended lifespan in C. elegans [196]. Phenformin, another biguanide complex I inhibitor that was withdrawn from human use because of lactic acidosis in patients, showed promising results in combination with the chemotherapeutic agent gemcitabine in the treatment of high-OXPHOS pancreatic ductal adenocarcinoma models [197]. We note that phenformin also increased the lifespan of worms [198] and that Arctigenin (in Phase I trial), a lignan from Arctium lappa., has been shown to extend lifespan, improve survival under oxidative stress, and decrease endogenous ROS levels in C. elegans [199]. Furthermore, some novel drugs targeting OXPHOS, such as GBS-01/Arctigenin, are in clinical trials [200].
Antibiotics have also been suggested in cancer therapy; for example, doxycycline was shown to act as a selective inhibitor of cancer stem cells [201,202]. Doxycycline also extends lifespan in C. elegans [203], reduces respiration, and activates mitochondrial unfolded protein response [204].
Aspirin’s well-known ability to reduce cancer risk [205] is understood to reduce oxidative stress and was reported to extend the lifespan of worms through a metabolic increase mediated by germline signaling [206].
Phenolic compounds synthesized by plants to increase their survival in response to environmental stresses are known to have antioxidative and anti-inflammatory effects. Resveratrol is a well-known example, which is present in grape, peanut, and berry fruits and associated with anti-obesity, cardioprotective, neuroprotective, antitumor, and antidiabetic properties [207]. Free-radical scavenging and antioxidative activities are well-known properties of resveratrol [208]. It is thought that many dietary compounds, including flavonols, function as antioxidants because they or their metabolites can alkylate Keap1. Keap1 alkylation leads to the activation of Nrf2 and its downstream target antioxidant enzymes [209]. The beneficial effects of resveratrol are also linked to mitochondrial biogenesis, and it has been suggested that resveratrol directly binds to complexes I [210], III [211], and V [212,213,214] and alters their activities (reviewed in [215]).
The antioxidant and ROS scavenging activities of resveratrol were tested by using free radical-producing chemicals juglone and paraquat as stressors in C. elegans. Worms treated with resveratrol under stressful conditions showed an expansion of lifespan [216]. The antioxidant curcumin (plant origin, widely used in Asia) has been used as a chemopreventive substance in cases of colorectal cancer [217]. Curcumin exerts its chemopreventive effect by inducing apoptosis of tumor cells through mitochondrially dependent pathways, such as loss of mitochondrial membrane potential, release of cytochrome C, changes in electron transport [218], activation of caspase pathways, and ROS production [219]. In addition, curcumin inhibits multiple cancer pathways [219]. Supplementation with curcumin caused an increased antioxidant capacity in worms and extended their lifespan [220]. Similarly, treatment with its derivative, curcumin–acetylsalicylate also resulted in lifespan extension [221].
6. Discussion
It has been suggested that ancient worms buried in the ocean floor, where sulfur–iron pyrites were abundant, may have sparked biodiversity [222]. Therefore, it may not be a coincidence that, to this day, FeS clusters are not only permissive for electron transport across species but also that the majority of human genes encoding ETC components have orthologs in C. elegans [131]. The worm provides an important test for human diseases of ETC function because ETC complex mutations affect all stages of C. elegans development, manifesting either developmental larval arrest, sterility, or reductions in lifespan. Table 1 summarizes human vs. nematode mutations in homologous genes encoding ETC complex subunits and their phenotypes. When comparing the type of mutant alleles in homologous genes, relatively few attempts have hitherto been made to reproduce the clinically relevant mutations in the nematode. Nonetheless, in three studies, mutant worms have recapitulated important aspects of the corresponding human disease. For example, worms carrying A352, T434M, and A443F substitutions in NDUFV1 homolog NUO-1 protein showed hallmarks of complex I dysfunction such as lactic acidosis and decreased NADH-dependent mitochondrial respiration. Furthermore, clinically relevant substitutions P211 and R244H in SDHB homolog SDHB-1 protein resulted in hypersensitivity to oxidative stress and aberrant, Warburg-like glycolytic activity, as also observed in SDHB mutant PHEO cell lines. In the future, genome editing techniques will allow for the generation of more mutants carrying clinically relevant mutations. Thus, worms can provide mechanistic insight.
Regarding similarities in the presentation of different ETC complex mutations in man and worm, we can see that both show measurable metabolic effects—notably, lactic acidosis. Human ACAD9 and COXVIA1 mutations manifest with muscle weakness; this phenotype has also been observed in several worm ETC mutants, such as nuo-1, nuo-6, clk-1, and atp-2 mutants, which show slow locomotion, defecation defects, and decreased pharynx pumping due to neuromuscular impairment (see details in Table 1).
A pleiotropic phenotype, which can be the consequence of many developmental and signaling defects (therefore, hard to translate to human health), is arrest at the L3 larval stage that can be observed in several ETC mutants. L3-to-L4 transition has a high energy demand, accompanied by an increase in oxygen consumption [135]. Transition to the L4 stage is not possible in nuo-1 and atp-2 mutants because of their decreased energy production.
Aging phenotypes can help our understanding of age-related diseases, as C. elegans has been extensively used as an aging model [223]. Indeed, many signaling pathways that regulate aging were first described in the worm, for example, insulin signaling or the mTOR pathway. Together, numerous genetic and environmental conditions are known to regulate aging, as reviewed elsewhere [224]. Loss of mitochondrial function is a hallmark of aging, as the ETC is impaired in old age [225]. The detrimental effects of different ETC mutations on lifespan have been widely studied in worms. Complex II mutations shorten the lifespan of worms, which reflects the dual role of the SDH complex: the improper function of the SDH complex results in a hindered succinate-to-fumarate conversion in the TCA-cycle, as well as a damaged mitochondrial electron transport function. Mutations in other complex components affect lifespan differently: when mutated or impaired, some complex I, III, and IV defects shorten the lifespan (for example, gas-1(fc21)); others, in contrast, extend it (nuo- 1(ua1), nuo-6(qm200), clk-1(e2519), isp-1(qm150), and atp-2(ua2)). Actually, the majority of mutations in genes encoding complex I, III, and V subunits, as well as RNAi specific for complex IV subunits, result in lifespan extension. Extension of lifespan was also observed in Drosophila when five genes encoding components of respiratory complexes I, III, IV, and V were treated by RNAi [226]. Interestingly, in mice, mutation of complex IV assembly factor Surf1 and heterozygous mutations in MCLK1, a factor involved in the synthesis of ubiquinone, increase lifespan [227,228].
One model to explain the lifespan-extending effect of ETC mutations in several genetic models is the “rate of living” hypothesis, originally proposed by Max Rubner in 1908, which argues that a lower basal metabolic rate caused by decreased ETC function increases life expectancy [229].
However, increased lifespan does not automatically mean a longer healthy life [230]. Analysis of healthspan is also possible in C. elegans by using different parameters in aging worms. For example, a study by Bansal et al. [231] examined four parameters, namely resistance to heat and oxidative stress and movement capacity in liquid and solid media in long-lived mutants, including clk-1(qm30). clk-1 mutants animals showed extended lifespan but not healthspan, as they exhibited an increased ratio of time spent in a frail state.
It is intriguing that mutations in genes encoding subunits of complex I and complex III have been reported to increase lifespan in worms in a ROS-dependent manner [141]. Increased longevity observed in nuo-6 and isp-1 partial loss-of-function mutants is due to elevated superoxide levels [232]. It was also shown that elevated ROS levels in isp-1 mutants cause the activation of stress response pathways such as the mitochondrial unfolded protein response, the SKN-1-mediated stress response, and the hypoxia response [233].
This idea is consistent with both hormesis and eustress, where a transient increase in ROS levels activates a stress response pathway. Complex I mutations can also extend the fly lifespan through a similar mechanism [234]. These discoveries underpin the concept of mitohormesis, which proposes that boosting ROS levels activates a stress response to compensate for initial damage (reviewed in [83]). Metformin, a non-specific inhibitor of complex I also extended lifespan of worms, also through a mitohormetic response: the metformin–ROS signal activated the PRDX-2 periredoxin pathway, which led to lifespan extension [235].
Although in vitro measurements have identified eleven ROS-generating sites on isolated mitochondria (associated with substrate catabolism and the ETC [236]), the main mtROS generators are complex I and complex III of the ETC. To better understand the role of complex I- and complex III-derived ROS in different physiological and pathological processes, recent research has focused on specific sites, where electrons leak, for example, the flavin site (IF) and quinone binding site (IQ) of complex I (reviewed in [237]). Certain conditions induce the production of ROS at the IQ site by reverse electron transport (RET). Better understanding of the regulation of RET-ROS is crucial, as it not only contributes to different physiological processes but is also linked to stroke and age-related diseases.
In C. elegans, RET was induced by inhibiting complex V using TCA-cycle metabolite alpha-ketoglutarate. Interestingly, alpha-ketoglutarate administration in worms resulted in lifespan extension, which is dependent on mTOR, although the exact mechanism has not been described [238]. ROS produced by RET has also been shown to extend lifespan in Drosophila [118].
To comprehend the role of mtROS in health and disease and potentially use it as a therapeutic tool in the future, we need to understand where (in which site) and when ROS are generated and how the intensity of the ROS signal influences the outcome. A recent study in C. elegans showed that elevating ROS levels experimentally during the L2 larval stage of development results in lifespan extension, meaning that exposure to oxidants during early development influences the duration of the adult lifespan [239]. These data also show that C. elegans, among other genetic models, continues to help our understanding of spatio-temporal contexts of ROS signaling.
Together, the above data demonstrate that C. elegans will continue to serve as a tractable genetic model in further understanding of signaling pathways both up- and downstream of ROS. In addition, this tractable nematode has emerged as an alternative platform in drug testing; thus. compounds acting on the ETC and/or influencing mtROS levels can also be examined using worms.
7. Conclusions
Mutations in highly conserved genes encoding components of the electron transport chain (ETC) provide valuable insights into the mechanisms of oxidative stress and mitochondrial ROS (mtROS) in a wide range of diseases, including cancer, neurodegenerative disorders, and aging. C. elegans provides an important test for human diseases of ETC function because ETC complex mutations affect all stages of worm development, manifesting either developmental larval arrest, sterility, or reductions/extensions in lifespan. Some studies have reproduced clinically relevant mutations in homologous complex I or complex II ETC subunits in C. elegans, and the mutant worms recapitulated human metabolic phenotypes such as lactic acidosis or aberrant, Warburg-like glycolytic activity. In the future, genome editing techniques will allow for the generation of more mutants carrying clinically relevant mutations; thus, worms can provide mechanistic insights.
Aging phenotypes can help our understanding of age-related diseases, as C. elegans has been extensively used as an aging model; however, increased lifespan does not automatically mean a longer healthy life, which is a challenging task to address. Analysis of healthspan is also possible in C. elegans by using different parameters in aging worms.
Although in vitro measurements have identified eleven ROS-generating sites on isolated mitochondria, the main mtROS generators are complex I and complex III of the ETC. It is intriguing that mutations in genes encoding subunits of complex I and complex III have been reported to increase lifespan in worms in an ROS-dependent manner. This idea is consistent with both hormesis and eustress, where a transient increase in ROS levels activates a stress response pathway. These discoveries underpin the concept of mitohormesis, which proposes that boosting ROS levels activates a stress response to compensate for initial damage.
C. elegans has great potential as a platform for testing ETC-targeting drug candidates, including OXPHOS inhibitors, which represent promising avenues in cancer therapeutics. Together, these data demonstrate that C. elegans will continue to serve as a tractable genetic model in further understanding of signaling pathways both up- and downstream of ROS.
Author Contributions
Writing—review and editing: F.Ő., A.N., Z.F. and K.T.-V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Fund (National Research, Development and Innovation Office) grant K134285 to K.T.-V., Z.F. was supported by the EKÖP-24 University Excellence Scholarship Program of the Ministry for Culture and Innovation of the National Research, Development and Innovation Fund (EKÖP-24-4-II-ELTE-249).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Anil Mehta and Gordon Stewart for suggestions and helpful discussions. We also thank the Pheo Para Cancer Charity for supporting our research that is partially summarized in this review.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef]
- Lemarie, A.; Grimm, S. Mitochondrial respiratory chain complexes: Apoptosis sensors mutated in cancer? Oncogene 2011, 30, 3985–4003. [Google Scholar] [CrossRef] [PubMed]
- Brandon, M.; Baldi, P.; Wallace, D.C. Mitochondrial mutations in cancer. Oncogene 2006, 25, 4647–4662. [Google Scholar] [CrossRef]
- Rea, S.L.; Graham, B.H.; Nakamaru-Ogiso, E.; Kar, A.; Falk, M.J. Bacteria; yeast; worms, and flies: Exploiting simple model organisms to investigate human mitochondrial diseases. Dev. Disabil. Res. Rev. 2010, 16, 200–218. [Google Scholar] [CrossRef]
- Wallace, D.C. Mouse models for mitochondrial disease. Am. J. Med. Genet. 2001, 106, 71–93. [Google Scholar] [CrossRef] [PubMed]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, O.A.; Mohiuddin, S.S. Biochemistry, Oxidative Phosphorylation; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Pagniez-Mammeri, H.; Loublier, S.; Legrand, A.; Bénit, P.; Rustin, P.; Slama, A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol. Genet. Metab. 2012, 105, 163–172. [Google Scholar] [CrossRef]
- Carroll, J.; Fearnley, I.M.; Skehel, J.M.; Shannon, R.J.; Hirst, J.; Walker, J.E. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006, 281, 32724–32727. [Google Scholar] [CrossRef]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 2010, 329, 448–451. [Google Scholar] [CrossRef]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Sazanov, L.A. Gaining mass: The structure of respiratory complex I-from bacterial towards mitochondrial versions. Curr. Opin. Struct. Biol. 2015, 33, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J. Mitochondrial complex I. Annu. Rev. Biochem. 2013, 82, 551–575. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef]
- Zanette, V.; do Valle, D.; Telles, B.A.; Robinson, A.J.; Monteiro, V.; Santos, M.L.S.F.; Souza, R.L.R.; Benincá, C. NDUFV1 mutations in complex I deficiency: Case reports and review of symptoms. Genet. Mol. Biol. 2021, 44, e20210149. [Google Scholar] [CrossRef]
- Marin, S.E.; Mesterman, R.; Robinson, B.; Rodenburg, R.J.; Smeitink, J.; Tarnopolsky, M.A. Leigh syndrome associated with mitochondrial complex I deficiency due to novel mutations In NDUFV1 and NDUFS2. Gene 2013, 516, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.L.; Hogan, V.E.; He, L.; Blakely, E.L.; Worgan, L.; Al-Dosary, M.; Saretzki, G.; Alston, C.L.; Morris, A.A.; Clarke, M.; et al. The p.M292T NDUFS2 mutation causes complex I-deficient Leigh syndrome in multiple families. Brain 2010, 133, 2952–2963. [Google Scholar] [CrossRef] [PubMed]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stöckler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Parmar, G.; Fong-McMaster, C.; Pileggi, C.A.; Patten, D.A.; Cuillerier, A.; Myers, S.; Wang, Y.; Hekimi, S.; Cuperlovic-Culf, M.; Harper, M.-E.; et al. Accessory subunit NDUFB4 participates in mitochondrial complex I supercomplex formation. J. Biol. Chem. 2024, 300, 105626. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Heidler, J.; Dibley, M.G.; Kremer, L.S.; Taylor, L.S.; Fratter, C.; French, C.E.; Glasgow, R.I.C.; Feichtinger, R.G.; Delon, I.; et al. Bi-allelic Mutations in NDUFA6 Establish Its Role in Early-Onset Isolated Mitochondrial Complex I Deficiency. Am. J. Hum. Genet. 2018, 103, 592–601. [Google Scholar] [CrossRef]
- Scholte, H.R.; Busch, H.F.; Bakker, H.D.; Bogaard, J.M.; Luyt-Houwen, I.E.; Kuyt, L.P. Riboflavin-responsive complex I deficiency. Biochim. Biophys. Acta 1995, 1271, 75–83. [Google Scholar] [CrossRef]
- Nouws, J.; Nijtmans, L.; Houten, S.M.; van den Brand, M.; Huynen, M.; Venselaar, H.; Hoefs, S.; Gloerich, J.; Kronick, J.; Hutchin, T.; et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010, 12, 283–294. [Google Scholar] [CrossRef]
- Gerards, M.; van den Bosch, B.J.C.; Danhauser, K.; Serre, V.; van Weeghel, M.; Wanders, R.J.A.; Nicolaes, G.A.F.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134 Pt 1, 210–219. [Google Scholar] [CrossRef]
- Dewulf, J.P.; Barrea, C.; Vincent, M.-F.; De Laet, C.; Van Coster, R.; Seneca, S.; Marie, S.; Nassogne, M.-C. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: Report on nine patients. Mol. Genet. Metab. 2016, 118, 185–189. [Google Scholar] [CrossRef]
- Yankovskaya, V.; Horsefield, R.; Törnroth, S.; Luna-Chavez, C.; Miyoshi, H.; Léger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Na, U.; Winge, D.R.; Rutter, J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef]
- Crona, J.; Taïeb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Ibrahim, A.; Chopra, S. Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumors. Arch. Pathol. Lab. Med. 2020, 144, 655–660. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Kang, Y.-K.; Nishida, T.; von Mehren, M. Gastrointestinal stromal tumours. Nat. Rev. Dis. Primers 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Pitsava, G.; Settas, N.; Faucz, F.R.; Stratakis, C.A.; Triad, C.; Syndrome, C.-S. 3PAS and Other Tumors Due to SDH Deficiency. Front. Endocrinol. 2021, 12, 680609. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, D.; Yang, K.; Xu, N.; Peng, J.; Zhu, Z. Research progress on the pathogenesis of the SDHB mutation and related diseases. Biomed. Pharmacother. 2023, 167, 115500. [Google Scholar] [CrossRef]
- Kim, E.; Rath, E.M.; Tsang, V.H.M.; Duff, A.P.; Robinson, B.G.; Church, W.B.; Benn, D.E.; Dwight, T.; Clifton-Bligh, R.J. Structural and functional consequences of succinate dehydrogenase subunit B mutations. Endocr. Relat. Cancer 2015, 22, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Shand, J.A.D.; van Schalkwyk, J.; Beavis, V.; Niyagama, A.; Taylor, S.; Eagleton, C. High rates of the SDHB p.Arg46Gln pathogenic variant predisposes New Zealand Māori to phaeochromocytoma/paraganglioma. Intern. Med. J. 2023, 53, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Biggar, M.; Park, B.; Xia, W.; Karalus, M.; Moss, D.; Rahman, H. Adrenalectomy and Abdominal Paraganglioma Surgery at an Ethnically Diverse New Zealand Center: Māori Ethnicity Frequent amongst Paraganglioma Patients. World J. Endocr. Surg. 2023, 14, 73–78. [Google Scholar] [CrossRef]
- Else, T.; Marvin, M.L.; Everett, J.N.; Gruber, S.B.; Arts, H.A.; Stoffel, E.M.; Auchus, R.J.; Raymond, V.M. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J. Clin. Endocrinol. Metab. 2014, 99, E1482–E1486. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, I.; Grunenwald, S.; Burnichon, N.; Khalifa, E.; Dumas, N.; Binet, M.-C.; Nolet, S.; Gimenez-Roqueplo, A.-P. A SDHC Founder Mutation Causes Paragangliomas (PGLs) in the French Canadians: New Insights on the SDHC-Related PGL. J. Clin. Endocrinol. Metab. 2016, 101, 4710–4718. [Google Scholar] [CrossRef]
- Jha, A.; de Luna, K.; Balili, C.A.; Millo, C.; Paraiso, C.A.; Ling, A.; Gonzales, M.K.; Viana, B.; Alrezk, R.; Adams, K.T.; et al. Clinical, Diagnostic, and Treatment Characteristics of SDHA-Related Metastatic Pheochromocytoma and Paraganglioma. Front. Oncol. 2019, 9, 53. [Google Scholar] [CrossRef]
- Schipani, A.; Nannini, M.; Astolfi, A.; Pantaleo, M.A. SDHA Germline Mutations in SDH-Deficient GISTs: A Current Update. Genes 2023, 14, 646. [Google Scholar] [CrossRef]
- Wang, Y.; Lilienfeldt, N.; Hekimi, S. Understanding coenzyme Q. Physiol. Rev. 2024, 104, 1533–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gumus, E.; Hekimi, S. A novel COQ7 mutation causing primarily neuromuscular pathology and its treatment options. Mol. Genet. Metab. Rep. 2022, 31, 100877. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, A.; Theuriet, J.; Fontaine, F.; Mosbach, V.; Lacoste, N.; Ribault, S.; Risson, V.; Carras, J.; Coudert, L.; Simonet, T.; et al. Homozygous COQ7 mutation: A new cause of potentially treatable distal hereditary motor neuropathy. Brain 2023, 146, 3470–3483. [Google Scholar] [CrossRef]
- Pettenuzzo, I.; Carli, S.; Sánchez-Cuesta, A.; Isidori, F.; Montanari, F.; Grippa, M.; Lanzoni, G.; Ambrosetti, I.; Di Pisa, V.; Cordelli, D.M.; et al. COQ7 defect causes prenatal onset of mitochondrial CoQ10 deficiency with cardiomyopathy and gastrointestinal obstruction. Eur. J. Hum. Genet. 2024, 32, 938–946. [Google Scholar] [CrossRef]
- Qiu, Y.; Xiong, Y.; Wang, L.; Zhu, M.; Tan, D.; Hong, D. Homozygous variant in COQ7 causes autosomal recessive hereditary spastic paraplegia. Ann. Clin. Transl. Neurol. 2024, 11, 1067–1074. [Google Scholar] [CrossRef]
- Fabra, M.A.; Paredes-Fuentes, A.J.; Carnerero, M.T.; de Ayala, D.J.M.F.; Luque, A.A.; Sánchez-Cuesta, A.; Staiano, C.; Sanchez-Pintos, P.; Couce, M.L.; Tomás, M.; et al. New variants expand the neurological phenotype of COQ7 deficiency. J. Inherit. Metab. Dis. 2024, 47, 1047–1068. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.C.; Pileggi, C.A.; Wang, Y.; Kernohan, K.; Hartley, T.; McMillan, H.J.; Sampaio, M.L.; Melkus, G.; Woulfe, J.; Parmar, G.; et al. Novel Homozygous Variant in COQ7 in Siblings With Hereditary Motor Neuropathy. Neurol. Genet. 2023, 9, e200048. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.A.; Xia, D.; Kim, H.; Deisenhofer, J.; Zhang, L.; Kachurin, A.M.; Yu, L. Structural basis of functions of the mitochondrial cytochrome bc1 complex. Biochim. Biophys. Acta 1998, 1365, 151–158. [Google Scholar] [CrossRef]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial complex III Rieske Fe-S protein processing and assembly. Cell Cycle 2018, 17, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Brandt, U.; Yu, L.; Yu, C.A.; Trumpower, B.L. The mitochondrial targeting presequence of the Rieske iron-sulfur protein is processed in a single step after insertion into the cytochrome bc1 complex in mammals and retained as a subunit in the complex. J. Biol. Chem. 1993, 268, 8387–8390. [Google Scholar] [CrossRef]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.-Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Blue, E.E.; Huang, S.J.; Khan, A.; Golden-Grant, K.; Boyd, B.; Rosenthal, E.A.; Gillentine, M.A.; Fleming, L.R.; Adams, D.R.; Wolfe, L.; et al. Dual diagnosis of UQCRFS1-related mitochondrial complex III deficiency and recessive GJA8-related cataracts. Rare 2024, 2, 100040. [Google Scholar] [CrossRef] [PubMed]
- Gaignard, P.; Menezes, M.; Schiff, M.; Bayot, A.; Rak, M.; de Baulny, H.O.; Su, C.-H.; Gilleron, M.; Lombes, A.; Abida, H.; et al. Mutations in CYC1, encoding cytochrome c1 subunit of respiratory chain complex III, cause insulin-responsive hyperglycemia. Am. J. Hum. Genet. 2013, 93, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Yanagisawa, K.; Kunishima, S.; Shiina, M.; Ogawa, Y.; Nakashima, M.; Hirato, J.; Imagawa, E.; Fujita, A.; Hamanaka, K.; et al. A novel CYCS mutation in the α-helix of the CYCS C-terminal domain causes non-syndromic thrombocytopenia. Clin. Genet. 2018, 94, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Che, F.; Zhao, J.; Zhao, Y.; Wang, Z.; Zhang, L.; Yang, Y. A Novel Heterozygous Pathogenic Variation in CYCS Gene Cause Autosomal Dominant Non-Syndromic Thrombocytopenia 4 in a Large Chinese Family. Front. Genet. 2021, 12, 783455. [Google Scholar] [CrossRef]
- De Rocco, D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Morison, I.M.; Bordé, E.M.C.; Cheesman, E.J.; Cheong, P.L.; Holyoake, A.J.; Fichelson, S.; Weeks, R.J.; Lo, A.; Davies, S.M.K.; Wilbanks, S.M.; et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 2008, 40, 387–389. [Google Scholar] [CrossRef]
- Giavi, K.; Glentis, S.; Bouchla, A.; Apostolidou, A.; Marinakis, N.M.; Kattamis, A.; Katsantoni, E.; Pappa, V. A Novel Variant of the CYCS Gene Alters Apoptosis of Megakaryocytes in a Family with Thrombocytopenia. Blood 2023, 142, 5409. [Google Scholar] [CrossRef]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B. Complex IV—The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef]
- Kadenbach, B. Regulation of cytochrome c oxidase contributes to health and optimal life. World J. Biol. Chem. 2020, 11, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Abu-Libdeh, B.; Douiev, L.; Amro, S.; Shahrour, M.; Ta-Shma, A.; Miller, C.; Elpeleg, O.; Saada, A. Mutation in the COX4I1 gene is associated with short stature, poor weight gain and increased chromosomal breaks, simulating Fanconi anemia. Eur. J. Hum. Genet. 2017, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Pillai, N.R.; AlDhaheri, N.S.; Ghosh, R.; Lim, J.; Streff, H.; Nayak, A.; Graham, B.H.; Hanchard, N.A.; Elsea, S.H.; Scaglia, F. Biallelic variants in COX4I1 associated with a novel phenotype resembling Leigh syndrome with developmental regression, intellectual disability, and seizures. Am. J. Med. Genet. A 2019, 179, 2138–2143. [Google Scholar] [CrossRef]
- Shteyer, E.; Saada, A.; Shaag, A.; Al-Hijawi, F.A.; Kidess, R.; Revel-Vilk, S.; Elpeleg, O. Exocrine pancreatic insufficiency, dyserythropoeitic anemia, and calvarial hyperostosis are caused by a mutation in the COX4I2 gene. Am. J. Hum. Genet. 2009, 84, 412–417. [Google Scholar] [CrossRef]
- Baertling, F.; Al-Murshedi, F.; Sánchez-Caballero, L.; Al-Senaidi, K.; Joshi, N.P.; Venselaar, H.; van der Brand, M.A.; Nijtmans, L.G.; Rodenburg, R.J. Mutation in mitochondrial complex IV subunit COX5A causes pulmonary arterial hypertension, lactic acidemia, and failure to thrive. Hum. Mutat. 2017, 38, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, G.; Makino, S.; Hayashi, M.; Abe, A.; Numakura, C.; Ueki, M.; Tanaka, A.; Ito, C.; Toshimori, K.; Ogawa, N.; et al. A mutation of COX6A1 causes a recessive axonal or mixed form of Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2014, 95, 294–300. [Google Scholar] [CrossRef]
- Inoue, M.; Uchino, S.; Iida, A.; Noguchi, S.; Hayashi, S.; Takahashi, T.; Fujii, K.; Komaki, H.; Takeshita, E.; Nonaka, I.; et al. COX6A2 variants cause a muscle-specific cytochrome c oxidase deficiency. Ann. Neurol. 2019, 86, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Massa, V.; Fernandez-Vizarra, E.; Alshahwan, S.; Bakhsh, E.; Goffrini, P.; Ferrero, I.; Mereghetti, P.; D’Adamo, P.; Gasparini, P.; Zeviani, M. Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus-encoded subunit of cytochrome c oxidase. Am. J. Hum. Genet. 2008, 82, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef]
- Indrieri, A.; van Rahden, V.A.; Tiranti, V.; Morleo, M.; Iaconis, D.; Tammaro, R.; D’Amato, I.; Conte, I.; Maystadt, I.; Demuth, S.; et al. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am. J. Hum. Genet. 2012, 91, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Hallmann, K.; Kudin, A.P.; Zsurka, G.; Kornblum, C.; Reimann, J.; Stüve, B.; Waltz, S.; Hattingen, E.; Thiele, H.; Nürnberg, P.; et al. Loss of the smallest subunit of cytochrome c oxidase, COX8A, causes Leigh-like syndrome and epilepsy. Brain 2016, 139 Pt 2, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Boyer, P.D. The ATP synthase--a splendid molecular machine. Annu. Rev. Biochem. 1997, 66, 717–749. [Google Scholar] [CrossRef]
- Walker, J.E. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 1992, 25, 253–324. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Valdebenito, G.E.; Chacko, A.R.; Duchen, M.R. The mitochondrial ATP synthase as an ATP consumer-a surprising therapeutic target. EMBO J. 2023, 42, e114141. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Benincá, C.; del Rio, L.F.; Shu, C.; Baghdasarian, S.; Zanette, V.; Gerle, C.; Jiko, C.; Khairallah, R.; Khan, S.; et al. Inhibition of ATP synthase reverse activity restores energy homeostasis in mitochondrial pathologies. EMBO J. 2023, 42, e111699. [Google Scholar] [CrossRef]
- Houstek, J.; Mrácek, T.; Vojtísková, A.; Zeman, J. Mitochondrial diseases and ATPase defects of nuclear origin. Biochim. Biophys. Acta 2004, 1658, 115–121. [Google Scholar] [CrossRef]
- Nasca, A.; Mencacci, N.E.; Invernizzi, F.; Zech, M.; Sarmiento, I.J.K.; Legati, A.; Frascarelli, C.; Bustos, B.I.; Romito, L.M.; Krainc, D.; et al. Variants in ATP5F1B are associated with dominantly inherited dystonia. Brain 2023, 146, 2730–2738. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, F.-J.; Renger, G.; Friedrich, T.; Kreslavski, V.D.; Zharmukhamedov, S.K.; Los, D.A.; Kuznetsov, V.V.; Allakhverdiev, S.I. Reactive oxygen species: Re-evaluation of generation, monitoring and role in stress-signaling in phototrophic organisms. Biochim. Biophys. Acta 2014, 1837, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A. Mitochondrial reactive oxygen species: Do they extend or shorten animal lifespan? Biochim. Biophys. Acta 2016, 1857, 1116–1126. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Mallikarjun, V.; Stefanatos, R.; Sanz, A. Regulation of Lifespan by the Mitochondrial Electron Transport Chain: Reactive Oxygen Species-Dependent and Reactive Oxygen Species-Independent Mechanisms. Antioxid. Redox Signal. 2013, 19, 1953–1969. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef]
- Lee, Y.M.; He, W.; Liou, Y.-C. The redox language in neurodegenerative diseases: Oxidative post-translational modifications by hydrogen peroxide. Cell Death Dis. 2021, 12, 58. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Castejon-Vega, B.; Cordero, M.D.; Sanz, A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants 2023, 12, 831. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Bayir, H.; Belousov, V.; Chang, C.J.; Davies, K.J.A.; Davies, M.J.; Dick, T.P.; Finkel, T.; Forman, H.J.; Janssen-Heininger, Y.; et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metab. 2022, 4, 651–662. [Google Scholar] [CrossRef]
- Scialo, F.; Sanz, A. Coenzyme Q redox signalling and longevity. Free Radic. Biol. Med. 2021, 164, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Back, P.; De Vos, W.H.; Depuydt, G.G.; Matthijssens, F.; Vanfleteren, J.R.; Braeckman, B.P. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free Radic. Biol. Med. 2012, 52, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.-L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf; stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88 Pt B, 290–301. [Google Scholar] [CrossRef]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef]
- Oláhová, M.; Taylor, S.R.; Khazaipoul, S.; Wang, J.; Morgan, B.A.; Matsumoto, K.; Blackwell, T.K.; Veal, E.A. A redox-sensitive peroxiredoxin that is important for longevity has tissue- and stress-specific roles in stress resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 19839–19844. [Google Scholar] [CrossRef]
- Chávez, V.; Mohri-Shiomi, A.; Maadani, A.; Vega, L.A.; Garsin, D.A. Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics 2007, 176, 1567–1577. [Google Scholar] [CrossRef]
- Inoue, H.; Hisamoto, N.; An, J.H.; Oliveira, R.P.; Nishida, E.; Blackwell, T.K.; Matsumoto, K. The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 2005, 19, 2278–2283. [Google Scholar] [CrossRef]
- Kondo, M.; Yanase, S.; Ishii, T.; Hartman, P.S.; Matsumoto, K.; Ishii, N. The p38 signal transduction pathway participates in the oxidative stress-mediated translocation of DAF-16 to Caenorhabditis elegans nuclei. Mech. Ageing Dev. 2005, 126, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Hsin, H.; Libina, N.; Kenyon, C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 2001, 28, 139–145. [Google Scholar] [CrossRef]
- Tullet, J.M.A.; Hertweck, M.; An, J.H.; Baker, J.; Hwang, J.Y.; Liu, S.; Oliveira, R.P.; Baumeister, R.; Blackwell, T.K. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 2008, 132, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Tissenbaum, H.A. DAF-16: FOXO in the Context of C. elegans. Curr. Top. Dev. Biol. 2018, 127, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Libina, N.; Berman, J.R.; Kenyon, C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 2003, 115, 489–502. [Google Scholar] [CrossRef]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef]
- Sies, H.; Mailloux, R.J.; Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 2024, 25, 701–719. [Google Scholar] [CrossRef]
- Kalous, K.S.; Wynia-Smith, S.L.; Smith, B.C. Sirtuin Oxidative Post-translational Modifications. Front. Physiol. 2021, 12, 763417. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Mazzeo, L.E.M.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Branicky, R.; Wang, Y.; Khaki, A.; Liu, J.-L.; Kramer-Drauberg, M.; Hekimi, S. Stimulation of RAS-dependent ROS signaling extends longevity by modulating a developmental program of global gene expression. Sci. Adv. 2022, 8, eadc9851. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, J.; Mai, Y.; Hong, Y.; Jia, Z.; Tian, G.; Liu, Y.; Liang, H.; Liu, J. Current advances and future trends of hormesis in disease. npj Aging 2024, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Nascarella, M.; Pressman, P.; Hayes, A.W.; Dhawan, G.; Kapoor, R.; Calabrese, V.; Agathokleous, E. Hormesis determines lifespan. Ageing Res. Rev. 2024, 94, 102181. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Hollunger, G. The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate-linked reduction of pyridine nucleotide. J. Biol. Chem. 1961, 236, 1534–1543. [Google Scholar] [CrossRef]
- Wright, J.J.; Biner, O.; Chung, I.; Burger, N.; Bridges, H.R.; Hirst, J. Reverse Electron Transfer by Respiratory Complex I Catalyzed in a Modular Proteoliposome System. J. Am. Chem. Soc. 2022, 144, 6791–6801. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef]
- Scialò, F.; Sriram, A.; Fernández-Ayala, D.; Gubina, N.; Lõhmus, M.; Nelson, G.; Logan, A.; Cooper, H.M.; Navas, P.; Enríquez, J.A.; et al. Mitochondrial ROS Produced via Reverse Electron Transport Extend Animal Lifespan. Cell Metab. 2016, 23, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Agüera, M.C.; Gao, L.; González-Rodríguez, P.; Pintado, C.O.; Arias-Mayenco, I.; García-Flores, P.; García-Pergañeda, A.; Pascual, A.; Ortega-Sáenz, P.; López-Barneo, J. Oxygen Sensing by Arterial Chemoreceptors Depends on Mitochondrial Complex I Signaling. Cell Metab. 2015, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.; Lu, B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants 2023, 12, 895. [Google Scholar] [CrossRef]
- Jiang, H.; Zuo, J.; Li, B.; Chen, R.; Luo, K.; Xiang, X.; Lu, S.; Huang, C.; Liu, L.; Tang, J.; et al. Drug-induced oxidative stress in cancer treatments: Angel or devil? Redox Biol. 2023, 63, 102754. [Google Scholar] [CrossRef] [PubMed]
- Barnes, L.D.; Robinson, A.K.; Williams, R.F.; Horowitz, P.M. Binding of colchicine to renal tubulin at 5 degrees C. Biochem. Biophys. Res. Commun. 1983, 116, 866–872. [Google Scholar] [CrossRef]
- Viña, J.; Borrás, C. Unlocking the biochemical secrets of longevity: Balancing healthspan and lifespan. FEBS Lett. 2024, 598, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Torres, T.C.; Moaddeli, D.; Averbukh, M.; Coakley, A.; Dutta, N.; Garcia, G.; Higuchi-Sanabria, R. Surveying Low-Cost Methods to Measure Lifespan and Healthspan in Caenorhabditis elegans. J. Vis. Exp. 2022, 183, e64091. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, L.; Liu, Y. Protocol for assessing the healthspan of Caenorhabditis elegans after potential anti-aging drug treatment. STAR Protoc. 2023, 4, 102285. [Google Scholar] [CrossRef] [PubMed]
- Rollins, J.A.; Howard, A.C.; Dobbins, S.K.; Washburn, E.H.; Rogers, A.N. Assessing Health Span in Caenorhabditis elegans: Lessons From Short-Lived Mutants. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Thorburn, D.R.; Wong, L.-J.; Vladutiu, G.D.; Haas, R.H.; Le, T.; Hoppel, C.; Sedensky, M.; Morgan, P.; Hahn, S.H. Quality improvement of mitochondrial respiratory chain complex enzyme assays using Caenorhabditis elegans. Genet. Med. 2011, 13, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Lemire, B.D. Stable heteroplasmy but differential inheritance of a large mitochondrial DNA deletion in nematodes. Biochem. Cell Biol. 2002, 80, 645–654. [Google Scholar] [CrossRef]
- Murfitt, R.R.; Vogel, K.; Sanadi, D.R. Characterization of the mitochondria of the free-living nematode, Caenorhabditis elegans. Comp. Biochem. Physiol. Part B Comp. Biochem. 1976, 53, 423–430. [Google Scholar] [CrossRef]
- Wadsworth, W.G.; Riddle, D.L. Developmental regulation of energy metabolism in Caenorhabditis elegans. Dev. Biol. 1989, 132, 167–173. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Lemire, B.D. The role of mitochondria in the life of the nematode, Caenorhabditis elegans. Biochim. Biophys. Acta 2003, 1638, 91–105. [Google Scholar] [CrossRef]
- Falk, M.J.; Rosenjack, J.R.; Polyak, E.; Suthammarak, W.; Chen, Z.; Morgan, P.G.; Sedensky, M.M. Subcomplex Ilambda specifically controls integrated mitochondrial functions in Caenorhabditis elegans. PLoS ONE 2009, 4, e6607. [Google Scholar] [CrossRef]
- Grad, L.I.; Lemire, B.D. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase deficiency, oxidative stress and vitamin-responsive lactic acidosis. Hum. Mol. Genet. 2004, 13, 303–314. [Google Scholar] [CrossRef]
- Johnson, D.; Nehrke, K. Mitochondrial fragmentation leads to intracellular acidification in Caenorhabditis elegans and mammalian cells. Mol. Biol. Cell 2010, 21, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.Y.; Sayles, L.C.; Grad, L.I.; Pilgrim, D.B.; Lemire, B.D. Mitochondrial respiratory chain deficiency in Caenorhabditis elegans results in developmental arrest and increased life span. J. Biol. Chem. 2001, 276, 32240–32246. [Google Scholar] [CrossRef] [PubMed]
- Suthammarak, W.; Somerlot, B.H.; Opheim, E.; Sedensky, M.; Morgan, P.G. Novel interactions between mitochondrial superoxide dismutases and the electron transport chain. Aging Cell 2013, 12, 1132–1140. [Google Scholar] [CrossRef]
- Kayser, E.B.; Morgan, P.G.; Sedensky, M.M. GAS-1: A mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology 1999, 90, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Niezgoda, J.; Morgan, P.G. Anesthetic considerations in patients with mitochondrial defects. Paediatr. Anaesth. 2013, 23, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Lemire, B.D. Riboflavin enhances the assembly of mitochondrial cytochrome c oxidase in C. elegans NADH-ubiquinone oxidoreductase mutants. Biochim. Biophys. Acta 2006, 1757, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Christodoulou, J.; Rahman, S. Disorders of riboflavin metabolism. J. Inherit. Metab. Dis. 2019, 42, 608–619. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.-H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.D.; Miranda, M.; Skinner, O.S.; Wiesenthal, P.P.; Wellner, S.M.; Jourdian, A.A.; Ruvkun, G.; Mootha, V.K. Hypoxia and intra-complex genetic suppressors rescue complex I mutants by a shared mechanism. Cell 2024, 187, 659–675.e18. [Google Scholar] [CrossRef]
- Chuaijit, S.; Boonyatistan, W.; Boonchuay, P.; Metheetrairut, C.; Suthammarak, W. Identification of a novel mitochondrial complex I assembly factor ACDH-12 in Caenorhabditis elegans. Mitochondrion 2019, 46, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Kayser, E.-B.; Sedensky, M.M.; Morgan, P.G. The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegans. Mech. Ageing Dev. 2004, 125, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Lemire, B.D.; Oyedotun, K.S. The Saccharomyces cerevisiae mitochondrial succinate:ubiquinone oxidoreductase. Biochim. Biophys. Acta 2002, 1553, 102–116. [Google Scholar] [CrossRef]
- Veloukas, T.; Karaoglanidis, G.S. Biological activity of the succinate dehydrogenase inhibitor fluopyram against Botrytis cinerea and fungal baseline sensitivity. Pest Manag. Sci. 2012, 68, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Duvenage, L.; Munro, C.A.; Gourlay, C.W. The potential of respiration inhibition as a new approach to combat human fungal pathogens. Curr. Genet. 2019, 65, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Schleker, A.S.S.; Rist, M.; Matera, C.; Damijonaitis, A.; Collienne, U.; Matsuoka, K.; Habash, S.S.; Twelker, K.; Gutbrod, O.; Saalwächter, C.; et al. Mode of action of fluopyram in plant-parasitic nematodes. Sci. Rep. 2022, 12, 11954. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Takahashi, K.; Tomita, S.; Keino, T.; Honda, S.; Yoshino, K.; Suzuki, K. A methyl viologen-sensitive mutant of the nematode Caenorhabditis elegans. Mutat. Res. 1990, 237, 165–171. [Google Scholar] [CrossRef]
- Adachi, H.; Fujiwara, Y.; Ishii, N. Effects of oxygen on protein carbonyl and aging in Caenorhabditis elegans mutants with long (age-1) and short (mev-1) life spans. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, B240–B244. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Wei, A.Y.; Sherman, T.A.; Foster, T.H.; Nehrke, K. Chromophore-Assisted Light Inactivation of Mitochondrial Electron Transport Chain Complex II in Caenorhabditis elegans. Sci. Rep. 2016, 6, 29695. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Miyazawa, M.; Onodera, A.; Yasuda, K.; Kawabe, N.; Kirinashizawa, M.; Yoshimura, S.; Maruyama, N.; Hartman, P.S.; Ishii, N. Mitochondrial reactive oxygen species generation by the SDHC V69E mutation causes low birth weight and neonatal growth retardation. Mitochondrion 2011, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Pujol, C.; Bratic-Hench, I.; Sumakovic, M.; Hench, J.; Mourier, A.; Baumann, L.; Pavlenko, V.; Trifunovic, A. Succinate dehydrogenase upregulation destabilize complex I and limits the lifespan of gas-1 mutant. PLoS ONE 2013, 8, e59493. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-W.; Wei, C.-C.; Liao, V.H.-C. Curcumin-mediated oxidative stress resistance in Caenorhabditis elegans is modulated by age-1, akt-1, pdk-1, osr-1, unc-43, sek-1, skn-1, sir-2.1, and mev-1. Free Radic. Res. 2014, 48, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.T.G.; Rodrigues, L.B.L.; Salgueiro, W.G.; de Castro Dal Forno, A.H.; Rodrigues, C.F.; Sacramento, M.; Franco, J.; Alves, D.; de Paula Oliveira, R.; Pinton, S.; et al. Organoselenotriazoles attenuate oxidative damage induced by mitochondrial dysfunction in mev-1 Caenorhabditis elegans mutants. J. Trace. Elem. Med. Biol. 2019, 53, 34–40. [Google Scholar] [CrossRef]
- Huang, J.; Lemire, B.D. Mutations in the C. elegans succinate dehydrogenase iron-sulfur subunit promote superoxide generation and premature aging. J. Mol. Biol. 2009, 387, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.M.; Damjanac, T.; Zhang, Y.; Chen, C.; Hu, J.; Maher, L.J. Modeling succinate dehydrogenase loss disorders in C. elegans through effects on hypoxia-inducible factor. PLoS ONE 2019, 14, e0227033. [Google Scholar] [CrossRef] [PubMed]
- Saskői, É.; Hujber, Z.; Nyírő, G.; Likó, I.; Mátyási, B.; Petővári, G.; Mészáros, K.; Kovács, A.L.; Patthy, L.; Supekar, S. The SDHB Arg230His mutation causing familial paraganglioma alters glycolysis in a new Caenorhabditis elegans model. Dis. Model. Mech. 2020, 13, dmm044925. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, M.A.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Huang, H.; Li, G.; He, Y.; Chen, J.; Yan, J.; Zhang, Q.; Li, L.; Cai, X. Cellular succinate metabolism and signaling in inflammation: Implications for therapeutic intervention. Front. Immunol. 2024, 15, 1404441. [Google Scholar] [CrossRef]
- Goncalves, J.; Wan, Y.; Guo, X.; Rha, K.; LeBoeuf, B.; Zhang, L.; Estler, K.; Garcia, L.R. Succinate Dehydrogenase-Regulated Phosphoenolpyruvate Carboxykinase Sustains Copulation Fitness in Aging C. elegans Males. iScience 2020, 23, 100990. [Google Scholar] [CrossRef]
- Hipólito, A.; Martins, F.; Mendes, C.; Lopes-Coelho, F.; Serpa, J. Molecular and Metabolic Reprogramming: Pulling the Strings Toward Tumor Metastasis. Front. Oncol. 2021, 11, 656851. [Google Scholar] [CrossRef]
- Cerón, J. Caenorhabditis elegans for research on cancer hallmarks. Dis. Model. Mech. 2023, 16, dmm050079. [Google Scholar] [CrossRef] [PubMed]
- Asencio, C.; Navas, P.; Cabello, J.; Schnabel, R.; Cypser, J.R.; Johnson, T.E.; Rodríguez-Aguilera, J.C. Coenzyme Q supports distinct developmental processes in Caenorhabditis elegans. Mech. Ageing Dev. 2009, 130, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Gavilán, A.; Asencio, C.; Cabello, J.; Rodríguez-Aguilera, J.C.; Schnabel, R.; Navas, P. C. elegans knockouts in ubiquinone biosynthesis genes result in different phenotypes during larval development. Biofactors 2005, 25, 21–29. [Google Scholar] [CrossRef]
- Hihi, A.K.; Gao, Y.; Hekimi, S. Ubiquinone is necessary for Caenorhabditis elegans development at mitochondrial and non-mitochondrial sites. J. Biol. Chem. 2002, 277, 2202–2206. [Google Scholar] [CrossRef]
- Gomez, F.; Saiki, R.; Chin, R.; Srinivasan, C.; Clarke, C.F. Restoring de novo coenzyme Q biosynthesis in Caenorhabditis elegans coq-3 mutants yields profound rescue compared to exogenous coenzyme Q supplementation. Gene 2012, 506, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.; Hihi, A.K.; Benard, C.Y.; Branicky, R.; Hekimi, S. Molecular mechanism of maternal rescue in the clk-1 mutants of Caenorhabditis elegans. J. Biol. Chem. 2003, 278, 49555–49562. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.; Larsen, P.L.; Clarke, C.F. A dietary source of coenzyme Q is essential for growth of long-lived Caenorhabditis elegans clk-1 mutants. Proc. Natl. Acad. Sci. USA 2001, 98, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Miyadera, H.; Amino, H.; Hiraishi, A.; Taka, H.; Murayama, K.; Miyoshi, H.; Sakamoto, K.; Ishii, N.; Hekimi, S.; Kiyoshi, K. Altered quinone biosynthesis in the long-lived clk-1 mutants of Caenorhabditis elegans. J. Biol. Chem. 2001, 276, 7713–7716. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Ewbank, J.J.; Barnes, T.M.; Lakowski, B.; Lussier, M.; Bussey, H.; Hekimi, S. Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 1997, 275, 980–983. [Google Scholar] [CrossRef]
- Liu, J.-L.; Yee, C.; Wang, Y.; Hekimi, S. A single biochemical activity underlies the pleiotropy of the aging-related protein CLK-1. Sci. Rep. 2017, 7, 859. [Google Scholar] [CrossRef] [PubMed]
- Gomez, F.; Monsalve, G.C.; Tse, V.; Saiki, R.; Weng, E.; Lee, L.; Srinivasan, C.; Frand, A.R.; Clarke, C.F. Delayed accumulation of intestinal coliform bacteria enhances life span and stress resistance in Caenorhabditis elegans fed respiratory deficient E. coli. BMC Microbiol. 2012, 12, 300. [Google Scholar] [CrossRef]
- Feng, J.; Bussière, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef]
- Jafari, G.; Wasko, B.M.; Tonge, A.; Schurman, N.; Dong, C.; Li, Z.; Peters, R.; Kayser, E.-B.; Pitt, J.N.; Morgan, P.G.; et al. Tether mutations that restore function and suppress pleiotropic phenotypes of the C. elegans isp-1(qm150) Rieske iron-sulfur protein. Proc. Natl. Acad. Sci. USA 2015, 112, E6148–E6157. [Google Scholar] [CrossRef] [PubMed]
- Gatti, D.L.; Meinhardt, S.W.; Ohnishi, T.; Tzagoloff, A. Structure and function of the mitochondrial bc1 complex. A mutational analysis of the yeast Rieske iron-sulfur protein. J. Mol. Biol. 1989, 205, 421–435. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, L.; Shulmeister, V.M.; Chi, Y.-I.; Kim, K.K.; Hung, L.-W.; Crofts, A.R.; Berry, E.A.; Kim, S.-H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Dillin, A.; Hsu, A.-L.; Arantes-Oliveira, N.; Lehrer-Graiwer, J.; Hsin, H.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Kenyon, C. Rates of behavior and aging specified by mitochondrial function during development. Science 2002, 298, 2398–2401. [Google Scholar] [CrossRef]
- Cristina, D.; Cary, M.; Lunceford, A.; Clarke, C.; Kenyon, C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Park, D.; Riddle, D.L. Increased longevity of some C. elegans mitochondrial mutants explained by activation of an alternative energy-producing pathway. Mech. Ageing Dev. 2011, 132, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Novack, G.V.; Galeano, P.; Castaño, E.M.; Morelli, L. Mitochondrial Supercomplexes: Physiological Organization and Dysregulation in Age-Related Neurodegenerative Disorders. Front. Endocrinol. 2020, 11, 600. [Google Scholar] [CrossRef] [PubMed]
- Vincelli, A.J.; Pottinger, D.S.; Zhong, F.; Hanske, J.; Rolland, S.G.; Conradt, B.; Pletneva, E.V. Recombinant expression, biophysical characterization, and cardiolipin-induced changes of two Caenorhabditis elegans cytochrome c proteins. Biochemistry 2013, 52, 653–666. [Google Scholar] [CrossRef]
- Lan, J.; Rollins, J.A.; Zang, X.; Wu, D.; Zou, L.; Wang, Z.; Ye, C.; Wu, Z.; Kapahi, P.; Rogers, A.N.; et al. Translational Regulation of Non-autonomous Mitochondrial Stress Response Promotes Longevity. Cell Rep. 2019, 28, 1050–1062.e6. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, B.; Bender, E.; Arnold, S.; Hüttemann, M.; Lee, I.; Kadenbach, B. Cytochrome C oxidase and the regulation of oxidative phosphorylation. Chembiochem 2001, 2, 392–403. [Google Scholar] [CrossRef]
- Suthammarak, W.; Yang, Y.-Y.; Morgan, P.G.; Sedensky, M.M. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 6425–6435. [Google Scholar] [CrossRef]
- Lee, S.S.; Lee, R.Y.N.; Fraser, A.G.; Kamath, R.S.; Ahringer, J.; Ruvkun, G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 2003, 33, 40–48. [Google Scholar] [CrossRef]
- Tsang, W.Y.; Lemire, B.D. Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 2002, 291, 8–16. [Google Scholar] [CrossRef]
- Payne, B.A.I.; Chinnery, P.F. Mitochondrial dysfunction in aging: Much progress but many unresolved questions. Biochim. Biophys. Acta 2015, 1847, 1347–1353. [Google Scholar] [CrossRef]
- Rea, S.L.; Ventura, N.; Johnson, T.E. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007, 5, e259. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Hwang, W.; Jeong, D.-E.; Ryu, Y.; Ha, C.M.; Lee, S.-J.V.; Liu, L.; He, Z.M. Genetic inhibition of an ATP synthase subunit extends lifespan in C. elegans. Sci. Rep. 2018, 8, 14836. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cárdenas, L.P.; Villanueva-Chimal, E.; Salinas, L.S.; José-Nuñez, C.; Tuena de Gómez Puyou, M.; Navarro, R.E. Caenorhabditis elegans ATPase inhibitor factor 1 (IF1) MAI-2 preserves the mitochondrial membrane potential (Δψm) and is important to induce germ cell apoptosis. PLoS ONE 2017, 12, e0181984. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.R.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, B.; Oshiro-Rapley, N.; Li, M.; Paulo, J.A.; Webster, C.M.; Mou, F.; Kacergis, M.C.; Talkowski, M.E.; Carr, C.E.; et al. An Ancient, Unified Mechanism for Metformin Growth Inhibition in C. elegans and Cancer. Cell 2016, 167, 1705–1718.e13. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Hadi, N.A.; Gicquel, T.; Kaoutari, A.E.; Diémé, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Cedillo, L.; Ahsan, F.M.; Li, S.; Stuhr, N.L.; Zhou, Y.; Zhang, Y.; Adedoja, A.; Murphy, L.M.; Yerevanian, A.; Emans, S.; et al. Ether lipid biosynthesis promotes lifespan extension and enables diverse pro-longevity paradigms in Caenorhabditis elegans. eLife 2023, 12, e82210. [Google Scholar] [CrossRef]
- Su, S.; Wink, M. Natural lignans from Arctium lappa as antiaging agents in Caenorhabditis elegans. Phytochemistry 2015, 117, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.; Segaran, A.; Lord, S. Targeting OXPHOS and the electron transport chain in cancer; Molecular and therapeutic implications. Semin Cancer Biol. 2022, 86 Pt 2, 851–859. [Google Scholar] [CrossRef]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Sotgia, F.; Lisanti, M.P. Doxycycline and therapeutic targeting of the DNA damage response in cancer cells: Old drug, new purpose. Oncoscience 2015, 2, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; Brooks, D.R.; Shepherd, S.; Sotgia, F.; Lisanti, M.P. Antibiotics that target mitochondria extend lifespan in C. elegans. Aging 2023, 15, 11764–11781. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.; Morgan, G.; Watkins, J.; Protty, M.; Mason, M.; Adams, R.; Dolwani, S.; Pickering, J.; Delon, C.; Longley, M. Aspirin and cancer treatment: Systematic reviews and meta-analyses of evidence: For and against. Br. J. Cancer 2024, 130, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-B.; Mu, X.-H.; Wan, Q.-L.; He, X.-M.; Wu, G.-S.; Luo, H.-R. Aspirin increases metabolism through germline signalling to extend the lifespan of Caenorhabditis elegans. PLoS ONE 2017, 12, e0184027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-X.; Li, C.-X.; Kakar, M.U.; Khan, M.S.; Wu, P.-F.; Amir, R.M.; Dai, D.-F.; Naveed, M.; Li, Q.-Y.; Saeed, M.; et al. Resveratrol (RV): A pharmacological review and call for further research. Biomed. Pharmacother. 2021, 143, 112164. [Google Scholar] [CrossRef] [PubMed]
- Khanduja, K.L.; Bhardwaj, A. Stable free radical scavenging and antiperoxidative properties of resveratrol compared in vitro with some other bioflavonoids. Indian J. Biochem. Biophys. 2003, 40, 416–422. [Google Scholar] [PubMed]
- Forman, H.J. Redox signaling: An evolution from free radicals to aging. Free Radic. Biol. Med. 2016, 97, 398–407. [Google Scholar] [CrossRef]
- Desquiret-Dumas, V.; Gueguen, N.; Leman, G.; Baron, S.; Nivet-Antoine, V.; Chupin, S.; Chevrollier, A.; Vessières, E.; Ayer, A.; Ferré, M.; et al. Resveratrol induces a mitochondrial complex I-dependent increase in NADH oxidation responsible for sirtuin activation in liver cells. J. Biol. Chem. 2013, 288, 36662–36675. [Google Scholar] [CrossRef]
- Zini, R.; Morin, C.; Bertelli, A.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Gledhill, J.R.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc. Natl. Acad. Sci. USA 2007, 104, 13632–13637. [Google Scholar] [CrossRef]
- Kipp, J.L.; Ramirez, V.D. Effect of estradiol; diethylstilbestrol, and resveratrol on F0F1-ATPase activity from mitochondrial preparations of rat heart, liver, and brain. Endocrine 2001, 15, 165–175. [Google Scholar] [CrossRef]
- Zheng, J.; Ramirez, V.D. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br. J. Pharmacol. 2000, 130, 1115–1123. [Google Scholar] [CrossRef]
- Lee, I. Regulation of Cytochrome c Oxidase by Natural Compounds Resveratrol, (-)-Epicatechin, and Betaine. Cells 2021, 10, 1346. [Google Scholar] [CrossRef]
- Rea, S.L.; Wu, D.; Cypser, J.R.; Vaupel, J.W.; Johnson, T.E. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat. Genet. 2005, 37, 894–898. [Google Scholar] [CrossRef]
- Pricci, M.; Girardi, B.; Giorgio, F.; Losurdo, G.; Ierardi, E.; Di Leo, A. Curcumin and Colorectal Cancer: From Basic to Clinical Evidences. Int. J. Mol. Sci. 2020, 21, 2364. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Chiang, I.-T.; Ding, K.; Chung, J.-G.; Lin, W.-J.; Lin, S.-S.; Hwang, J.-J. Curcumin-induced apoptosis in human hepatocellular carcinoma j5 cells: Critical role of Ca(+2)-dependent pathway. Evid. Based Complement. Altern. Med. 2012, 2012, 512907. [Google Scholar] [CrossRef]
- Ataei, M.; Roufogalis, B.D.; Majeed, M.; Shah, M.A.; Sahebkar, A. Curcumin Nanofibers: A Novel Approach to Enhance the Anticancer Potential and Bioavailability of Curcuminoids. Curr. Med. Chem. 2023, 30, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Du, P.; Liu, X.; Xu, X.; Ge, Y.; Zhang, C. Curcumin supplementation increases longevity and antioxidant capacity in Caenorhabditis elegans. Front. Pharmacol. 2023, 14, 1195490. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, J.; Bu, L.-L.; Liao, D.-F.; Cheng, S.-W.; Zheng, X.-L. Curcumin Acetylsalicylate Extends the Lifespan of Caenorhabditis elegans. Molecules 2021, 26, 6609. [Google Scholar] [CrossRef] [PubMed]
- Hantsoo, K.; Gomes, M.; Brenner, D.; Cornwell, J.; Palinkas, C.M.; Malkin, S. Trends in estuarine pyrite formation point to an alternative model for Paleozoic pyrite burial. Geochim. Cosmochim. Acta 2024, 374, 51–71. [Google Scholar] [CrossRef]
- Spanoudakis, E.; Tavernarakis, N. Age-associated anatomical and physiological alterations in Caenorhabditis elegans. Mech. Ageing Dev. 2023, 213, 111827. [Google Scholar] [CrossRef]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Copeland, J.M.; Cho, J.; Lo, T.; Hur, J.H.; Bahadorani, S.; Arabyan, T.; Rabie, J.; Soh, J.; Walker, D.W. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 2009, 19, 1591–1598. [Google Scholar] [CrossRef]
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Rubner, M. Das Problem der Lebensdauer und seine Beziehungen zu Wachstum und Ernährung; De Gruyter: Berlin, Germany, 1908. [Google Scholar] [CrossRef]
- Garmany, A.; Terzic, A. Global Healthspan-Lifespan Gaps Among 183 World Health Organization Member States. JAMA Netw. Open 2024, 7, e2450241. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Zhu, L.J.; Yen, K.; Tissenbaum, H.A. Uncoupling lifespan and healthspan in Caenorhabditis elegans longevity mutants. Proc. Natl. Acad. Sci. USA 2015, 112, E277–E286. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Bowman, M.J.; Winn, M.E.; Senchuk, M.M.; Van Raamsdonk, J.M. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic Biol. Med. 2017, 108, 362–373. [Google Scholar] [CrossRef]
- Owusu-Ansah, E.; Song, W.; Perrimon, N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 2013, 155, 699–712. [Google Scholar] [CrossRef] [PubMed]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Vitale, M.; Sanz, A.; Scialò, F. Mitochondrial redox signaling: A key player in aging and disease. Aging 2023, 15, 2817–2818. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Bazopoulou, D.; Knoefler, D.; Zheng, Y.; Ulrich, K.; Oleson, B.J.; Xie, L.; Kim, M.; Kaufmann, A.; Lee, Y.-T.; Dou, Y.; et al. Developmental ROS individualizes organismal stress resistance and lifespan. Nature 2019, 576, 301–305. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).