

Lipid Oxidation at the Crossroads: Oxidative Stress and Neurodegeneration Explored in Caenorhabditis elegans

 , , and
, , and
Abstract
:1. Introduction
1.1. Brief Overview of Neurodegenerative Diseases Caused by Protein Aggregation
1.2. Comprehensive View of the Protein Homeostasis Network in NDDs
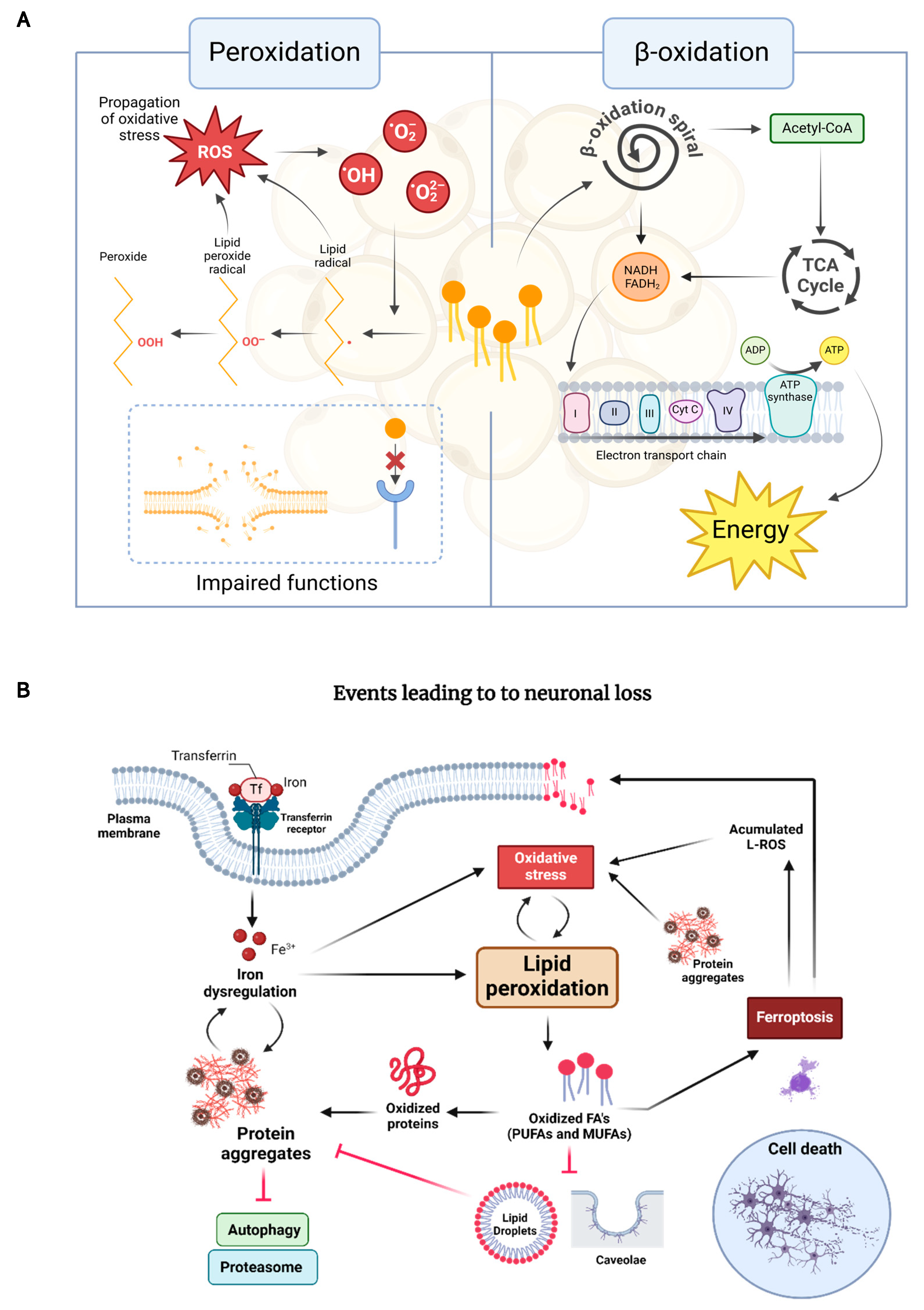
1.3. The Interplay Between Lipid Oxidation, Proteostasis, and Neuronal Death in NDDs
1.4. Key Genes Linking Lipid Dysregulation to NDDs
2. The Role of Lipid Peroxidation in Neurodegeneration: Insights from C. elegans Models
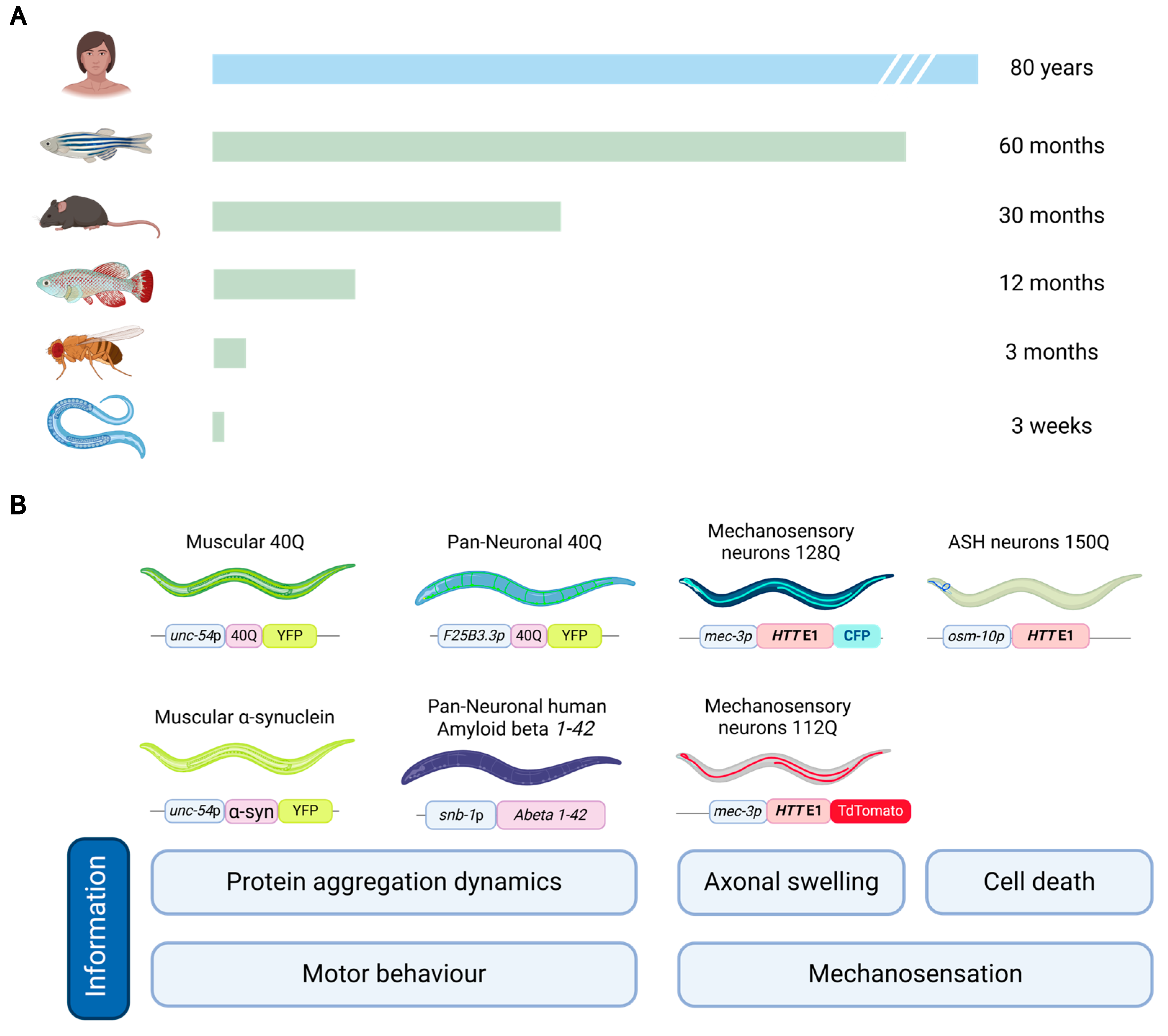
2.1. Advantages and Challenges of Using C. elegans for Lipid Peroxidation Studies
2.2. Current Understanding of Lipid Peroxidation in Neurodegenerative Diseases from C. elegans Models

{kind=link}
{kind=link}
| Metabolic Processes | Pathology | Key Findings | Intervention | Strain Generation | Applied Technologies | References |
|---|---|---|---|---|---|---|
| Oxidative stress | Alzheimer’s Disease | Decrease in lipid deposition and reduction in Aβ aggregation and oxidative stress. Rise in ACh levels (preventing cholinergic neuronal degeneration). | Benzofuran derivatives and chalcones | Conventional genetics | Microscopy | [124] |
| Oxidative stress | Alzheimer’s disease and polyQs-induced toxicity | Recalibration of lipid metabolism with an increase in the expression of genes involved in fatty acid β-oxidation, restoration of innate immune, and detoxification responses. Activation of SKN-1 leading to stress resistance. Increase in life span and improvement of age-related physical fitness together with the rescue of HD- and AD-like behavioral deficits. | AbaPep#07 (derived from abalone hemocyanin) | Conventional genetics | Microscopy, RNA-seq, RT-PCR, and GC–MS | [135] |
| Oxidative stress | Polyglutamine-induced neurotoxicity | Alleviation of lipid peroxidation and ROS production. Increases in the survival rates of polyQ nematodes intoxicated by paraquat by boosting antioxidant defenses. | Epimedium polysaccharide (EbPS-A1) | Conventional genetics | Microscopy and spectrophotometry | [123] |
| Oxidative stress | Parkinson’s disease | Restoration of lipid content in transgenic worms expressing α-synuclein. Recovery of DAergic neurons after 6-OHDA-induced neurodegeneration and a significant decrease in α-synuclein aggregation. Reduction in intracellular ROS through the activation of DAF-16 transcription factor (sod-3, hsp16.1, hsp16.2, and hsp12.6). | HLEA-P1 compound- Decanoic acid | Conventional genetics | Microscopy, spectrophotometry, and RT-qPCR | [132] |
| Oxidative stress | Parkinson’s disease | Reduction in malondialdehyde content (inhibition of lipid peroxidation) and increase in SOD and GPx activities. Mitigation of oxidative stress by regulating apoptosis and restoring the function of the cholinergic system. | Astragalan | Conventional genetics | Microscopy, spectrophotometry, and RT-PCR | [136] |
| Oxidative stress | Parkinson’s disease | Alleviation of lipid level changes, α-synuclein aggregation, improvement of locomotory behavior, and augmentation of dopamine levels. Increase in mRNA expression of daf-16, sod-1, sod-3, and ctl-2 and downregulation of sod-2, resulting in lifespan extension. | Tambulin | Conventional genetics | Microscopy, spectrophotometry, LC-MS/MS, and qPCR | [137] |
| Oxidative stress | Parkinson’s disease | Restoration of lipid deposition through upregulation of fat-7 and enhanced gcs-1-mediated glutathione synthesis. Potential activation mediated by HSF-1 and DAF-16 transcription factors. | 2-butoxy tetrahydrofuran (2-BTHF) | Conventional genetics, transgenesis | Molecular biology, in silico molecular coupling, RNA Seq, RT-qPCR, and microscopy | [138] |
| Oxidative stress | Parkinson’s disease | RAC1/ced-10 mutants displayed an increased unsaturation index, suggesting an increase in the content of polyunsaturated fatty acid (PUFA) and lipo-oxidative damage. ced-10 mutation produces a decrease in dopaminergic function and an elevation in the number of autophagic vesicles. | RAC1/ced-10 gene | Conventional genetics | Microscopy, synchrotron radiation, µFourier-transform infrared spectroscopy (SR-µFTIR), and UHPLC-MS | [117] |
| Oxidative stress | Parkinson’s disease | Significant decrease in fat content and reduction in alpha-synuclein aggregation. Positive regulation of sod-1, sod-2, sod-3, gst-4, gst-7, and ctl-2. Decrease in protein carbonyl content. | Shatavarin IV | Conventional genetics | Microscopy, RT-PCR, and LC-MS/MS | [139] |
| Oxidative stress | Parkinson’s disease | Increase in lipid accumulation as lipid droplets and protection against 6-hydroxydopamine (6-OHDA)-induced degeneration. | High-glucose and high-fructose diets | Transgenesis via extrachromosomal arrays, fluorescent protein tagging, and conventional genetics | Microscopy and qPCR | [140] |
| Oxidative stress | Parkinson’s disease | Recovery of lipid deposition and reduction in α-synuclein aggregation. Neuroprotection, food-sensing improvement, and lifespan extension. Upregulation of cat-2 (DA-synthesis) and sod-3 (free-radical scavenging) and downregulation of egl-1 (apoptosis). | H. leucospilota body wall and cuveirian tubule extracts | Conventional genetics | Microscopy, RT-PCR, and H-NMR | [133] |
| Oxidative stress | Neurodegeneration mediated by glucotoxicity | The combination of compounds prevented the rise in ROS and the accumulation of methylglyoxal-derived advanced glycation end products and safeguarded neuronal function, maintaining lifespan at levels similar to the wild-type strain. | Sulforaphane (SFN) and vitamin E (alpha-tocopherol) | Conventional genetics | Microscopy and LC/MS-MS | [129] |
| Lipid peroxidation | Parkinson’s disease | Reduction in lipid peroxidation and the increment of lipid depositions trigger the induction of the ubiquitin-like proteasome and autophagy flux and reduce oxidative stress by upregulation of sod-3 expression. | Cannabidiol | Transgenesis and conventional genetics | Microscopy and immunofluorescence | [122] |
| Lipid peroxidation | Parkinson’s disease | Neuroprotective effect due to the mobilization of fatty acids and triglycerides in excess. | Lipid droplets | Conventional genetics and transgenesis CRISPR | Molecular biology, single nucleotide polymorphism (SNP) mapping, microscopy, and HPLC | [118] |
| Ferroptosis | Parkinson’s disease and ferroptosis-mediated neurodegeneration | Ferrostatin-1 elevated lipid peroxidation, mitigated increased mortality, and reduced GPX4 activity and morphological damage to dopaminergic neurons. | Ferrostatin-1 | Conventional genetics | Microscopy and spectrophotometry | [128] |
| Ferroptosis | Ferroptosis-mediated neurodegeneration | DGLA triggers neurodegeneration upon conversion to dihydroxyeicosadienoic acid through the action of CYP-EH (CYP, cytochrome P450; EH, epoxide hydrolase), representing a new class of lipid metabolites that induce neurodegeneration via ferroptosis. | Dihomo-γ-linolenic acid (DGLA) | Transgenesis, fluorescent protein tagging, and conventional genetics | Microscopy and LC/MS-MS | [125] |
| Ferroptosis | Ferroptosis-mediated neurodegeneration | Ferrostatin-1 provides protection by displacing polyunsaturated fatty acids (PUFAs) from cellular membranes, thus decreasing the buildup of lipid-derived ROS. Furthermore, it notably reduces germ cell death and sterility caused by DGLA. Additionally, oleic acid demonstrates protective effects, which are linked to the nuclear hormone receptor NHR-49/PPAR-α. | Ferrostatin-1, Oleic acid | Conventional genetics and transgenesis | Fluorescence microscopy | [130,131] |
| Ferroptosis | Ferroptosis-mediated neurodegeneration | Lip-1 significantly lowers lipid peroxidation markers, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), reducing age-related ferroptotic cell death in intestinal cells and enhancing healthspan. | Liproxstatin-1 (Lip-1) | Conventional genetics | Spectrophotometry, microscopy, X-ray fluorescence microscopy, and LC-inductively coupled plasma MS | [127] |
| β-oxidation | ALS and Huntington’s disease | Restoration of lipid homeostasis, lipid accumulation, and energy balance through mitochondrial β-oxidation by upregulation of acdh-1 and kat-1 (fatty acid metabolism and β-oxidation). | L. rhamnosus HA-114 | Conventional genetics and transgenesis | Microscopy and RNA-Seq analysis (LC-MS) | [134] |
3. Potential Interventions and Future Perspectives
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paulsen, J.S.; Nance, M.; Kim, J.; Carlozzi, N.; Panegyres, P.; Erwin, C.; Goh, A.; McCusker, E.; Williams, J.K. A Review of Quality of Life after Predictive Testing for and Earlier Identification of Neurodegenerative Diseases. Prog. Neurobiol. 2013, 110, 2–28. [Google Scholar] [CrossRef] [PubMed]
- Nassan, M.; Videnovic, A. Circadian Rhythms in Neurodegenerative Disorders. Nat. Rev. Neurol. 2022, 18, 7–24. [Google Scholar] [CrossRef]
- van Lonkhuizen, P.J.C.; Frank, W.; Heemskerk, A.-W.; van Duijn, E.; de Bot, S.T.; Mühlbäck, A.; Landwehrmeyer, G.B.; Chavannes, N.H.; Meijer, E.; the HEALTHE-RND consortium. Quality of Life, Health-Related Quality of Life, and Associated Factors in Huntington’s Disease: A Systematic Review. J. Neurol. 2023, 270, 2416–2437. [Google Scholar] [CrossRef] [PubMed]
- Leite Silva, A.B.R.; Gonçalves de Oliveira, R.W.; Diógenes, G.P.; de Castro Aguiar, M.F.; Sallem, C.C.; Lima, M.P.P.; de Albuquerque Filho, L.B.; Peixoto de Medeiros, S.D.; Penido de Mendonça, L.L.; de Santiago Filho, P.C.; et al. Premotor, Nonmotor and Motor Symptoms of Parkinson’s Disease: A New Clinical State of the Art. Ageing Res. Rev. 2023, 84, 101834. [Google Scholar] [CrossRef]
- Zhao, N.; Yang, Y.; Zhang, L.; Zhang, Q.; Balbuena, L.; Ungvari, G.S.; Zang, Y.-F.; Xiang, Y.-T. Quality of Life in Parkinson’s Disease: A Systematic Review and Meta-Analysis of Comparative Studies. CNS Neurosci. Ther. 2021, 27, 270–279. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Cermenati, G.; Mitro, N.; Audano, M.; Melcangi, R.C.; Crestani, M.; De Fabiani, E.; Caruso, D. Lipids in the Nervous System: From Biochemistry and Molecular Biology to Patho-Physiology. Biochim. Biophys. Acta 2015, 1851, 51–60. [Google Scholar] [CrossRef] [PubMed]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The Structure of the Nervous System of the Nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Chen, B.L.; Hall, D.H.; Chklovskii, D.B. Wiring Optimization Can Relate Neuronal Structure and Function. Proc. Natl. Acad. Sci. USA 2006, 103, 4723–4728. [Google Scholar] [CrossRef]
- Varshney, L.R.; Chen, B.L.; Paniagua, E.; Hall, D.H.; Chklovskii, D.B. Structural Properties of the Caenorhabditis elegans Neuronal Network. PLoS Comput. Biol. 2011, 7, e1001066. [Google Scholar] [CrossRef]
- Gómez-Escribano, A.P.; Mora-Martínez, C.; Roca, M.; Walker, D.S.; Panadero, J.; Sequedo, M.D.; Saini, R.; Knölker, H.-J.; Blanca, J.; Burguera, J.; et al. Changes in Lipid Metabolism Driven by Steroid Signalling Modulate Proteostasis in C. elegans. EMBO Rep. 2023, 24, e55556. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bahramimehr, F.; Shahhamzehei, N.; Fu, H.; Lin, S.; Wang, H.; Li, C.; Efferth, T.; Hong, C. Anti-Aging Effects of Medicinal Plants and Their Rapid Screening Using the Nematode Caenorhabditis elegans. Phytomedicine 2024, 129, 155665. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The Metabolite α-Ketoglutarate Extends Lifespan by Inhibiting ATP Synthase and TOR. Nature 2014, 510, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Sarkar, A.; Chabattula, S.C.; Verma, P.R.P.; Nazir, A.; Gupta, P.K.; Ruokolainen, J.; Kesari, K.K.; Singh, S.K. Food-Grade Quercetin-Loaded Nanoemulsion Ameliorates Effects Associated with Parkinson’s Disease and Cancer: Studies Employing a Transgenic C. elegans Model and Human Cancer Cell Lines. Antioxidants 2022, 11, 1378. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Goedert, M. Alzheimer’s and Parkinson’s Diseases: The Prion Concept in Relation to Assembled Aβ, Tau, and α-Synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Min, J.; Staropoli, J.F.; Collin, E.; Bi, S.; Feng, X.; Barone, R.; Cao, Y.; O’Malley, L.; Xin, W.; et al. SOD1, ANG, TARDBP and FUS Mutations in Amyotrophic Lateral Sclerosis: A United States Clinical Testing Lab Experience. Amyotroph. Lateral Scler. 2012, 13, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of Human Beta-Amyloid Peptide in Transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef]
- Lakso, M.; Vartiainen, S.; Moilanen, A.-M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic Neuronal Loss and Motor Deficits in Caenorhabditis elegans Overexpressing Human α-Synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Springer, W.; Hoppe, T.; Schmidt, E.; Baumeister, R. A Caenorhabditis elegans Parkin Mutant with Altered Solubility Couples α-Synuclein Aggregation to Proteotoxic Stress. Hum. Mol. Genet. 2005, 14, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Ved, R.; Saha, S.; Westlund, B.; Perier, C.; Burnam, L.; Sluder, A.; Hoener, M.; Rodrigues, C.M.; Alfonso, A.; Steer, C.; et al. Similar Patterns of Mitochondrial Vulnerability and Rescue Induced by Genetic Modification of Alpha-Synuclein, Parkin, and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 2005, 280, 42655–42668. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic Loss and Inclusion Body Formation in Alpha-Synuclein Mice: Implications for Neurodegenerative Disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Lord, A.; Kalimo, H.; Eckman, C.; Zhang, X.-Q.; Lannfelt, L.; Nilsson, L.N.G. The Arctic Alzheimer Mutation Facilitates Early Intraneuronal Abeta Aggregation and Senile Plaque Formation in Transgenic Mice. Neurobiol. Aging 2006, 27, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.; Weil, A.; Hill, K.; Hussain, I.; Richardson, J.C.; Cusdin, F.S.; Chen, Y.H.; Randall, A.D. Transgenic Mice Over-Expressing Human Beta-Amyloid Have Functional Nicotinic Alpha 7 Receptors. Neuroscience 2006, 137, 795–805. [Google Scholar] [CrossRef]
- Magrané, J.; Rosen, K.M.; Smith, R.C.; Walsh, K.; Gouras, G.K.; Querfurth, H.W. Intraneuronal Beta-Amyloid Expression Downregulates the Akt Survival Pathway and Blunts the Stress Response. J. Neurosci. 2005, 25, 10960–10969. [Google Scholar] [CrossRef]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. 2022, 37, 2327–2335. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Sittler, A.; Schweiger, K.; Heiser, V.; Lurz, R.; Hasenbank, R.; Bates, G.P.; Lehrach, H.; Wanker, E.E. Self-Assembly of Polyglutamine-Containing Huntingtin Fragments into Amyloid-like Fibrils: Implications for Huntington’s Disease Pathology. Proc. Natl. Acad. Sci. USA 1999, 96, 4604–4609. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Folstein, S.E. Huntington’s Disease: A Disorder of Families; Johns Hopkins University Press: Baltimore, MD, USA, 1989; p. 251. ISBN 978-0-8018-3860-6. [Google Scholar]
- Heinsen, H.; Strik, M.; Bauer, M.; Luther, K.; Ulmar, G.; Gangnus, D.; Jungkunz, G.; Eisenmenger, W.; Götz, M. Cortical and Striatal Neuron Number in Huntington’s Disease. Acta Neuropathol. 1994, 88, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Huntington’s Disease, 3rd ed.; Bates, G., Harper, P., Jones, L., Bates, G., Harper, P., Jones, L., Eds.; Oxford Monographs on Medical Genetics; Oxford University Press: Oxford, UK, 2002; ISBN 978-0-19-851060-4. [Google Scholar]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; ISBN 978-0-9944381-6-4. [Google Scholar]
- Shin, C.; Lim, Y.; Lim, H.; Ahn, T.-B. Plasma Short-Chain Fatty Acids in Patients With Parkinson’s Disease. Mov. Disord. 2020, 35, 1021–1027. [Google Scholar] [CrossRef]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-Synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef]
- Eliezer, D.; Kutluay, E.; Bussell, R.; Browne, G. Conformational Properties of Alpha-Synuclein in Its Free and Lipid-Associated States. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Ageing as a Primary Risk Factor for Parkinson’s Disease: Evidence from Studies of Non-Human Primates. Nat. Rev. Neurosci. 2011, 12, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sidhu, J.; Lui, F.; Tsao, J.W. Alzheimer Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Moda, F.; Ciullini, A.; Dellarole, I.L.; Lombardo, A.; Campanella, N.; Bufano, G.; Cazzaniga, F.A.; Giaccone, G. Secondary Protein Aggregates in Neurodegenerative Diseases: Almost the Rule Rather than the Exception. Front. Biosci.-Landmark 2023, 28, 255. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like Aggregates Sequester Numerous Metastable Proteins with Essential Cellular Functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic Protein Aggregates Interfere with Nucleocytoplasmic Transport of Protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef]
- Hipp, M.S.; Hartl, F.U. Interplay of Proteostasis Capacity and Protein Aggregation: Implications for Cellular Function and Disease. J. Mol. Biol. 2024, 436, 168615. [Google Scholar] [CrossRef] [PubMed]
- Milakovic, T.; Johnson, G.V.W. Mitochondrial Respiration and ATP Production Are Significantly Impaired in Striatal Cells Expressing Mutant Huntingtin*. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Montiel, T.; Quiroz-Báez, R.; Massieu, L. β-Amyloid Neurotoxicity Is Exacerbated during Glycolysis Inhibition and Mitochondrial Impairment in the Rat Hippocampus in Vivo and in Isolated Nerve Terminals: Implications for Alzheimer’s Disease. Exp. Neurol. 2002, 176, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson Phenotype in Aged PINK1-Deficient Mice Is Accompanied by Progressive Mitochondrial Dysfunction in Absence of Neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Distinct Subcellular Autophagy Impairments in Induced Neurons from Patients with Huntington’s Disease | Brain | Oxford Academic. Available online: https://academic.oup.com/brain/article/145/9/3035/6479694 (accessed on 7 November 2024).
- Nechushtai, L.; Frenkel, D.; Pinkas-Kramarski, R. Autophagy in Parkinson’s Disease. Biomolecules 2023, 13, 1435. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Jia, H.; Kast, R.J.; Steffan, J.S.; Thomas, E.A. Selective Histone Deacetylase (HDAC) Inhibition Imparts Beneficial Effects in Huntington’s Disease Mice: Implications for the Ubiquitin-Proteasomal and Autophagy Systems. Hum. Mol. Genet. 2012, 21, 5280–5293. [Google Scholar] [CrossRef]
- Wilson, D.M.; Cookson, M.R.; Bosch, L.V.D.; Zetterberg, H.; Holtzman, D.M.; Dewachter, I. Hallmarks of Neurodegenerative Diseases. Cell 2023, 186, 693–714. [Google Scholar] [CrossRef]
- Ciechanover, A.; Brundin, P. The Ubiquitin Proteasome System in Neurodegenerative Diseases: Sometimes the Chicken, Sometimes the Egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised Autophagy and Neurodegenerative Diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular Quality Control by the Ubiquitin-Proteasome System and Autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.-H.; Chen, Y.-H.; Huang, T.-Y. Ubiquitin-Mediated Regulation of Autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Coleman, M.; Zhang, L.; Zheng, X.; Yue, Z. Autophagy in Axonal and Dendritic Degeneration. Trends Neurosci. 2013, 36, 418–428. [Google Scholar] [CrossRef]
- Vijayan, V.; Verstreken, P. Autophagy in the Presynaptic Compartment in Health and Disease. J. Cell Biol. 2017, 216, 1895–1906. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef]
- Chadwick, S.R.; Lajoie, P. Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front. Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered Cholesterol and Fatty Acid Metabolism in Huntington Disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-A.; Choi, D.-I.; Choi, J.-Y.; Kim, S.-O.; Cho, K.-A.; Lee, J.-B.; Yun, S.-J.; Lee, S.-C. Methyl-β-Cyclodextrin up-Regulates Collagen I Expression in Chronologically Aged Skin via Its Anti-Caveolin-1 Activity. Oncotarget 2014, 6, 1942–1953. [Google Scholar] [CrossRef]
- Salani, B.; Passalacqua, M.; Maffioli, S.; Briatore, L.; Hamoudane, M.; Contini, P.; Cordera, R.; Maggi, D. IGF-IR Internalizes with Caveolin-1 and PTRF/Cavin in Hacat Cells. PLoS ONE 2010, 5, e14157. [Google Scholar] [CrossRef] [PubMed]
- Moldavski, O.; Amen, T.; Levin-Zaidman, S.; Eisenstein, M.; Rogachev, I.; Brandis, A.; Kaganovich, D.; Schuldiner, M. Lipid Droplets Are Essential for Efficient Clearance of Cytosolic Inclusion Bodies. Dev. Cell 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Caro, A.A.; Cederbaum, A.I. Role of Cytochrome P450 in Phospholipase A2- and Arachidonic Acid-Mediated Cytotoxicity. Free Radic. Biol. Med. 2006, 40, 364–375. [Google Scholar] [CrossRef]
- Stadtman, E.R. Oxidation of Free Amino Acids and Amino Acid Residues in Proteins by Radiolysis and by Metal-Catalyzed Reactions. Annu. Rev. Biochem. 1993, 62, 797–821. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Protein Damage and Degradation by Oxygen Radicals. I. General Aspects. J. Biol. Chem. 1987, 262, 9895–9901. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, M.; Inoue, K.; Nojima, S. Effect of Ferrous Ion and Ascorbate-Induced Lipid Peroxidation on Liposomal Membranes. Biochim. Biophys. Acta 1981, 646, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.N.; Agarwal, S. Liposomes as Membrane Model for Study of Lipid Peroxidation. Free Radic. Biol. Med. 1988, 4, 51–72. [Google Scholar] [CrossRef]
- Elkin, E.R.; Harris, S.M.; Loch-Caruso, R. Trichloroethylene Metabolite S-(1,2-Dichlorovinyl)-l-Cysteine Induces Lipid Peroxidation-Associated Apoptosis via the Intrinsic and Extrinsic Apoptosis Pathways in a First-Trimester Placental Cell Line. Toxicol. Appl. Pharmacol. 2018, 338, 30–42. [Google Scholar] [CrossRef]
- Chaudhary, P.; Sharma, R.; Sharma, A.; Vatsyayan, R.; Yadav, S.; Singhal, S.S.; Rauniyar, N.; Prokai, L.; Awasthi, S.; Awasthi, Y.C. Mechanisms of 4-Hydroxy-2-Nonenal Induced Pro- and Anti-Apoptotic Signaling. Biochemistry 2010, 49, 6263–6275. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS Induced Lipid Peroxidation and Their Role in Ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef]
- Olešová, D.; Dobešová, D.; Majerová, P.; Brumarová, R.; Kvasnička, A.; Kouřil, Š.; Stevens, E.; Hanes, J.; Fialová, Ľ.; Michalicová, A.; et al. Changes in Lipid Metabolism Track with the Progression of Neurofibrillary Pathology in Tauopathies. J. Neuroinflammation 2024, 21, 78. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Alterations in Mitochondrial Membrane Fluidity by Lipid Peroxidation Products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Fruhwirth, G.O.; Hermetter, A. Mediation of Apoptosis by Oxidized Phospholipids. In Lipids in Health and Disease; Quinn, P.J., Wang, X., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 351–367. ISBN 978-1-4020-8831-5. [Google Scholar]
- Borisenko, G.G.; Iverson, S.L.; Ahlberg, S.; Kagan, V.E.; Fadeel, B. Milk Fat Globule Epidermal Growth Factor 8 (MFG-E8) Binds to Oxidized Phosphatidylserine: Implications for Macrophage Clearance of Apoptotic Cells. Cell Death Differ. 2004, 11, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef]
- Matsura, T.; Togawa, A.; Kai, M.; Nishida, T.; Nakada, J.; Ishibe, Y.; Kojo, S.; Yamamoto, Y.; Yamada, K. The Presence of Oxidized Phosphatidylserine on Fas-Mediated Apoptotic Cell Surface. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2005, 1736, 181–188. [Google Scholar] [CrossRef]
- Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Djukic, M.; Quinn, P.J.; Pitt, B.R.; Kagan, V.E. Oxidation of Phosphatidylserine: A Mechanism for Plasma Membrane Phospholipid Scrambling during Apoptosis? Biochem. Biophys. Res. Commun. 2004, 324, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Savill, J.; Fadok, V. Corpse Clearance Defines the Meaning of Cell Death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Vales, A.; Mitulovic, G.; Blumer, M.; Schmid, R.; Witztum, J.L.; Binder, B.R.; Leitinger, N. Oxidized Membrane Vesicles and Blebs from Apoptotic Cells Contain Biologically Active Oxidized Phospholipids That Induce Monocyte-Endothelial Interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrøm, L.; Wiethoff, S.; Houlden, H. Chapter 22—Genetics of Neurodegenerative Diseases: An Overview. In Handbook of Clinical Neurology; Kovacs, G.G., Alafuzoff, I., Eds.; Neuropathology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 309–323. [Google Scholar]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-Avidity Binding to Beta-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Colton, C.A.; Brown, C.M.; Cook, D.; Needham, L.K.; Xu, Q.; Czapiga, M.; Saunders, A.M.; Schmechel, D.E.; Rasheed, K.; Vitek, M.P. APOE and the Regulation of Microglial Nitric Oxide Production: A Link between Genetic Risk and Oxidative Stress. Neurobiol. Aging 2002, 23, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Arbones-Mainar, J.M.; Johnson, L.A.; Torres-Perez, E.; Garcia, A.E.; Perez-Diaz, S.; Raber, J.; Maeda, N. Metabolic Shifts toward Fatty-Acid Usage and Increased Thermogenesis Are Associated with Impaired Adipogenesis in Mice Expressing Human APOE4. Int. J. Obes. 2016, 40, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Di Nottia, M.; Masciullo, M.; Verrigni, D.; Petrillo, S.; Modoni, A.; Rizzo, V.; Di Giuda, D.; Rizza, T.; Niceta, M.; Torraco, A.; et al. DJ-1 Modulates Mitochondrial Response to Oxidative Stress: Clues from a Novel Diagnosis of PARK7. Clin. Genet. 2017, 92, 18–25. [Google Scholar] [CrossRef]
- Skou, L.D.; Johansen, S.K.; Okarmus, J.; Meyer, M. Pathogenesis of DJ-1/PARK7-Mediated Parkinson’s Disease. Cells 2024, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Galano, M.; Ezzat, S.; Papadopoulos, V. SCP2 Variant Is Associated with Alterations in Lipid Metabolism, Brainstem Neurodegeneration, and Testicular Defects. Hum. Genom. 2022, 16, 32. [Google Scholar] [CrossRef]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory Regulators in Parkinson’s Disease: iNOS, Lipocortin-1, and Cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Sairam, K.; Saravanan, K.S.; Banerjee, R.; Mohanakumar, K.P. Non-Steroidal Anti-Inflammatory Drug Sodium Salicylate, but Not Diclofenac or Celecoxib, Protects against 1-Methyl-4-Phenyl Pyridinium-Induced Dopaminergic Neurotoxicity in Rats. Brain Res. 2003, 966, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 Is Instrumental in Parkinson’s Disease Neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Brückner, M.K.; Rozemuller, A.J.M.; Veerhuis, R.; Eikelenboom, P.; Arendt, T. Cyclin D1 and Cyclin E Are Co-Localized with Cyclo-Oxygenase 2 (COX-2) in Pyramidal Neurons in Alzheimer Disease Temporal Cortex. J. Neuropathol. Exp. Neurol. 2002, 61, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.V.; O’Banion, M.K. Downregulation of Neuronal Cyclooxygenase-2 Expression in End Stage Alzheimer’s Disease. Neurobiol. Aging 2001, 22, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Yasojima, K.; Tourtellotte, W.W.; McGeer, E.G.; McGeer, P.L. Marked Increase in Cyclooxygenase-2 in ALS Spinal Cord. Neurology 2001, 57, 952–956. [Google Scholar] [CrossRef] [PubMed]
- Almer, G.; Teismann, P.; Stevic, Z.; Halaschek–Wiener, J.; Deecke, L.; Kostic, V.; Przedborski, S. Increased Levels of the Pro-Inflammatory Prostaglandin PGE2 in CSF from ALS Patients. Neurology 2002, 58, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Shureiqi, I.; Chen, D.; Lee, J.J.; Yang, P.; Newman, R.A.; Brenner, D.E.; Lotan, R.; Fischer, S.M.; Lippman, S.M. 15-LOX-1: A Novel Molecular Target of Nonsteroidal Anti-Inflammatory Drug-Induced Apoptosis in Colorectal Cancer Cells. JNCI J. Natl. Cancer Inst. 2000, 92, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Xiao, Y.; Ma, S.; Liu, Q.; Dang, S.; Jin, M.; Shi, Y.; Wan, B.; Zhang, Y. Inhibition of 12/15-Lipoxygenase by Baicalein Induces Microglia PPARβ/δ: A Potential Therapeutic Role for CNS Autoimmune Disease. Cell Death Dis. 2013, 4, e569. [Google Scholar] [CrossRef]
- Succol, F.; Praticò, D. A Role for 12/15 Lipoxygenase in the Amyloid β Precursor Protein Metabolism. J. Neurochem. 2007, 103, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Li, J.-G.; Giannopoulos, P.F.; Blass, B.E.; Childers, W.; Abou-Gharbia, M.; Praticò, D. Pharmacologic Blockade of 12/15-Lipoxygenase Ameliorates Memory Deficits, Aβ and Tau Neuropathology in the Triple-Transgenic Mice. Mol. Psychiatry 2015, 20, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, A.; Bodhicharla, R.; Winter, J.; Anbalagan, C.; Morgan, K.; Searle, M.; Nazir, A.; Adenle, A.; Fineberg, A.; Brady, D.; et al. A Fluorescence Resonance Energy Transfer Assay For Monitoring α- Synclein Aggregation in a Caenorhabditis elegans Model For Parkinson’s Disease. CNS Neurol. Disord.-Drug Targets 2015, 14, 1054–1068. [Google Scholar] [CrossRef]
- Lee, A.L.; Ung, H.M.; Sands, L.P.; Kikis, E.A. A New Caenorhabditis elegans Model of Human Huntingtin 513 Aggregation and Toxicity in Body Wall Muscles. PLoS ONE 2017, 12, e0173644. [Google Scholar] [CrossRef] [PubMed]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-Mediated Dysfunction and Apoptotic Death of a Caenorhabditis elegans Sensory Neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Satyal, S.H.; Schmidt, E.; Kitagawa, K.; Sondheimer, N.; Lindquist, S.; Kramer, J.M.; Morimoto, R.I. Polyglutamine Aggregates Alter Protein Folding Homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2000, 97, 5750–5755. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The Threshold for Polyglutamine-Expansion Protein Aggregation and Cellular Toxicity Is Dynamic and Influenced by Aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef]
- Zhu, T.-Y.; Li, S.-T.; Liu, D.-D.; Zhang, X.; Zhou, L.; Zhou, R.; Yang, B. Single-Worm Quantitative Proteomics Reveals Aging Heterogeneity in Isogenic Caenorhabditis elegans. Aging Cell 2024, 23, e14055. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.-H.; Wang, T.-Y.; Park, H.; Lomenick, B.; Chou, T.-F.; Sternberg, P.W. Single-Tissue Proteomics in Caenorhabditis elegans Reveals Proteins Resident in Intestinal Lysosome-Related Organelles. Proc. Natl. Acad. Sci. USA 2024, 121, e2322588121. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.J.; Schwager, F.; Pintard, L.; Gotta, M.; Goldstein, B. A Single-Cell Biochemistry Approach Reveals PAR Complex Dynamics during Cell Polarization. Dev. Cell 2017, 42, 416–434.e11. [Google Scholar] [CrossRef] [PubMed]
- Shamim, A.; Mahmood, T.; Ahsan, F.; Kumar, A.; Bagga, P. Lipids: An Insight into the Neurodegenerative Disorders. Clin. Nutr. Exp. 2018, 20, 1–19. [Google Scholar] [CrossRef]
- Muñoz-Juan, A.; Benseny-Cases, N.; Guha, S.; Barba, I.; Caldwell, K.A.; Caldwell, G.A.; Agulló, L.; Yuste, V.J.; Laromaine, A.; Dalfó, E. Caenorhabditis elegans RAC1/Ced-10 Mutants as a New Animal Model to Study Very Early Stages of Parkinson’s Disease. Prog. Neurobiol. 2024, 234, 102572. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liang, J.; Lam, S.M.; Yavuz, A.; Shui, G.; Ding, M.; Huang, X. Neuronal Lipolysis Participates in PUFA-mediated Neural Function and Neurodegeneration. EMBO Rep. 2020, 21, e50214. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Devkota, R.; Kaper, D.; Bodhicharla, R.; Henricsson, M.; Borén, J.; Pilon, M. A Genetic Titration of Membrane Composition in Caenorhabditis elegans Reveals Its Importance for Multiple Cellular and Physiological Traits. Genetics 2021, 219, iyab093. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, F.; Liu, Y.; Wang, N.; Zhao, L.; Zhou, Y.; Yang, H.; Li, H. Neuroprotective Effects of Cannabidiol on Dopaminergic Neurodegeneration and α-Synuclein Accumulation in C. elegans Models of Parkinson’s Disease. NeuroToxicology 2022, 93, 128–139. [Google Scholar] [CrossRef]
- Xiang, Y.; Zhang, J.; Li, H.; Wang, Q.; Xiao, L.; Weng, H.; Zhou, X.; Ma, C.W.; Ma, F.; Hu, M.; et al. Epimedium Polysaccharide Alleviates Polyglutamine-Induced Neurotoxicity in Caenorhabditis elegans by Reducing Oxidative Stress. Rejuvenation Res. 2017, 20, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Modukuri, R.K.; Jadiya, P.; Dodda, R.P.; Kumar, M.; Sridhar, B.; Kumar, V.; Haque, R.; Siddiqi, M.I.; Nazir, A. Benzofuran–Chalcone Hybrids as Potential Multifunctional Agents against Alzheimer’s Disease: Synthesis and in Vivo Studies with Transgenic Caenorhabditis elegans. ChemMedChem 2014, 9, 2671–2684. [Google Scholar] [CrossRef]
- Sarparast, M.; Pourmand, E.; Hinman, J.; Vonarx, D.; Reason, T.; Zhang, F.; Paithankar, S.; Chen, B.; Borhan, B.; Watts, J.L.; et al. Dihydroxy-Metabolites of Dihomo-γ-Linolenic Acid Drive Ferroptosis-Mediated Neurodegeneration. ACS Cent. Sci. 2023, 9, 870–882. [Google Scholar] [CrossRef]
- Bao, C.; Liu, C.; Liu, Q.; Hua, L.; Hu, J.; Li, Z.; Xu, S. Liproxstatin-1 Alleviates LPS/IL-13-Induced Bronchial Epithelial Cell Injury and Neutrophilic Asthma in Mice by Inhibiting Ferroptosis. Int. Immunopharmacol. 2022, 109, 108770. [Google Scholar] [CrossRef]
- Jenkins, N.L.; James, S.A.; Salim, A.; Sumardy, F.; Speed, T.P.; Conrad, M.; Richardson, D.R.; Bush, A.I.; McColl, G. Changes in Ferrous Iron and Glutathione Promote Ferroptosis and Frailty in Aging Caenorhabditis elegans. eLife 2020, 9, e56580. [Google Scholar] [CrossRef] [PubMed]
- Ferreyra, M.R.; Romero, V.L.; Fernandez-Hubeid, L.E.; Gonzales-Moreno, C.; Aschner, M.; Virgolini, M.B. Ferrostatin-1 Mitigates Cellular Damage in a Ferroptosis-like Environment in Caenorhabditis elegans. Toxicol. Sci. 2024, 200, 357–368. [Google Scholar] [CrossRef]
- Schlotterer, A.; Masri, B.; Humpert, M.; Krämer, B.K.; Hammes, H.-P.; Morcos, M. Sulforaphane and Vitamin E Protect From Glucotoxic Neurodegeneration and Lifespan Reduction in C. elegans. Exp. Clin. Endocrinol. Diabetes 2020, 129, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditis elegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Reznik, E.; Santer, M.; Fongheiser, M.A.; Smith, N.; Hirschhorn, T.; Zandkarimi, F.; Soni, R.K.; Dafré, A.L.; Miranda-Vizuete, A.; et al. Ferroptosis Inhibition by Oleic Acid Mitigates Iron-Overload-Induced Injury. Cell Chem. Biol. 2024, 31, 249–264.e7. [Google Scholar] [CrossRef] [PubMed]
- Sanguanphun, T.; Sornkaew, N.; Malaiwong, N.; Chalorak, P.; Jattujan, P.; Niamnont, N.; Sobhon, P.; Meemon, K. Neuroprotective Effects of a Medium Chain Fatty Acid, Decanoic Acid, Isolated from H. Leucospilota against Parkinsonism in C. elegans PD Model. Front. Pharmacol. 2022, 13, 1004568. [Google Scholar] [CrossRef] [PubMed]
- Malaiwong, N.; Chalorak, P.; Jattujan, P.; Manohong, P.; Niamnont, N.; Suphamungmee, W.; Sobhon, P.; Meemon, K. Anti-Parkinson Activity of Bioactive Substances Extracted from Holothuria leucospilota. Biomed. Pharmacother. 2019, 109, 1967–1977. [Google Scholar] [CrossRef] [PubMed]
- Labarre, A.; Guitard, E.; Tossing, G.; Forest, A.; Bareke, E.; Labrecque, M.; Tétreault, M.; Ruiz, M.; Alex Parker, J. Fatty Acids Derived from the Probiotic Lacticaseibacillus Rhamnosus HA-114 Suppress Age-Dependent Neurodegeneration. Commun. Biol. 2022, 5, 1340. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Huang, Z.; Xiao, Y.; Liu, M.; Liu, H.; Yu, Y.; Liang, M.; Luo, N.; Li, K.; et al. Abalone Peptide Increases Stress Resilience and Cost-free Longevity via SKN-1-governed Transcriptional Metabolic Reprogramming in C. elegans. Aging Cell 2023, 23, e14046. [Google Scholar] [CrossRef]
- Li, H.; Shi, R.; Ding, F.; Wang, H.; Han, W.; Ma, F.; Hu, M.; Ma, C.W.; Huang, Z. Astragalus Polysaccharide Suppresses 6-Hydroxydopamine-Induced Neurotoxicity in Caenorhabditis elegans. Oxid. Med. Cell. Longev. 2016, 2016, 4856761. [Google Scholar] [CrossRef] [PubMed]
- Pandey, T.; Sammi, S.R.; Nooreen, Z.; Mishra, A.; Ahmad, A.; Bhatta, R.S.; Pandey, R. Anti-Ageing and Anti-Parkinsonian Effects of Natural Flavonol, Tambulin from Zanthoxyllum aramatum Promotes Longevity in Caenorhabditis elegans. Exp. Gerontol. 2019, 120, 50–61. [Google Scholar] [CrossRef]
- Promtang, S.; Sanguanphun, T.; Chalorak, P.; Pe, L.S.; Niamnont, N.; Sobhon, P.; Meemon, K. 2-Butoxytetrahydrofuran, Isolated from Holothuria Scabra, Attenuates Aggregative and Oxidative Properties of α-Synuclein and Alleviates Its Toxicity in a Transgenic Caenorhabditis elegans Model of Parkinson’s Disease. ACS Chem. Neurosci. 2024, 15, 2182–2197. [Google Scholar] [CrossRef] [PubMed]
- Smita, S.S.; Raj Sammi, S.; Laxman, T.S.; Bhatta, R.S.; Pandey, R. Shatavarin IV Elicits Lifespan Extension and Alleviates Parkinsonism in Caenorhabditis elegans. Free Radic. Res. 2017, 51, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Morton, K.S.; Hartman, J.H.; Heffernan, N.; Ryde, I.T.; Kenny-Ganzert, I.W.; Meng, L.; Sherwood, D.R.; Meyer, J.N. Chronic High-Sugar Diet in Adulthood Protects Caenorhabditis elegans from 6-OHDA-Induced Dopaminergic Neurodegeneration. BMC Biol. 2023, 21, 252. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Houldsworth, A. Role of Oxidative Stress in Neurodegenerative Disorders: A Review of Reactive Oxygen Species and Prevention by Antioxidants. Brain Commun. 2024, 6, fcad356. [Google Scholar] [CrossRef] [PubMed]
- Wilding, A.-S.; Patte-Mensah, C.; Taleb, O.; Brun, S.; Kemmel, V.; Mensah-Nyagan, A.-G. Protective Effect of 4-Phenylbutyrate against Proteolipid Protein Mutation-Induced Endoplasmic Reticulum Stress and Oligodendroglial Cell Death. Neurochem. Int. 2018, 118, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019, 20, 4256. [Google Scholar] [CrossRef]
- Alessenko, A.V.; Albi, E. Exploring Sphingolipid Implications in Neurodegeneration. Front. Neurol. 2020, 11, 437. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.; Karunatilleke, N.C.; Brickenden, A.; Choy, W.-Y.; Duennwald, M.L. Oxidative Stress-Induced Misfolding and Inclusion Formation of Nrf2 and Keap1. Antioxid. Basel Switz. 2022, 11, 243. [Google Scholar] [CrossRef]
- Van Assche, R.; Broeckx, V.; Boonen, K.; Maes, E.; De Haes, W.; Schoofs, L.; Temmerman, L. Integrating -Omics: Systems Biology as Explored Through C. elegans Research. J. Mol. Biol. 2015, 427, 3441–3451. [Google Scholar] [CrossRef]
- Zhang, S.; Li, F.; Zhou, T.; Wang, G.; Li, Z. Caenorhabditis elegans as a Useful Model for Studying Aging Mutations. Front. Endocrinol. 2020, 11, 554994. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wu, Y.; Brown, M.; Link, C.D. Caenorhabditis elegans Model for Initial Screening and Mechanistic Evaluation of Potential New Drugs for Aging and Alzheimer’s Disease. In Methods of Behavior Analysis in Neuroscience; Buccafusco, J.J., Ed.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; ISBN 978-1-4200-5234-3. [Google Scholar]
- Ventura, N.; Rea, S.; Henderson, S.T.; Condo, I.; Johnson, T.E.; Testi, R. Reduced Expression of Frataxin Extends the Lifespan of Caenorhabditis elegans. Aging Cell 2005, 4, 109–112. [Google Scholar] [CrossRef]
- Vázquez-Manrique, R.P.; González-Cabo, P.; Ros, S.; Aziz, H.; Baylis, H.A.; Palau, F. Reduction of Caenorhabditis elegans Frataxin Increases Sensitivity to Oxidative Stress, Reduces Lifespan, and Causes Lethality in a Mitochondrial Complex II Mutant. FASEB J. 2006, 20, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Ahringer, J. Reverse Genetics. WormBook 2006. [Google Scholar] [CrossRef]
- Evans, T. Transformation and Microinjection. WormBook 2006. [Google Scholar] [CrossRef]
- Ferguson, G.D.; Bridge, W.J. The Glutathione System and the Related Thiol Network in Caenorhabditis elegans. Redox Biol. 2019, 24, 101171. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.; Snoek, L.B.; De Bono, M.; Kammenga, J.E. Worms under Stress: C. elegans Stress Response and Its Relevance to Complex Human Disease and Aging. Trends Genet. 2013, 29, 367–374. [Google Scholar] [CrossRef]
- Possik, E.; Pause, A. Measuring Oxidative Stress Resistance of Caenorhabditis elegans in 96-Well Microtiter Plates. J. Vis. Exp. JoVE 2015, 99, e52746. [Google Scholar] [CrossRef]
- Wang, Y.L.; Grooms, N.W.; Jaklitsch, E.L.; Schulting, L.G.; Chung, S.H. High-Throughput Submicron-Resolution Microscopy of Caenorhabditis elegans Populations under Strong Immobilization by Cooling Cultivation Plates. iScience 2023, 26, 105999. [Google Scholar] [CrossRef]
- Pulak, R. Techniques for Analysis, Sorting, and Dispensing of C. elegans on the COPAS Flow-Sorting System. Methods Mol. Biol. Clifton NJ 2006, 351, 275–286. [Google Scholar] [CrossRef]
- Sánchez-Martínez, J.D.; Cifuentes, A.; Valdés, A. Omics Approaches to Investigate the Neuroprotective Capacity of a Citrus Sinensis (Sweet Orange) Extract in a Caenorhabditis elegans Alzheimer’s Model. Food Res. Int. 2023, 172, 113128. [Google Scholar] [CrossRef]
- Scott, A.; Willis, C.R.; Muratani, M.; Higashitani, A.; Etheridge, T.; Szewczyk, N.J.; Deane, C.S. Caenorhabditis elegans in Microgravity: An Omics Perspective. iScience 2023, 26, 107189. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tortajada-Pérez, J.; Carranza, A.d.V.; Trujillo-del Río, C.; Collado-Pérez, M.; Millán, J.M.; García-García, G.; Vázquez-Manrique, R.P. Lipid Oxidation at the Crossroads: Oxidative Stress and Neurodegeneration Explored in Caenorhabditis elegans. Antioxidants 2025, 14, 78. https://doi.org/10.3390/antiox14010078
Tortajada-Pérez J, Carranza AdV, Trujillo-del Río C, Collado-Pérez M, Millán JM, García-García G, Vázquez-Manrique RP. Lipid Oxidation at the Crossroads: Oxidative Stress and Neurodegeneration Explored in Caenorhabditis elegans. Antioxidants. 2025; 14(1):78. https://doi.org/10.3390/antiox14010078
Chicago/Turabian StyleTortajada-Pérez, Julia, Andrea del Valle Carranza, Cristina Trujillo-del Río, Mar Collado-Pérez, José María Millán, Gema García-García, and Rafael Pascual Vázquez-Manrique. 2025. "Lipid Oxidation at the Crossroads: Oxidative Stress and Neurodegeneration Explored in Caenorhabditis elegans" Antioxidants 14, no. 1: 78. https://doi.org/10.3390/antiox14010078
APA StyleTortajada-Pérez, J., Carranza, A. d. V., Trujillo-del Río, C., Collado-Pérez, M., Millán, J. M., García-García, G., & Vázquez-Manrique, R. P. (2025). Lipid Oxidation at the Crossroads: Oxidative Stress and Neurodegeneration Explored in Caenorhabditis elegans. Antioxidants, 14(1), 78. https://doi.org/10.3390/antiox14010078