
Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. SA and Dox Treatment in Cell Culture
2.2. Cell Viability Assay
2.3. Hoechst 33342 Dye Staining
2.4. Measurement of Intracellular ROS
2.5. Analysis of Mitochondrial Membrane Potential (MMP) and Mitochondrial ROS Production
2.6. Western Blot Analysis
2.7. Nuclear Fractionation of H9c2 Cardiomyoblasts
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Transfection of Nrf2 Small Interfering RNA (Nrf2 siRNA)
2.10. Statistical Analysis
3. Results
3.1. SA Prevents Dox-Induced Cytotoxicity in H9c2 Cardiomyoblasts
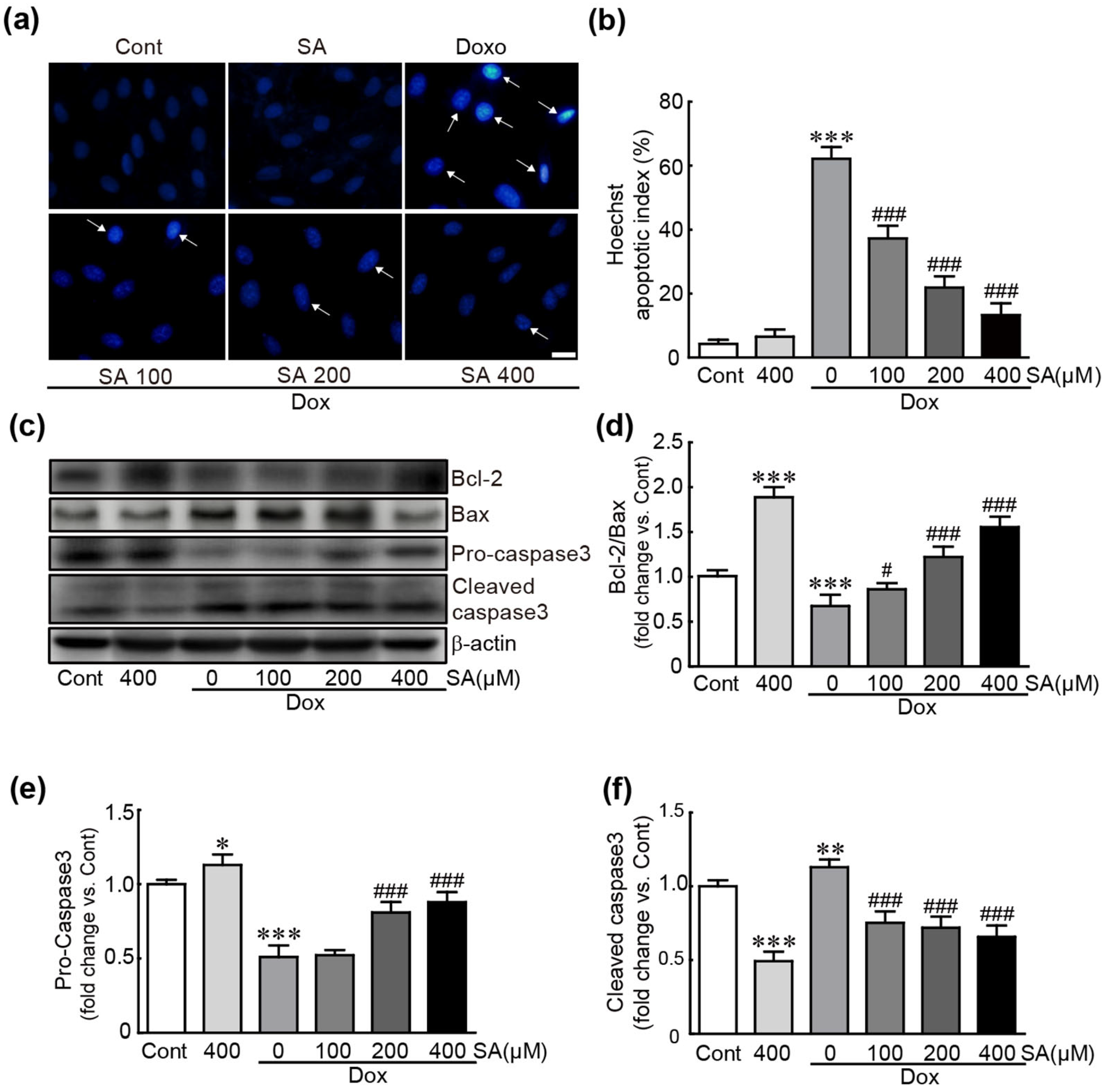
3.2. SA Prevents Dox-Induced Apoptotic Cell Death in H9c2 Cardiomyoblasts
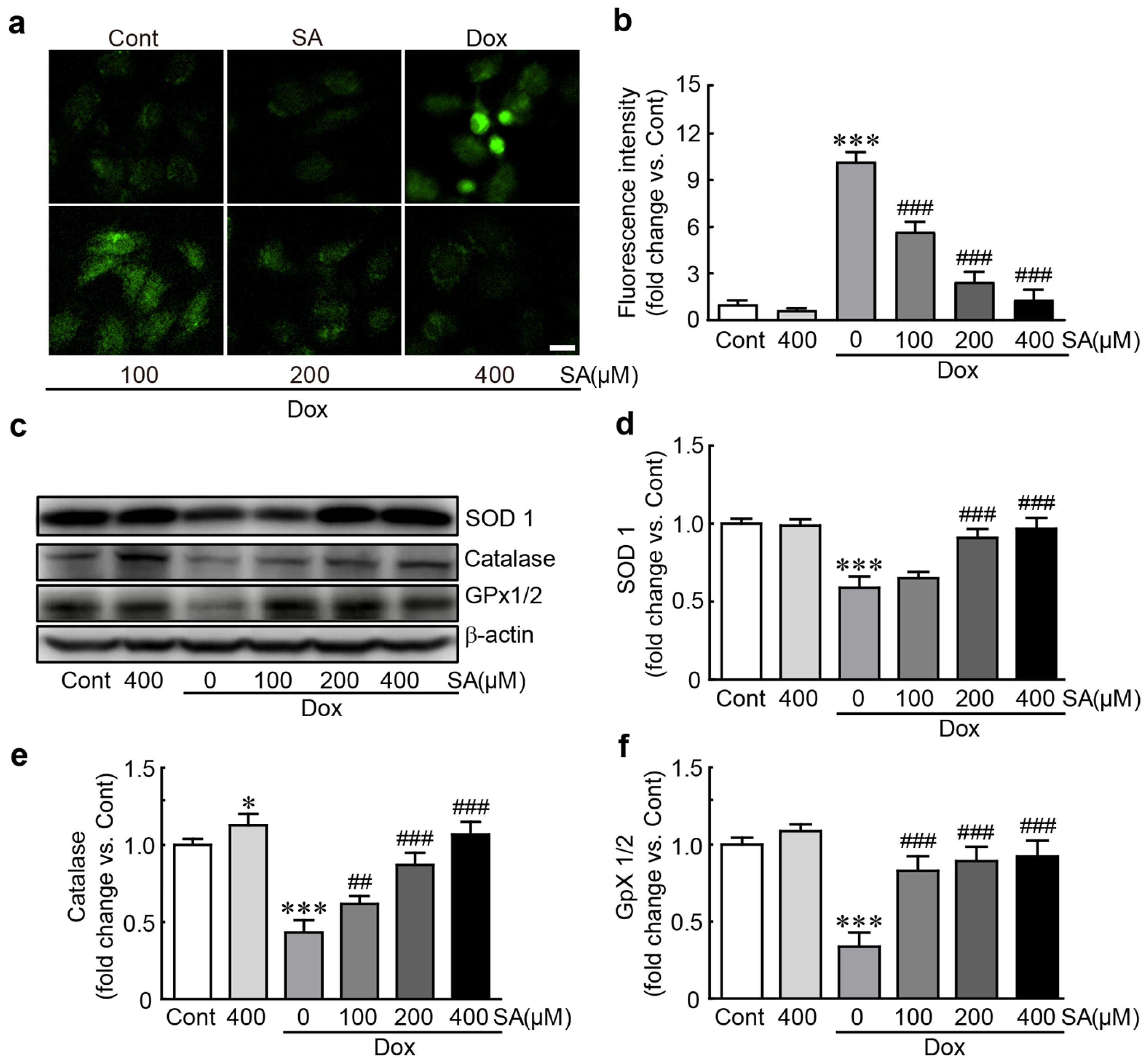
3.3. SA Protects H9c2 Cardiomyoblasts Against Dox-Induced Oxidative Stress
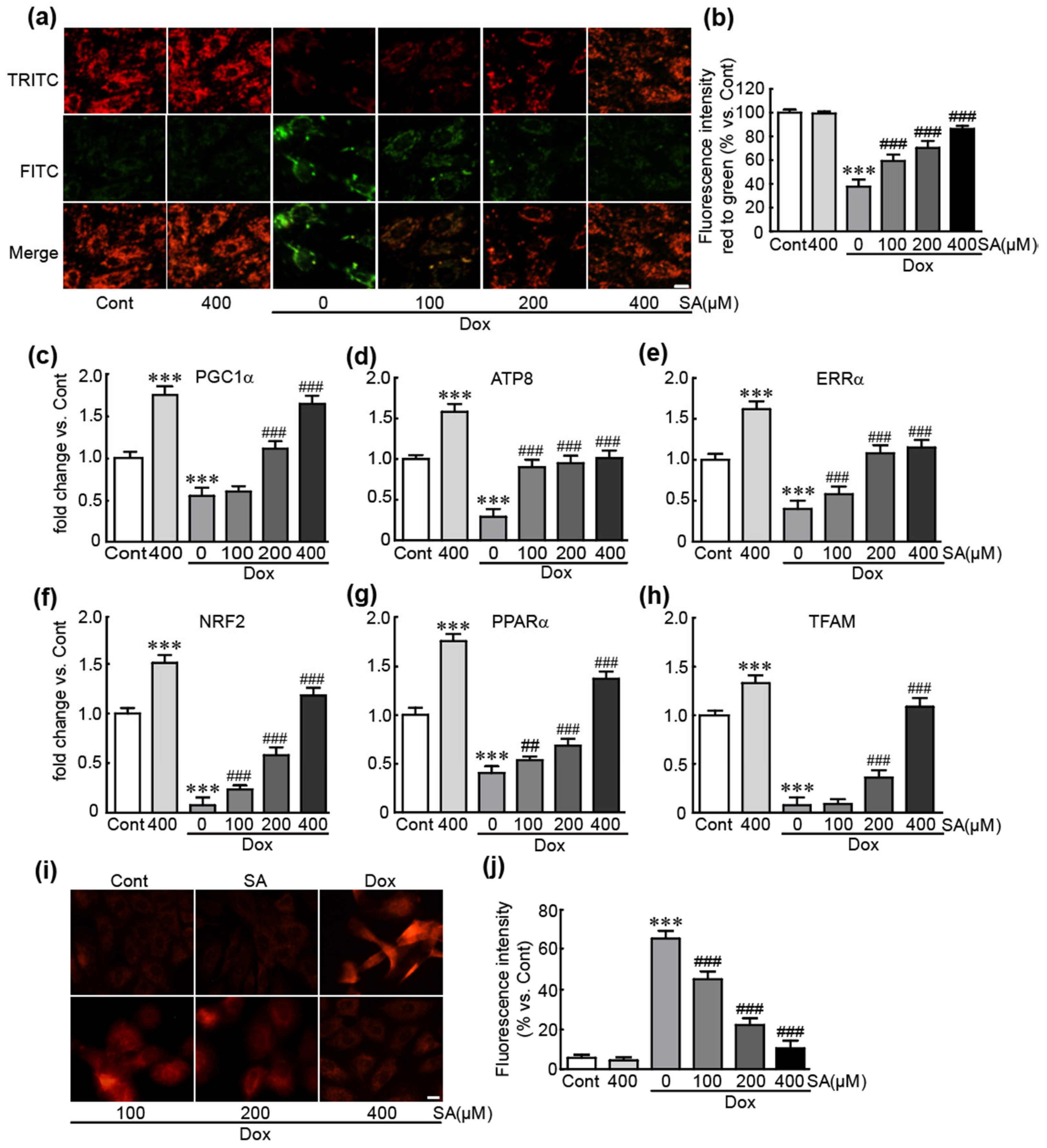
3.4. SA Inhibits Dox-Induced Mitochondrial Oxidative Stress and Mitochondrial Dysfunction in H9c2 Cardiomyoblasts
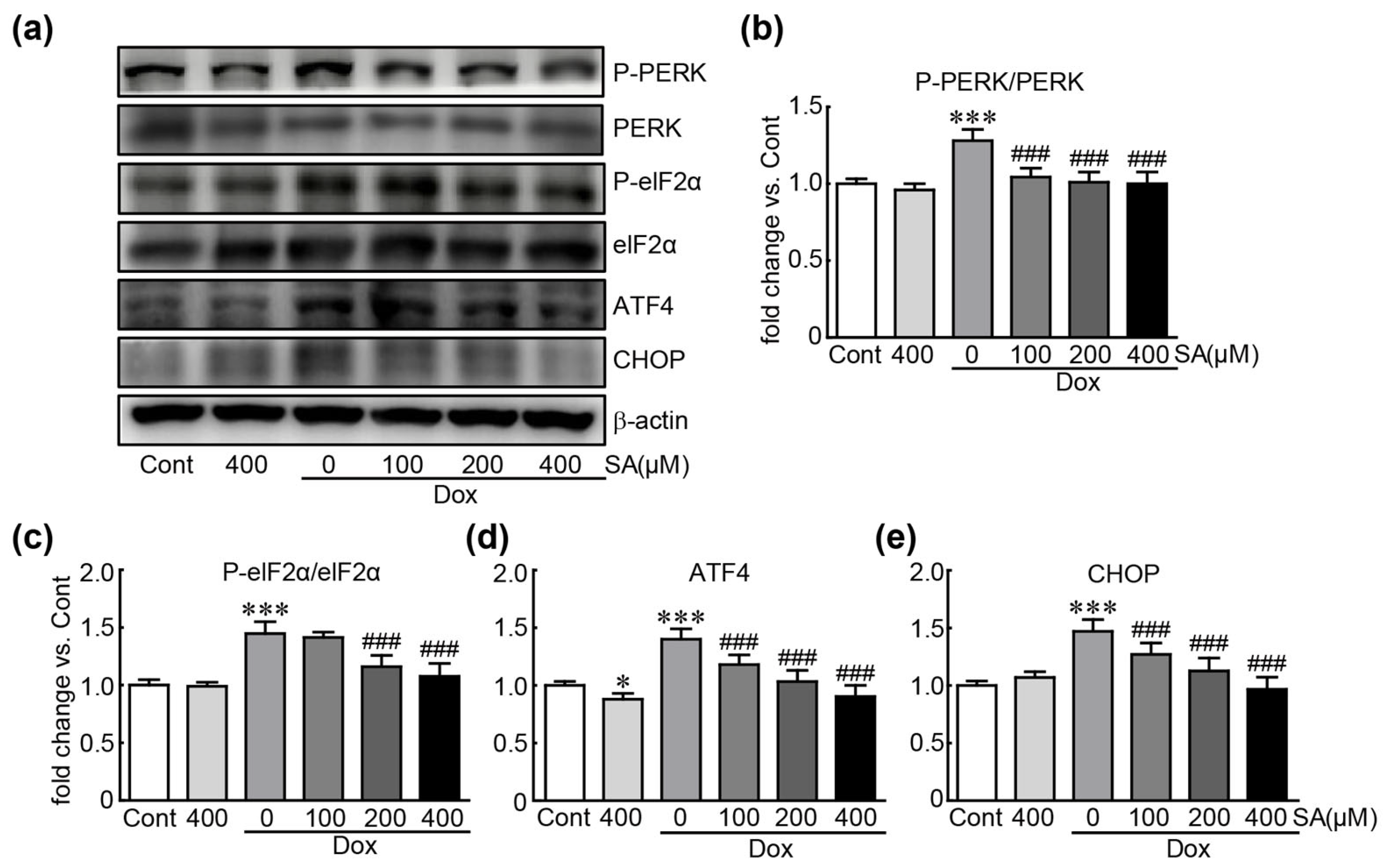
3.5. SA Prevents Dox-Induced ER Stress in H9c2 Cardiomyoblasts
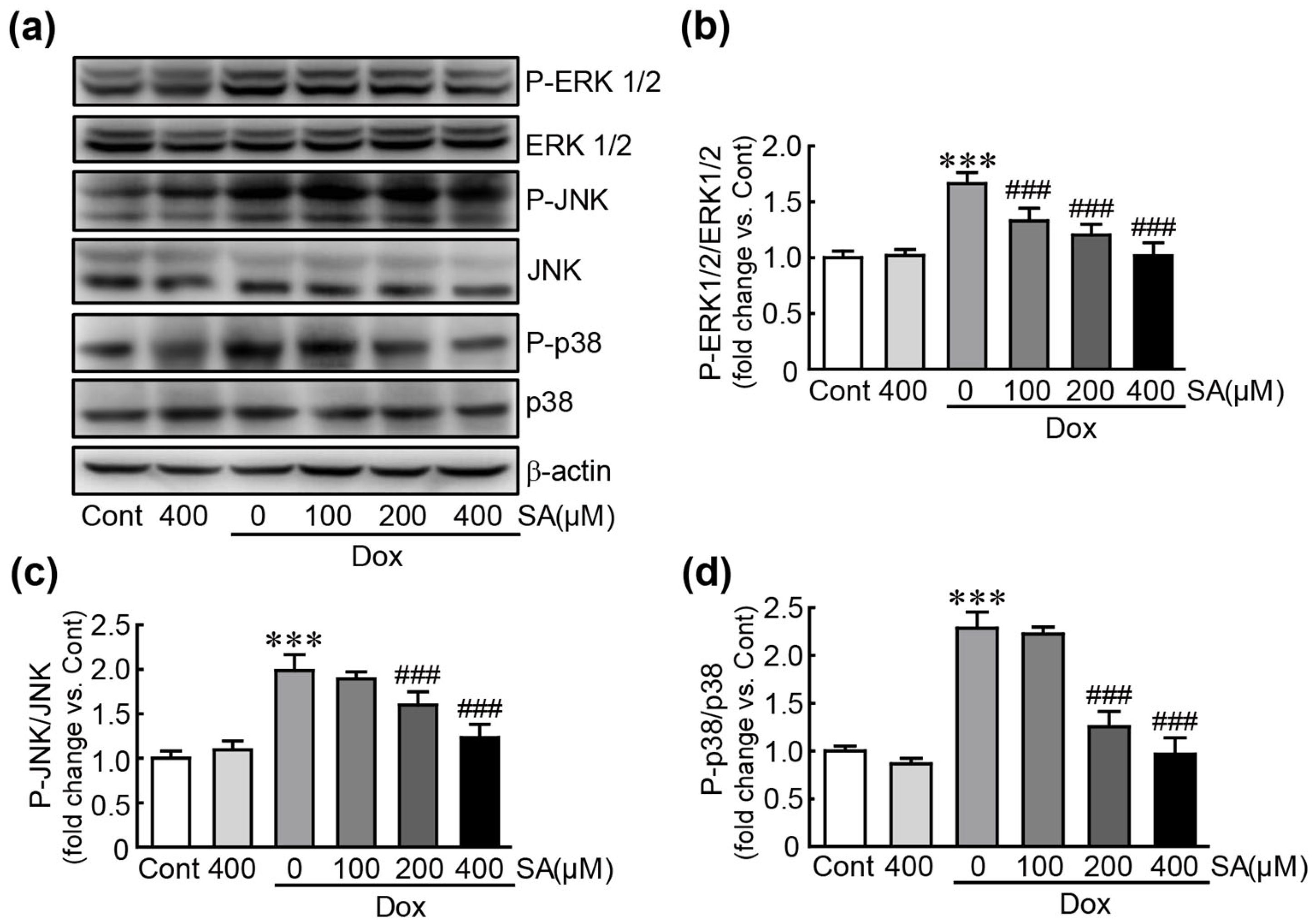
3.6. SA Inhibits the MAPK Signaling Pathway in Dox-Treated H9c2 Cardiomyoblasts
3.7. SA Activates the Nrf2 Signaling Pathway in Dox-Treated H9c2 Cardiomyoblasts
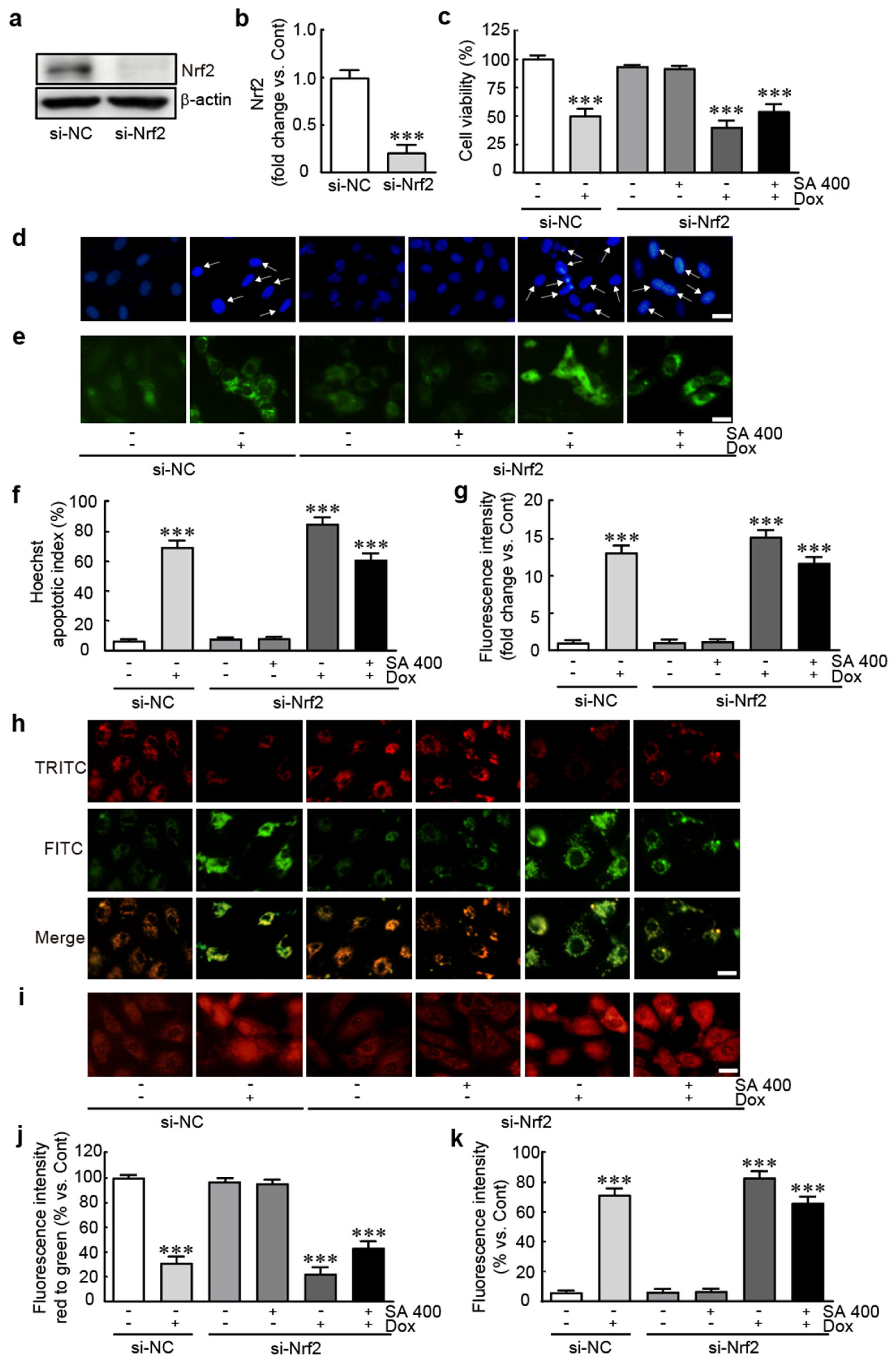
3.8. The Protective Effect of SA Is Abrogated in Nrf2-Silenced Dox-Treated H9c2 Cardiomyoblasts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kciuk, M.; Gielecinska, A.; Mujwar, S.; Kolat, D.; Kaluzinska-Kolat, Z.; Celik, I.; Kontek, R. Doxorubicin-An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef]
- van der Zanden, S.Y.; Qiao, X.H.; Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021, 288, 6095–6111. [Google Scholar] [CrossRef]
- Cheong, A.; McGrath, S.; Robinson, T.; Maliki, R.; Spurling, A.; Lock, P.; Rephaeli, A.; Nudelman, A.; Parker, B.S.; Pepe, S.; et al. A switch in mechanism of action prevents doxorubicin-mediated cardiac damage. Biochem. Pharmacol. 2021, 185, 114410. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Bacchiani, G.; Tedeschi, I.; Meroni, C.A.; Veglia, F.; Civelli, M.; Lamantia, G.; Colombo, N.; Curigliano, G.; et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015, 131, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; Rossi, F.; Piegari, E.; Quaini, F.; Berrino, L.; Urbanek, K.; De Angelis, A. Doxorubicin targets multiple players: A new view of an old problem. Pharmacol. Res. 2018, 127, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Sardao, V.A.; Oliveira, P.J.; Holy, J.; Oliveira, C.R.; Wallace, K.B. Morphological alterations induced by doxorubicin on H9c2 myoblasts: Nuclear, mitochondrial, and cytoskeletal targets. Cell Biol. Toxicol. 2009, 25, 227–243. [Google Scholar] [CrossRef]
- Rushton, M.; Crawley, F.; Sulpher, J.; Johnson, C.; Dent, S. Cardiotoxicity in breast cancer patients: A single center, retrospective review. Prog. Pediatr. Cardiol. 2015, 39, 67–69. [Google Scholar] [CrossRef]
- Dempke, W.C.M.; Zielinski, R.; Winkler, C.; Silberman, S.; Reuther, S.; Priebe, W. Anthracycline-induced cardiotoxicity—Are we about to clear this hurdle? Eur. J. Cancer 2023, 185, 94–104. [Google Scholar] [CrossRef]
- van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst. Rev. 2011, 2011, CD003917. [Google Scholar] [CrossRef]
- Niciforovic, N.; Abramovic, H. Sinapic Acid and Its Derivatives: Natural Sources and Bioactivity. Compr. Rev. Food Sci. Food Saf. 2014, 13, 34–51. [Google Scholar] [CrossRef]
- Pandi, A.; Kalappan, V.M. Pharmacological and therapeutic applications of Sinapic acid-an updated review. Mol. Biol. Rep. 2021, 48, 3733–3745. [Google Scholar] [CrossRef] [PubMed]
- Koriem, K.M.M.; Gad, I.B. Sinapic acid restores blood parameters, serum antioxidants, and liver and kidney functions in obesity. J. Diabetes Metab. Disord. 2022, 21, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Yan, R.; Wang, S.; Li, G.; Kang, X.; Hu, Y.; Lin, M.; Shan, W.; Zhao, Y.; Wang, Z.; et al. Sinapic Acid Reduces Oxidative Stress and Pyroptosis via Inhibition of BRD4 in Alcoholic Liver Disease. Front. Pharmacol. 2021, 12, 668708. [Google Scholar] [CrossRef] [PubMed]
- Chen, C. Sinapic Acid and Its Derivatives as Medicine in Oxidative Stress-Induced Diseases and Aging. Oxid. Med. Cell. Longev. 2016, 2016, 3571614. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef]
- Shi, S.; Chen, Y.; Luo, Z.; Nie, G.; Dai, Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun. Signal. 2023, 21, 61. [Google Scholar] [CrossRef]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Bin Jardan, Y.A.; Ansari, M.A.; Raish, M.; Alkharfy, K.M.; Ahad, A.; Al-Jenoobi, F.I.; Haq, N.; Khan, M.R.; Ahmad, A. Sinapic Acid Ameliorates Oxidative Stress, Inflammation, and Apoptosis in Acute Doxorubicin-Induced Cardiotoxicity via the NF-kappaB-Mediated Pathway. Biomed. Res. Int. 2020, 2020, 3921796. [Google Scholar] [CrossRef]
- Raish, M.; Ahmad, A.; Bin Jardan, Y.A.; Shahid, M.; Alkharfy, K.M.; Ahad, A.; Ansari, M.A.; Abdelrahman, I.A.; Al-Jenoobi, F.I. Sinapic acid ameliorates cardiac dysfunction and cardiomyopathy by modulating NF-kappaB and Nrf2/HO-1 signaling pathways in streptozocin induced diabetic rats. Biomed. Pharmacother. 2022, 145, 112412. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef]
- Sritharan, S.; Sivalingam, N. A comprehensive review on time-tested anticancer drug doxorubicin. Life Sci. 2021, 278, 119527. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Lin, X.; Wu, G.; Wang, S.; Huang, J. Bibliometric and visual analysis of doxorubicin-induced cardiotoxicity. Front. Pharmacol. 2023, 14, 1255158. [Google Scholar] [CrossRef] [PubMed]
- Meesa, M.; Yellu, N.R. Impact of Sinapic acid on Ertugliflozin Pharmacokinetics and Pharmacodynamics in Type-2 Diabetic Rats. J. Young Pharm. 2023, 15, 485–490. [Google Scholar] [CrossRef]
- Dwivedi, P.S.R.; Shastry, C.S. System biology mediated assessment of molecular mechanism for sinapic acid against breast cancer: Via network pharmacology and molecular dynamic simulation. Sci. Rep. 2023, 13, 21982. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef]
- Zhu, C.; Bai, Y.; Qiu, J.; Chen, G.; Guo, X.; Xu, R. CYP2J2-derived epoxyeicosatrienoic acids protect against doxorubicin-induced cardiotoxicity by reducing oxidative stress and apoptosis via activation of the AMPK pathway. Heliyon 2024, 10, e23526. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Li, W.; Wang, X.; Liu, T.; Zhang, Q.; Cao, J.; Jiang, Y.; Sun, Q.; Li, C.; Wang, W.; Wang, Y. Harpagoside Protects Against Doxorubicin-Induced Cardiotoxicity via P53-Parkin-Mediated Mitophagy. Front. Cell Dev. Biol. 2022, 10, 813370. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N.; Fyhrquist, F.Y.; Saijonmaa, O.J.; Fuster, V.; Kovacic, J.C. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 196–211. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol. 2013, 61, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Fa, H.; Xiao, D.; Chang, W.; Ding, L.; Yang, L.; Wang, Y.; Wang, M.; Wang, J. MicroRNA-194-5p Attenuates Doxorubicin-Induced Cardiomyocyte Apoptosis and Endoplasmic Reticulum Stress by Targeting P21-Activated Kinase 2. Front. Cardiovasc. Med. 2022, 9, 815916. [Google Scholar] [CrossRef]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.R.; Chae, H.J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef]
- Lou, H.; Danelisen, I.; Singal, P.K. Involvement of mitogen-activated protein kinases in adriamycin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1925–H1930. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.M.; Xu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-kappaB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sun, X.; Wang, Z.; Chen, M.; He, Y.; Zhang, H.; Han, D.; Zheng, L. Resveratrol protects against doxorubicin-induced cardiotoxicity by attenuating ferroptosis through modulating the MAPK signaling pathway. Toxicol. Appl. Pharmacol. 2024, 482, 116794. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Byer, S.H.; Holder, R.; Wu, L.; Burkey, K.; Shah, Z. Wnt10b protects cardiomyocytes against doxorubicin-induced cell death via MAPK modulation. PLoS ONE 2023, 18, e0277747. [Google Scholar] [CrossRef]
- Ngo, V.; Duennwald, M.L. Nrf2 and Oxidative Stress: A General Overview of Mechanisms and Implications in Human Disease. Antioxidants 2022, 11, 2345. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Chen, Q.M. Nrf2 for cardiac protection: Pharmacological options against oxidative stress. Trends Pharmacol. Sci. 2021, 42, 729–744. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tungalag, T.; Kang, H.-S.; Yang, D.K. Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway. Antioxidants 2025, 14, 337. https://doi.org/10.3390/antiox14030337
Tungalag T, Kang H-S, Yang DK. Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway. Antioxidants. 2025; 14(3):337. https://doi.org/10.3390/antiox14030337
Chicago/Turabian StyleTungalag, Tsendsuren, Hyung-Sub Kang, and Dong Kwon Yang. 2025. "Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway" Antioxidants 14, no. 3: 337. https://doi.org/10.3390/antiox14030337
APA StyleTungalag, T., Kang, H.-S., & Yang, D. K. (2025). Sinapic Acid Ameliorates Doxorubicin-Induced Cardiotoxicity in H9c2 Cardiomyoblasts by Inhibiting Oxidative Stress Through Activation of the Nrf2 Signaling Pathway. Antioxidants, 14(3), 337. https://doi.org/10.3390/antiox14030337