

Exploring the Mechanism of β-Cyclodextrin-Encased Phenolic Acids Functionalized with TPP for Antioxidant Activity and Targeting

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Compound 1a
2.3. General Method for TPP Antioxidant Synthesis (2a–c)
2.4. Conjugation of TPP Antioxidant Within β-CD Matrix (3a–c)
2.5. Determination of Entrapment Efficiency
2.6. ABTS and DPPH Scavenging Assays
2.7. Cell Viability
2.8. Quantum Chemical Calculations
2.9. Free Energy Calculations of TPP Conjugates Across Membranes
3. Results and Discussion
3.1. Chemistry
3.2. Entrapment Efficiency and Antioxidant Activity
3.3. Cell Viability Studies
3.4. Molecular Reactivity Analysis
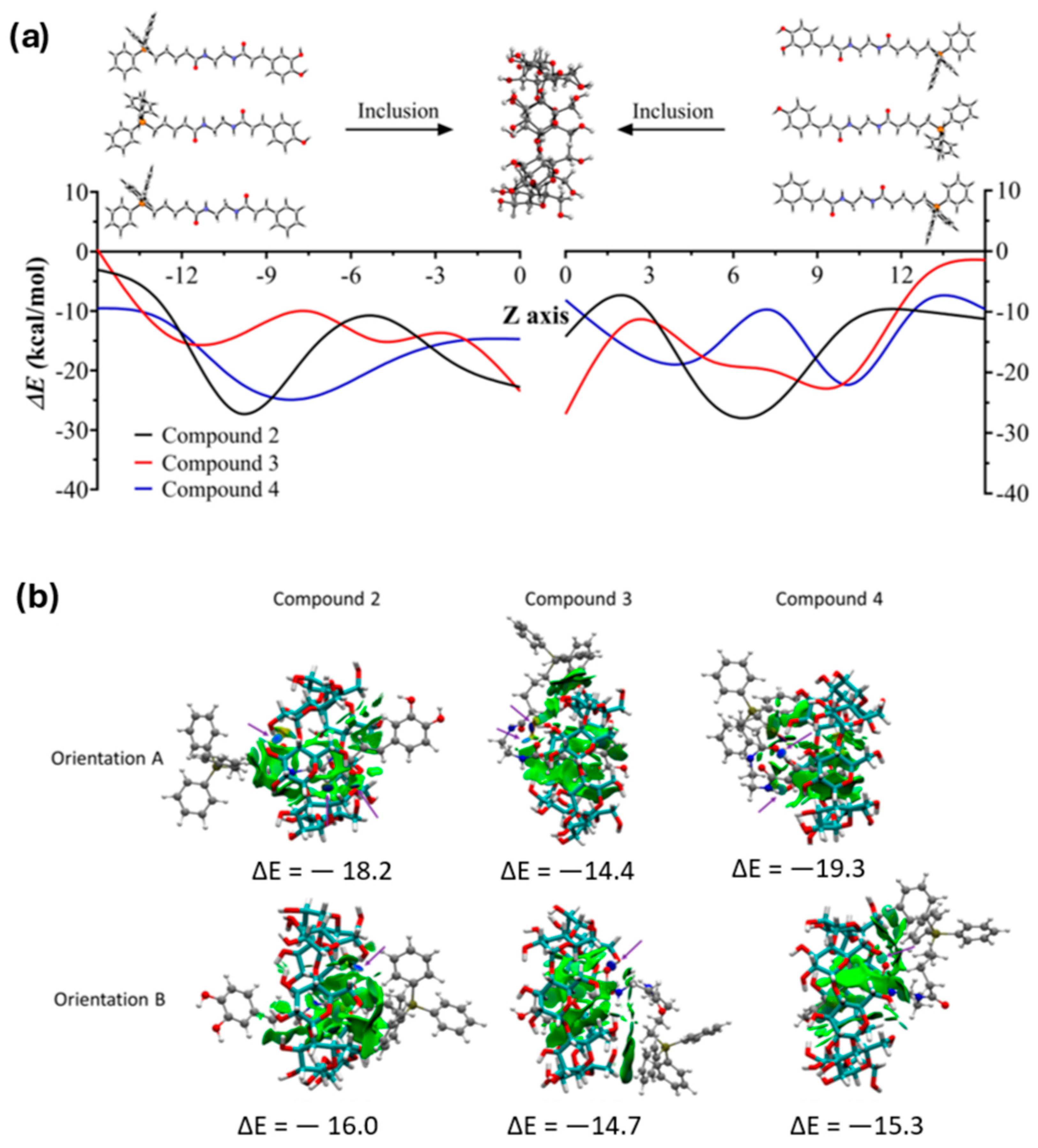
3.5. Complexation Energies and Kinetic Stability
3.6. Interaction of TPP Conjugates with Mitochondria Model Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rudrapal, M.; Khairnar, S.J.; Khan, J.; Bin Dukhyil, A.; Ansari, M.A.; Alomary, M.N.; Alshabrmi, F.M.; Palai, S.; Deb, P.K.; Devi, R. Dietary Polyphenols and Their Role in Oxidative Stress-Induced Human Diseases: Insights Into Protective Effects, Antioxidant Potentials and Mechanism(s) of Action. Front. Pharmacol. 2022, 13, 806470. [Google Scholar] [CrossRef]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Feng, C.; Yang, M.; Lan, M.; Liu, C.; Zhang, Y.; Huang, B.; Liu, H.; Zhou, Y. ROS: Crucial Intermediators in the Pathogenesis of Intervertebral Disc Degeneration. Oxidative Med. Cell. Longev. 2017, 2017, 5601593. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- de Sa Junior, P.L.; Câmara, D.A.D.; Porcacchia, A.S.; Fonseca, P.M.M.; Jorge, S.D.; Araldi, R.P.; Ferreira, A.K. The Roles of ROS in Cancer Heterogeneity and Therapy. Oxid. Med. Cell. Longev. 2017, 2017, 2467940. [Google Scholar] [CrossRef] [PubMed]
- Stromsnes, K.; Lagzdina, R.; Olaso-Gonzalez, G.; Gimeno-Mallench, L.; Gambini, J. Pharmacological properties of polyphenols: Bioavailability, mechanisms of action, and biological effects in in vitro studies, animal models, and humans. Biomedicines 2021, 9, 1074. [Google Scholar] [CrossRef] [PubMed]
- El Oirdi, M. Harnessing the Power of Polyphenols: A New Frontier in Disease Prevention and Therapy. Pharmaceuticals 2024, 17, 692. [Google Scholar] [CrossRef]
- Antony, A.; Farid, M. Effect of Temperatures on Polyphenols during Extraction. Appl. Sci. 2022, 12, 2107. [Google Scholar] [CrossRef]
- Martínez-Cuazitl, A.; Gómez-García, M.d.C.; Pérez-Mora, S.; Rojas-López, M.; Delgado-Macuil, R.J.; Ocampo-López, J.; Vázquez-Zapién, G.J.; Mata-Miranda, M.M.; Pérez-Ishiwara, D.G. Polyphenolic Compounds Nanostructurated with Gold Nanoparticles Enhance Wound Repair. Int. J. Mol. Sci. 2023, 24, 17138. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, K.; Liu, D.; Yang, Y.; Xi, H.; Xie, W.; Diao, K.; Rao, Z.; Wang, D.; Yang, W. Polyphenol-based polymer nanoparticles for inhibiting amyloid protein aggregation: Recent advances and perspectives. Front. Nutr. 2024, 11, 1408620. [Google Scholar] [CrossRef]
- Rosales, T.K.O.; Fabi, J.P. Valorization of polyphenolic compounds from food industry by-products for application in polysaccharide-based nanoparticles. Front. Nutr. 2023, 10, 1144677. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhang, T.; Chai, X.; Duan, X.; He, D.; Yu, H.; Liu, X.; Tao, Z. Encapsulation Efficiency and Functional Stability of Cinnamon Essential Oil in Modified β-cyclodextrins: In Vitro and In Silico Evidence. Foods 2023, 12, 45. [Google Scholar] [CrossRef]
- Li, J.; Xu, F.; Dai, Y.; Zhang, J.; Shi, Y.; Lai, D.; Sriboonvorakul, N.; Hu, J. A Review of Cyclodextrin Encapsulation and Intelligent Response for the Release of Curcumin. Polymers 2022, 14, 5421. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1–31. [Google Scholar] [CrossRef]
- Junaković, E.P.; Šandor, K.; Terzić, S.; Vujnović, A.; Andrišić, M.; Benić, M.; Fajdić, D.; Sinković, S.; Pehnec, M.; Žarković, I. Influence of Encapsulation of Propolis Extract with 2-Hydroxypropyl-β-cyclodextrin (HP-β-CD) on Polyphenolic Contents during In Vitro Simulation of Digestion. Appl. Sci. 2023, 13, 9357. [Google Scholar] [CrossRef]
- He, Z.-X.; Wang, Z.-H.; Zhang, H.-H.; Pan, X.; Su, W.-R.; Liang, D.; Wu, C.-B. Doxycycline and hydroxypropyl-β-cyclodextrin complex in poloxamer thermal sensitive hydrogel for ophthalmic delivery. Acta Pharm. Sin. B 2011, 1, 254–260. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, X.; Cui, Y.; Huang, Y.; Huang, Z.; Wang, G.; Liang, R.; Pan, X.; Tao, L.; Wu, C. Hydroxypropyl-β-cyclodextrin as anti-hygroscopicity agent inamorphous lactose carriers for dry powder inhalers. Powder Technol. 2019, 358, 29–38. [Google Scholar] [CrossRef]
- Kulkarni, C.A.; Fink, B.D.; Gibbs, B.E.; Chheda, P.R.; Wu, M.; Sivitz, W.I.; Kerns, R.J. A Novel Triphenylphosphonium Carrier to Target Mitochondria without Uncoupling Oxidative Phosphorylation. J. Med. Chem. 2021, 64, 662–676. [Google Scholar] [CrossRef]
- Yu, H.; Li, J.-M.; Deng, K.; Zhou, W.; Wang, C.-X.; Wang, Q.; Li, K.-H.; Zhao, H.-Y.; Huang, S.-W. Tumor acidity activated triphenylphosphonium-based mitochondrial targeting nanocarriers for overcoming drug resistance of cancer therapy. Theranostics 2019, 9, 7033–7050. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, Y.; Zhang, P.; Zhang, H. Computational Insights into Cyclodextrin Inclusion Complexes with the Organophosphorus Flame Retardant DOPO. Molecules 2024, 29, 2244. [Google Scholar] [CrossRef]
- Bezzina, B.; Djémil, R.; Khatmi, D.E.; Humbel, S.; Carissan, Y. Computational insights about the dynamic behavior for the inclusion process of deprotonated and neutral aspirin in β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 115–127. [Google Scholar] [CrossRef]
- Simsek, T.; Simsek, S.; Mayer, C.; Rasulev, B. Experimental and Computational Study on the Inclusion Complexes of β-Cyclodextrin with Selected Food Phenolic Compounds. Chemrxiv 2019. [Google Scholar] [CrossRef]
- Tyagi, V.; Vasquez-Montes, V.; Freites, J.A.; Kyrychenko, A.; Tobias, D.J.; Ladokhin, A.S. Effects of cardiolipin on the conformational dynamics of membrane-anchored bcl-xl. Int. J. Mol. Sci. 2021, 22, 9388. [Google Scholar] [CrossRef]
- Liu, E.Y.; Jung, S.; Yi, H. Improved Protein Conjugation with Uniform, Macroporous Poly(acrylamide-co-acrylic acid) Hydrogel Microspheres via EDC/NHS Chemistry. Langmuir 2016, 32, 11043–11054. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Suhagia, B.; Shah, S.; Rathod, I.; Parmar, V. Preparation and characterization of etoricoxib-β-cyclodextrin complexes prepared by the kneading method. Acta Pharm. 2007, 57, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Nenadis, N.; Wang, L.F.; Tsimidou, M.; Zhang, H.Y. Estimation of scavenging activity of phenolic compounds using the ABTS•+ assay. J. Agric. Food Chem. 2004, 52, 4669–4674. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant determinations by the use of a stable free radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA, v16.1.0.15350. Discovery Studio Modeling Environment. Dassault Systèmes: San Diego, CA, USA, 2017.
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision B.01; Gaussian, Inc.: Wallingford, UK, 2016. [Google Scholar]
- Yañez, O.; Osorio, M.I.; Areche, C.; Vasquez-Espinal, A.; Bravo, J.; Sandoval-Aldana, A.; Pérez-Donoso, J.M.; González-Nilo, F.; Matos, M.J.; Osorio, E.; et al. Theobroma cacao L. compounds: Theoretical study and molecular modeling as inhibitors of main SARS-CoV-2 protease. Biomed. Pharmacother. 2021, 140, 111764. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Espinal, A.; Yañez, O.; Osorio, E.; Areche, C.; García-Beltrán, O.; Ruiz, L.M.; Cassels, B.K.; Tiznado, W. Structure–antioxidant activity relationships in boldine and glaucine: A DFT study. New J. Chem. 2021, 45, 590–596. [Google Scholar] [CrossRef]
- Vásquez-Espinal, A.; Yañez, O.; Osorio, E.; Areche, C.; García-Beltrán, O.; Ruiz, L.M.; Cassels, B.K.; Tiznado, W. Theoretical Study of the Antioxidant Activity of Quercetin Oxidation Products. Front. Chem. 2019, 7, 818. [Google Scholar] [CrossRef]
- Osorio, E.; Pérez, E.G.; Areche, C.; Ruiz, L.M.; Cassels, B.K.; Flórez, E.; Tiznado, W. Why is quercetin a better antioxidant than taxifolin? Theoretical study of mechanisms involving activated forms. J. Mol. Model. 2013, 19, 2165–2172. [Google Scholar] [CrossRef]
- Pino-Rios, R.; Yañez, O.; Inostroza, D.; Ruiz, L.; Cardenas, C.; Fuentealba, P.; Tiznado, W. Proposal of a simple and effective local reactivity descriptor through a topological analysis of an orbital-weighted fukui function. J. Comput. Chem. 2017, 38, 481–488. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness; Wiley: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Lewars, E.G. Computational Chemistry-Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: Dordrecht, The Netherlands; New York, NY, USA, 2011. [Google Scholar] [CrossRef]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley & Sons: New York, NY, USA, 2002. [Google Scholar] [CrossRef]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Fukui, K. Role of frontier orbitals in chemical reactions. Science 1982, 218, 747–754. [Google Scholar] [CrossRef]
- Fukui, K. The Role of Frontier Orbitals in Chemical Reactions (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1982, 21, 801–809. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Ayers, P.W.; Levy, M. Perspective on Density functional approach to the frontier-electron theory of chemical reactivity. In Theoretical Chemistry Accounts: New Century Issue; Cramer, C.J., Truhlar, D.G., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 353–360. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef]
- Guendouzi, A.; Mekelleche, S.M.; Brahim, H.; Litim, K. Quantitative conformational stability host-guest complex of Carvacrol and Thymol with β-cyclodextrin: A theoretical investigation. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 143–155. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical Sampling Distributions in Monte Carlo Free-Energy Estimation: Umbrella Sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenbergl, J.M. The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E., III; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Yu, Y.; Krämer, A.; Venable, R.M.; Simmonett, A.C.; MacKerell, A.D., Jr.; Klauda, J.B.; Pastor, R.W.; Brooks, B.R. Semi-automated Optimization of the CHARMM36 Lipid Force Field to Include Explicit Treatment of Long-Range Dispersion. J. Chem. Theory Comput. 2021, 17, 1562–1580. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM general force field (CGENFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Mayne, C.G.; Saam, J.; Schulten, K.; Tajkhorshid, E.; Gumbart, J.C. Rapid parameterization of small molecules using the force field toolkit. J. Comput. Chem. 2013, 34, 2757–2770. [Google Scholar] [CrossRef]
- Hackett, J.C. Chemical Reactivity Theory: A Density Functional View. J. Am. Chem. Soc. 2010, 132, 7558. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef]
- López, P.; Méndez, F. Fukui Function as a Descriptor of the Imidazolium Protonated Cation Resonance Hybrid Structure. Org. Lett. 2004, 6, 1781–1783. [Google Scholar] [CrossRef]
- Litim, A.; Belhocine, Y.; Benlecheb, T.; Ghoniem, M.G.; Kabouche, Z.; Ali, F.A.M.; Abdulkhair, B.Y.; Seydou, M.; Rahali, S. DFT-D4 Insight into the Inclusion of Amphetamine and Methamphetamine in Cucurbit[7]uril: Energetic, Structural and Biosensing Properties. Molecules 2021, 26, 7479. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, D.; Shen, Z.; Li, S.; Li, H. Multi-Body Interactions in Molecular Docking Program Devised with Key Water Molecules in Protein Binding Sites. Molecules 2018, 23, 2321. [Google Scholar] [CrossRef] [PubMed]
- Spicher, S.; Grimme, S. Efficient Computation of Free Energy Contributions for Association Reactions of Large Molecules. J. Phys. Chem. Lett. 2020, 11, 6606–6611. [Google Scholar] [CrossRef]
- Menzel, J.P.; Kloppenburg, M.; Belić, J.; de Groot, H.J.M.; Visscher, L.; Buda, F. Efficient workflow for the investigation of the catalytic cycle of water oxidation catalysts: Combining GFN-xTB and density functional theory. J. Comput. Chem. 2021, 42, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Spicher, S.; Grimme, S. Single-Point Hessian Calculations for Improved Vibrational Frequencies and Rigid-Rotor-Harmonic-Oscillator Thermodynamics. J. Chem. Theory Comput. 2021, 17, 1701–1714. [Google Scholar] [CrossRef]
- Arnarez, C.; Marrink, S.J.; Periole, X. Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chem. Sci. 2016, 7, 4435–4443. [Google Scholar] [CrossRef]
- Boyd, K.J.; Alder, N.N.; May, E.R. Molecular Dynamics Analysis of Cardiolipin and Monolysocardiolipin on Bilayer Properties. Biophys. J. 2018, 114, 2116–2127. [Google Scholar] [CrossRef]
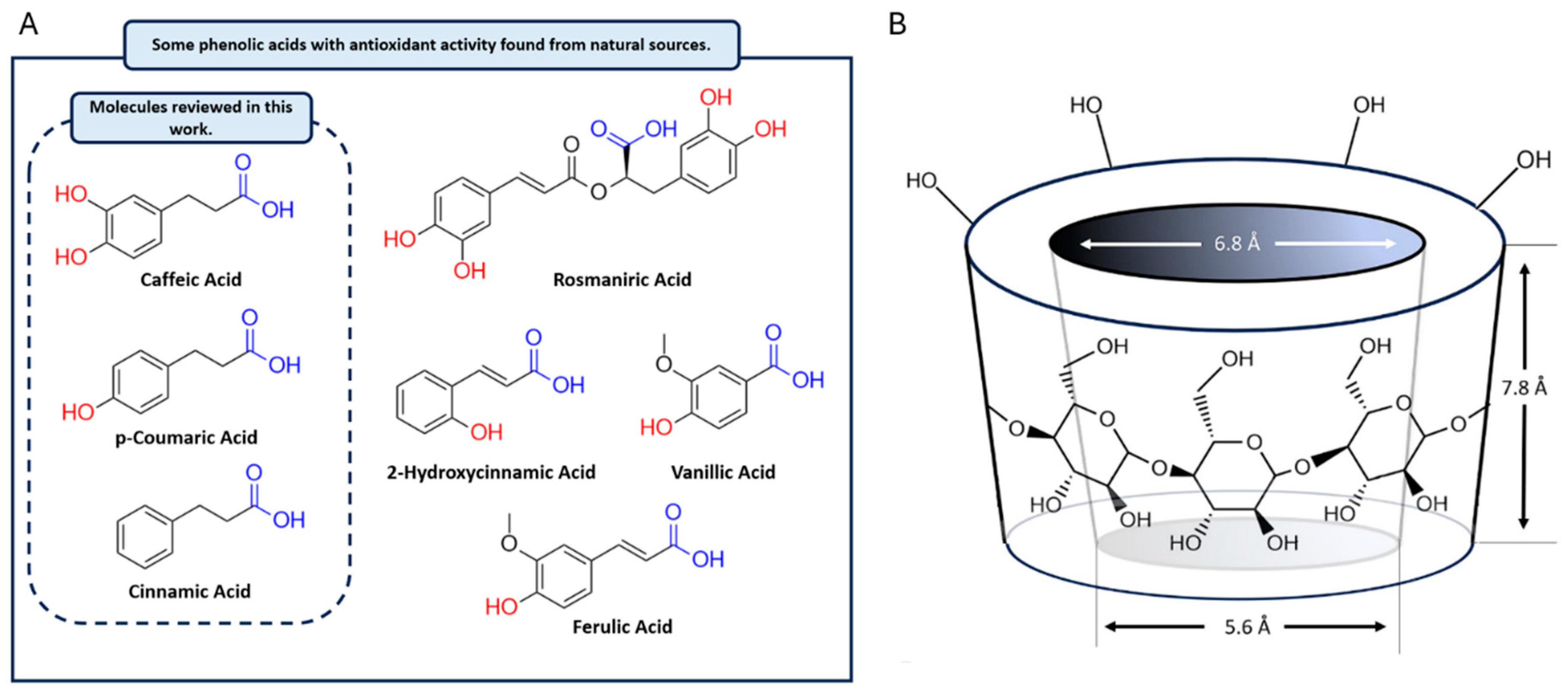
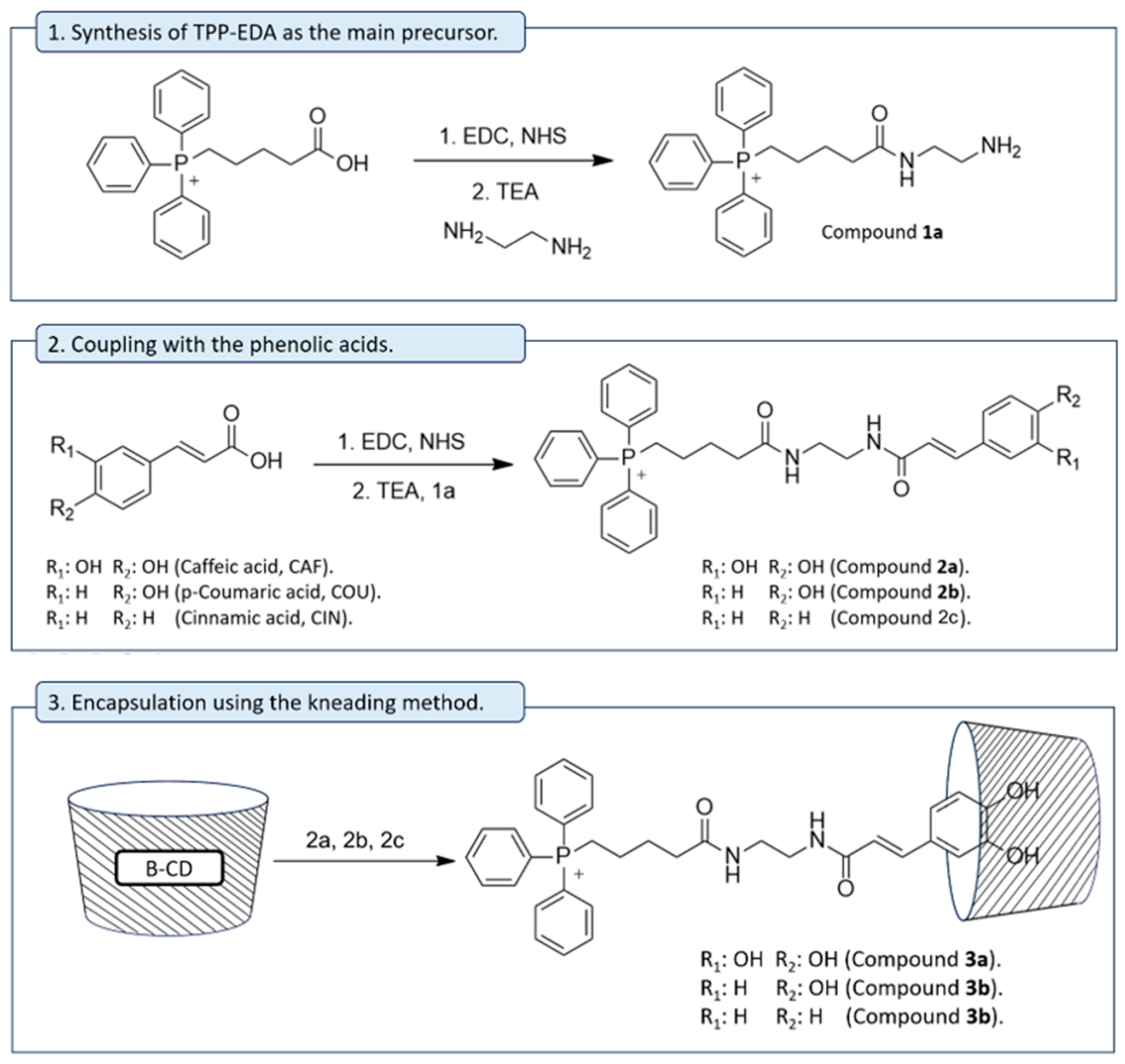
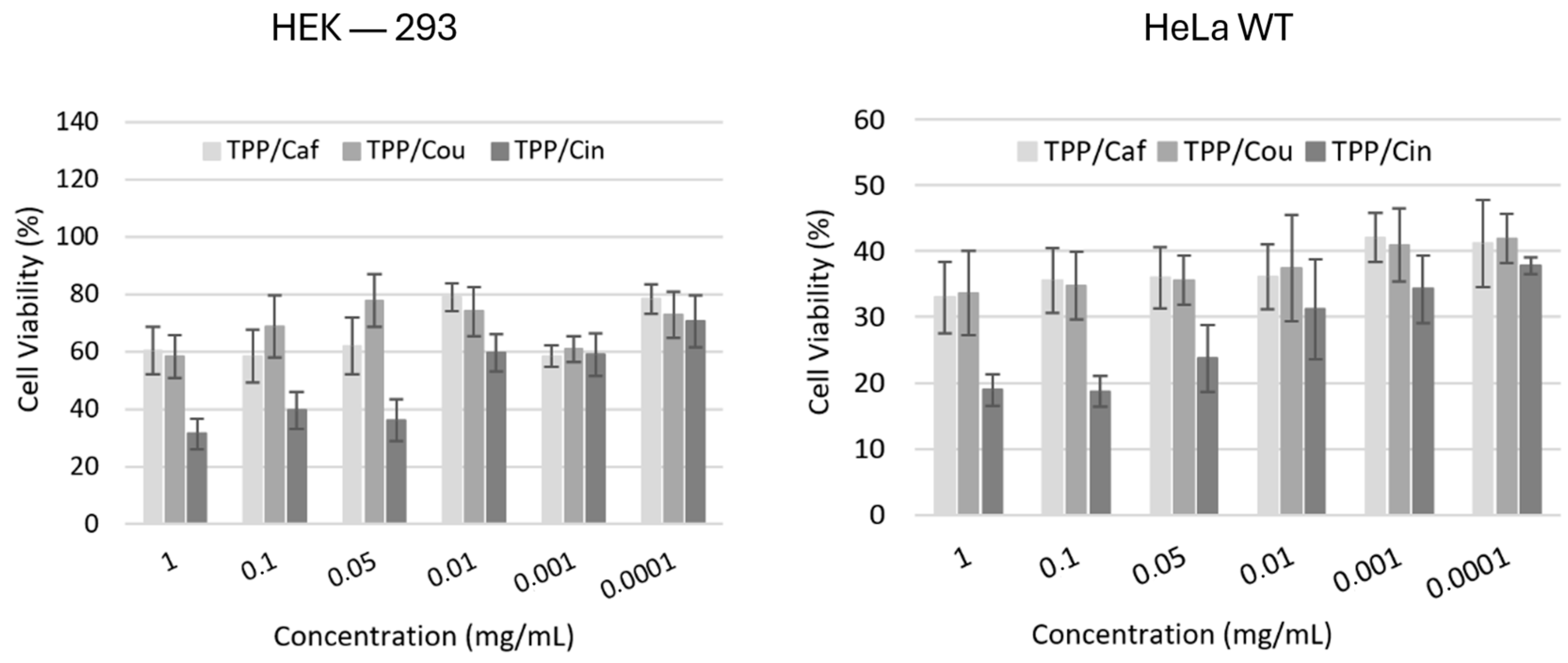
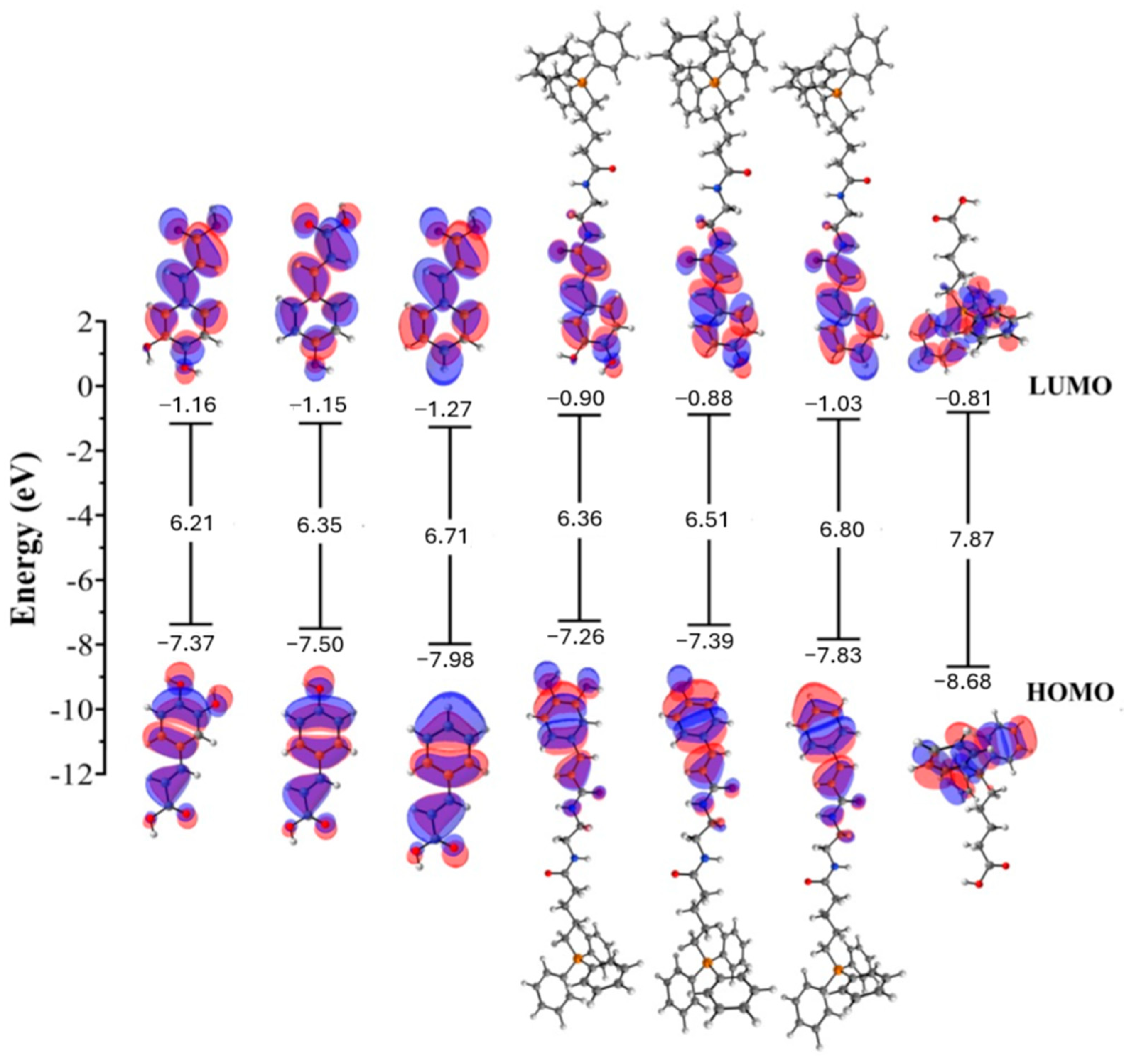


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Koopmans’ Theorem | Reference | |
|---|---|---|
| Electronegativity (χ) | [38,39,40] | |
| Electrophilicity (ω) | [41] | |
| Global hardness (η) | [38,42,43,44,45,46] | |
| Ionization potential (I) | [37] | |
| Electron Donator (A) | [37] | |
| HOMO–LUMO Gap (ΔEgap) | [37] | |
| Radical Fukui function (f 0) | [47,48,49,50] |
| n° | IUPAC Name | Molar Mass (g/mol) | Structure |
|---|---|---|---|
| 1 | (5−((aminoethyl)amino) −5−oxopentyl)triphenylphosphate(III) bromide a | 485.40 | 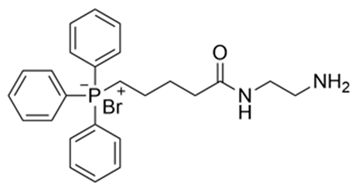 |
| 2 | (E)−(5−((2−(3−(3,4−dihydroxyphenyl)acrylamido)ethyl)amino)-5-oxopentyl)triphenylphosphate(III) bormide b | 647.54 | 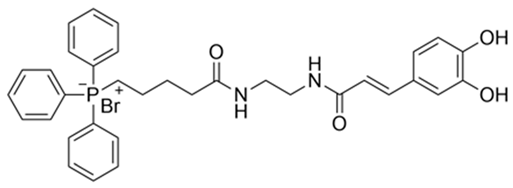 |
| 3 | (E)−(5−((2−(3−(4−hydroxyphenyl)acrylamido)ehtyl)amino) −5−oxopentyl)triphenylphosphate(III) bormide c | 631.54 | 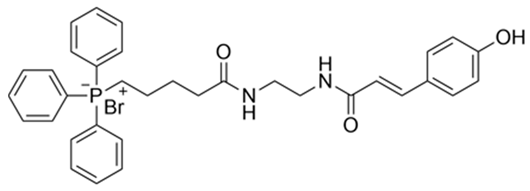 |
| 4 | (E)−(5−((2−cinnamamidoethyl)amino)-5-oxopentyl)triphenylphosphate(III) bormide d | 615.54 | 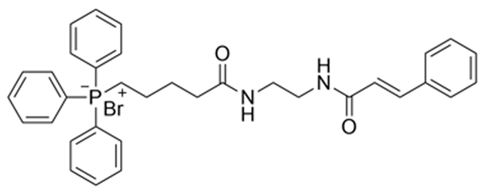 |
| ABTS Radical Scavenging Activity (%) | |||||
|---|---|---|---|---|---|
| Sample Concentration (mM) | |||||
| Sample Name | 0.01 | 0.05 | 0.1 | 0.25 | 0.5 |
| CAF | 66.5 ± 1.8 | ≥99 | ≥99 | ≥99 | ≥99 |
| COU | 19.0 ± 0.5 | 34.6 ± 1.4 | 61.3 ± 2.0 | 71.7 ± 8.1 | 99.7 ± 0.3 |
| CIN | 10.6 ± 0.7 | 8.9 ± 1.7 | 9.9 ± 3.3 | 10.2 ± 2.3 | 11.4 ± 1.9 |
| 4-TPP/CAF | 49.1 ± 2.7 | 99.7 ± 0.3 | ≥99 | ≥99 | ≥99 |
| 4-TPP/COU | 8.2 ± 2.4 | 16.4 ± 1.4 | 24.7 ± 1.3 | 48.8 ± 7.9 | 73.6 ± 11.1 |
| 4-TPP/CIN | 13.1 ± 1.9 | 14.3 ± 0.7 | 27.88 ± 4.7 | 35.7 ± 1.9 | 44.5 ± 5.3 |
| 4-TPP/CAF/B-CD | 34.5 ± 1.0 | 98.7 ± 0.5 | ≥99 | ≥99 | ≥99 |
| 4-TPP/COU/B-CD | 21.3 ± 3.1 | 36.7 ± 2.0 | 44.1 ± 1.1 | 51.6 ± 0.2 | 57.3 ± 1.2 |
| 4-TPP/CIN/B-CD | 10.1 ± 1.5 | 12.1 ± 3.4 | 6.7 ± 0.8 | 12.7 ± 0.3 | 13.4 ± 1.9 |
| DPPH Radical Scavenging Activity (%) | |||||
|---|---|---|---|---|---|
| Sample Concentration (mM) | |||||
| Sample Name | 0.01 | 0.05 | 0.1 | 0.25 | 0.5 |
| CAF | 27.8 ± 9.2 | 68.9 ± 1.1 | 90.2 ± 3.8 | 95.5 ± 0.2 | 95.5 ± 0.1 |
| COU | 47.6 ± 1.4 | 54.25 ± 1.0 | 60.9 ± 0.6 | 65.2 ± 3.0 | 67.7 ± 1.6 |
| CIN | 8.6 ± 0.7 | 6.6 ± 0.8 | 7.3 ± 0.5 | 6.2 ± 1.3 | 8.1 ± 0.7 |
| 4-TPP/CAF | 58.4 ± 1.9 | 95.4 ± 0.2 | 95.7 ± 0.1 | 95.6 ± 0.1 | 95.7 ± 0.2 |
| 4-TPP/COU | 12.2 ± 1.8 | 13.1 ± 1.2 | 19.1 ± 3.1 | 38.1 ± 3.7 | 65.2 ± 2.1 |
| 4-TPP/CIN | 9.6 ± 1.5 | 7.9 ± 1.7 | 8.4 ± 1.7 | 8.5 ± 2.1 | 10.6 ± 0.9 |
| 4-TPP/CAF/B-CD | 31.1 ± 2.0 | 92.3 ± 1.7 | 95.3 ± 0.1 | 95.4 ± 0.1 | 95.2 ± 0.2 |
| 4-TPP/COU/B-CD | 11.9 ± 0.8 | 18.4 ± 1.4 | 28.6 ± 1.6 | 41.2 ± 7.8 | 62.3 ± 6.5 |
| 4-TPP/CIN/B-CD | 10.8 ± 2.5 | 10.2 ± 0.9 | 9.6 ± 3.5 | 9.1 ± 0.8 | 11.5 ± 1.3 |
| Compound | HOMO | ΔEgap | χ | η | ω | I | A | |
|---|---|---|---|---|---|---|---|---|
| Caffeic acid | −7.37 | −1.16 | 6.21 | 4.26 | 3.10 | 2.93 | 7.37 | 1.16 |
| ρ-coumaric acid | −7.50 | −1.15 | 6.35 | 4.32 | 3.18 | 2.94 | 7.50 | 1.15 |
| Cinnamic acid | −7.98 | −1.27 | 6.71 | 4.63 | 3.35 | 3.19 | 7.98 | 1.27 |
| Compound 2 | −7.26 | −0.90 | 6.36 | 4.08 | 3.18 | 2.62 | 7.26 | 0.90 |
| Compound 3 | −7.39 | −0.88 | 6.51 | 4.13 | 3.25 | 2.63 | 7.39 | 0.88 |
| Compound 4 | −7.83 | −1.03 | 6.80 | 4.43 | 3.40 | 2.89 | 7.83 | 1.03 |
| TPP | −8.68 | −0.81 | 7.87 | 4.74 | 3.93 | 2.86 | 8.68 | 0.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbarbaro, C.; Márquez-Miranda, V.; Leal, M.; Pino-Rios, R.; Olivares, P.; González, M.; Díaz-Franulic, I.; González-Nilo, F.; Yáñez, O.; Duarte, Y. Exploring the Mechanism of β-Cyclodextrin-Encased Phenolic Acids Functionalized with TPP for Antioxidant Activity and Targeting. Antioxidants 2025, 14, 465. https://doi.org/10.3390/antiox14040465
Sbarbaro C, Márquez-Miranda V, Leal M, Pino-Rios R, Olivares P, González M, Díaz-Franulic I, González-Nilo F, Yáñez O, Duarte Y. Exploring the Mechanism of β-Cyclodextrin-Encased Phenolic Acids Functionalized with TPP for Antioxidant Activity and Targeting. Antioxidants. 2025; 14(4):465. https://doi.org/10.3390/antiox14040465
Chicago/Turabian StyleSbarbaro, Christopher, Valeria Márquez-Miranda, Matías Leal, Ricardo Pino-Rios, Pedro Olivares, Makarena González, Ignacio Díaz-Franulic, Fernando González-Nilo, Osvaldo Yáñez, and Yorley Duarte. 2025. "Exploring the Mechanism of β-Cyclodextrin-Encased Phenolic Acids Functionalized with TPP for Antioxidant Activity and Targeting" Antioxidants 14, no. 4: 465. https://doi.org/10.3390/antiox14040465
APA StyleSbarbaro, C., Márquez-Miranda, V., Leal, M., Pino-Rios, R., Olivares, P., González, M., Díaz-Franulic, I., González-Nilo, F., Yáñez, O., & Duarte, Y. (2025). Exploring the Mechanism of β-Cyclodextrin-Encased Phenolic Acids Functionalized with TPP for Antioxidant Activity and Targeting. Antioxidants, 14(4), 465. https://doi.org/10.3390/antiox14040465