
The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NADPH Oxidase Family
2.1. NOX4
2.2. The Role of NOX4 in Angiogenesis
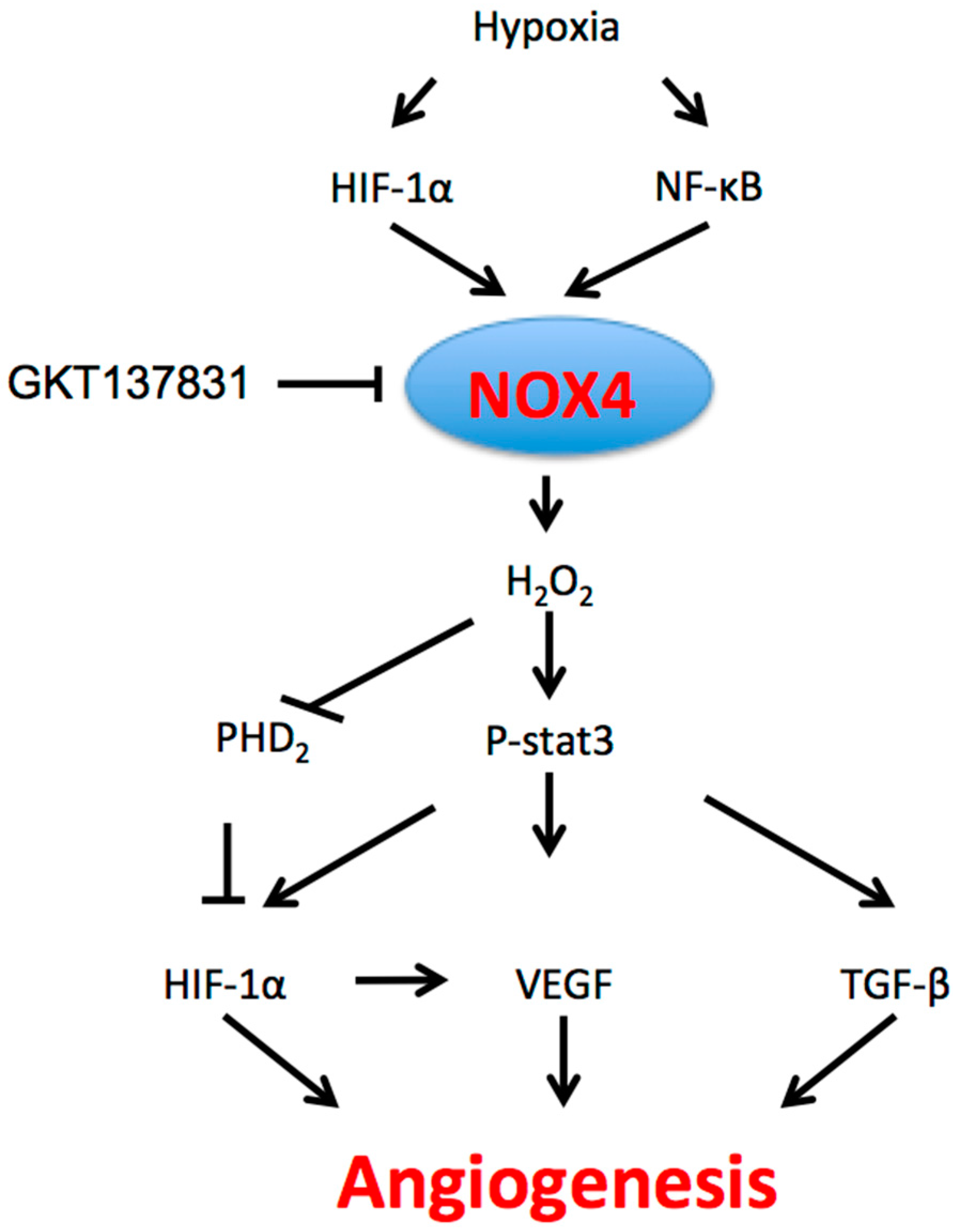
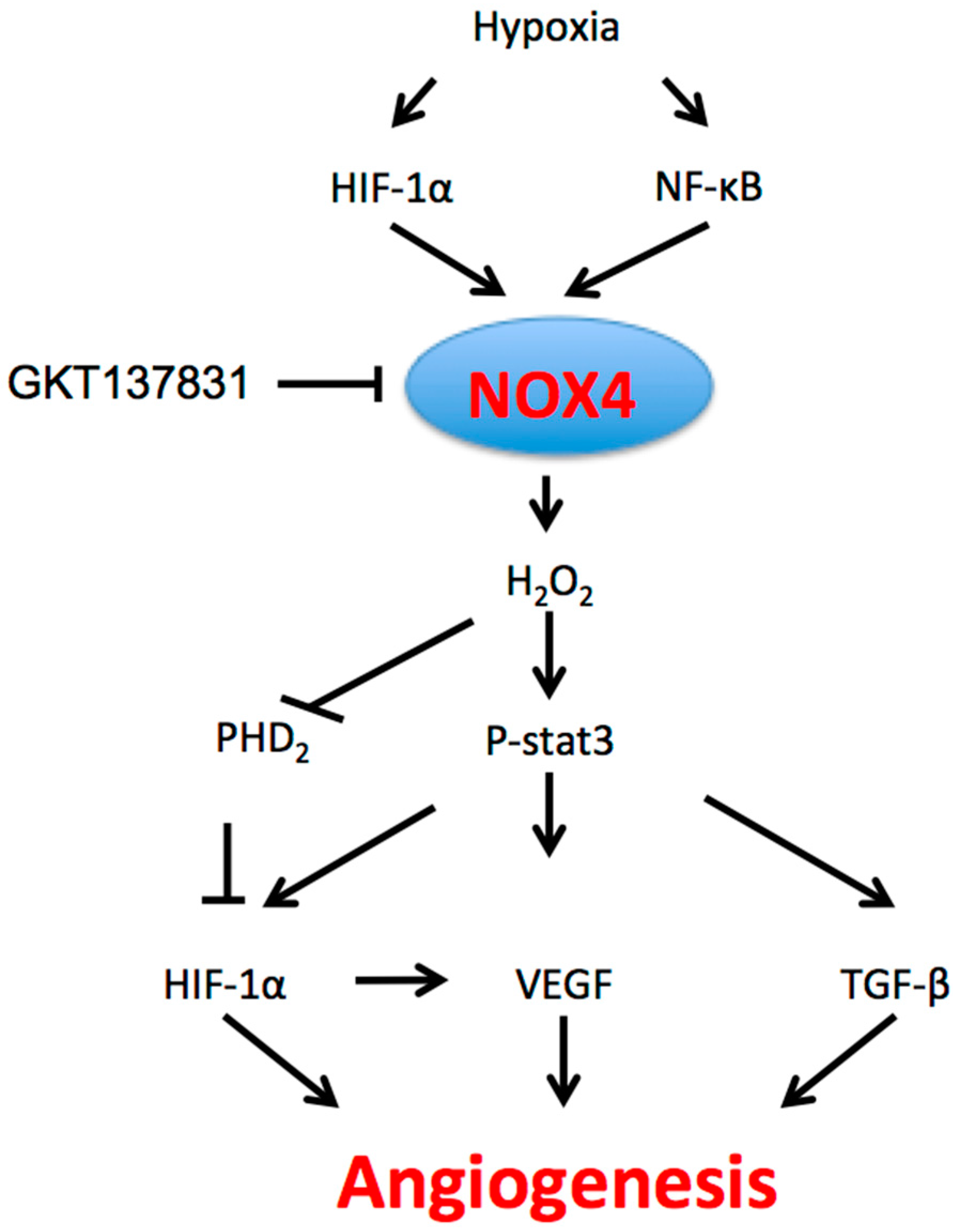
2.3. NOX4 Signaling Pathways and Regulation in Angiogenesis
2.3.1. Hypoxia
2.3.2. Ischemia
2.3.3. VEGF
2.3.4. TRAIL
2.3.5. TGF-β1
2.4. Role of NOX4-Mediated Angiogenesis in Cancer
3. The Thioredoxin System
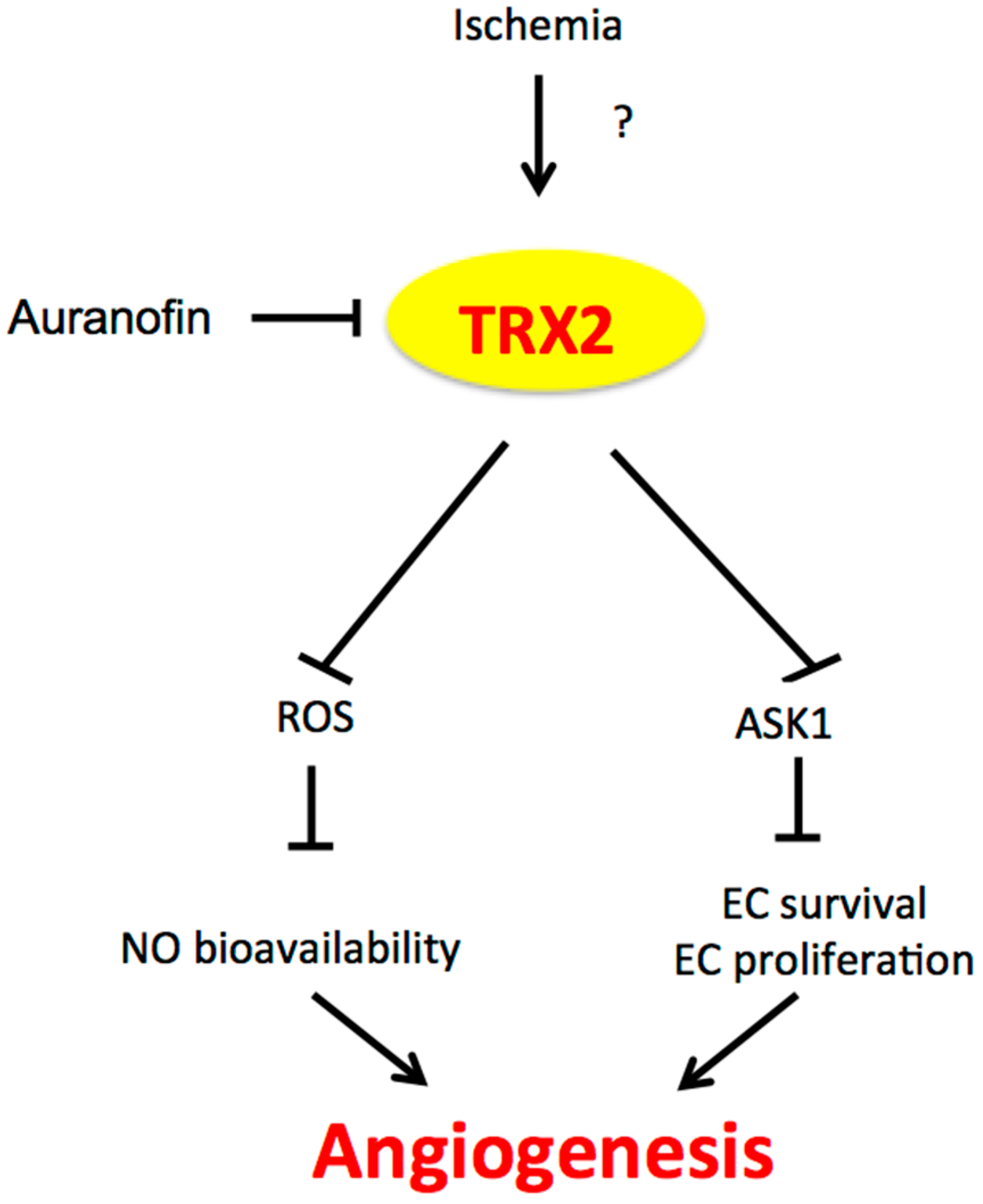
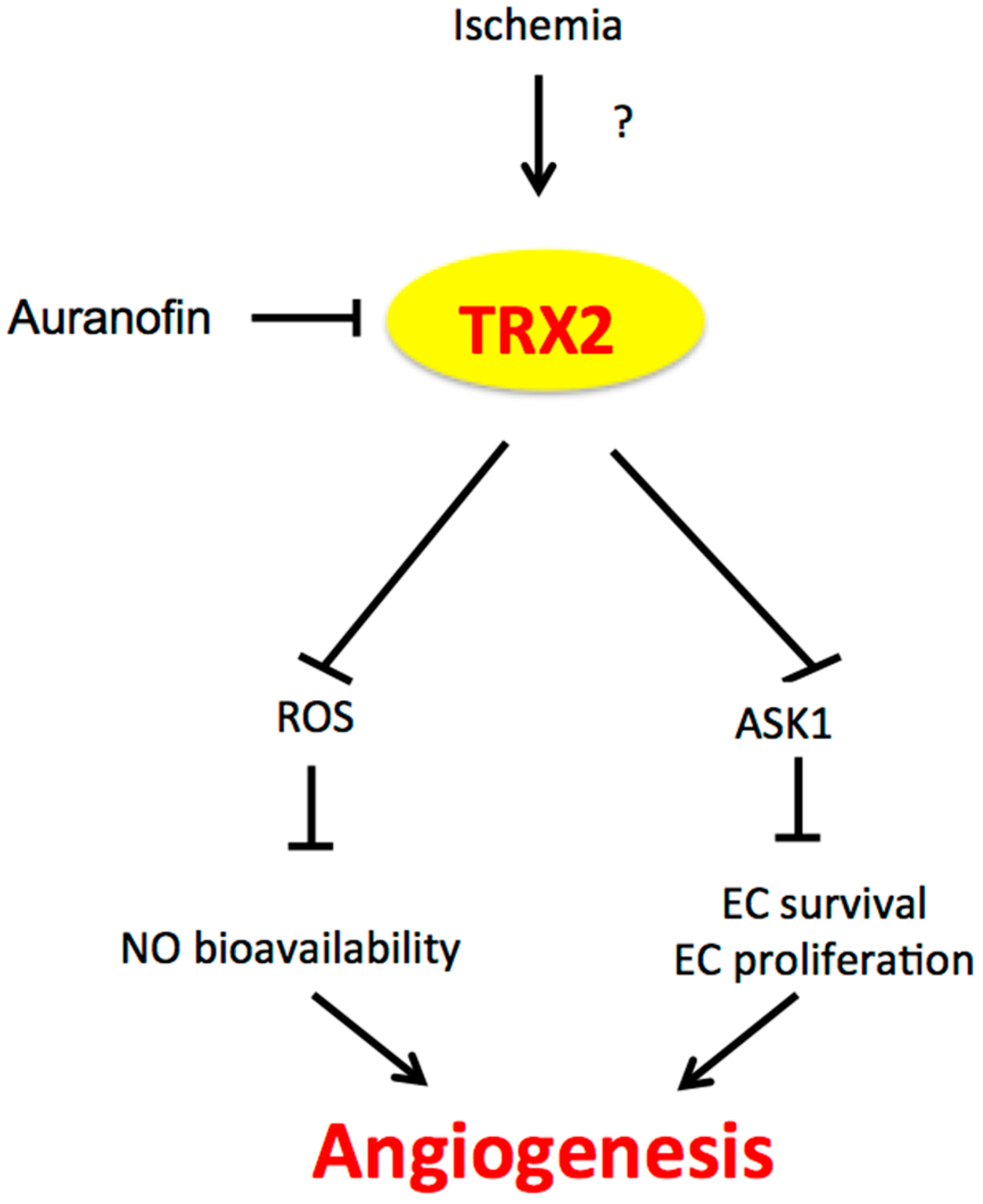
3.1. TRX1/2 and Angiogenesis
3.2. Role of TRX1/2-Mediated Angiogenesis in Cancer
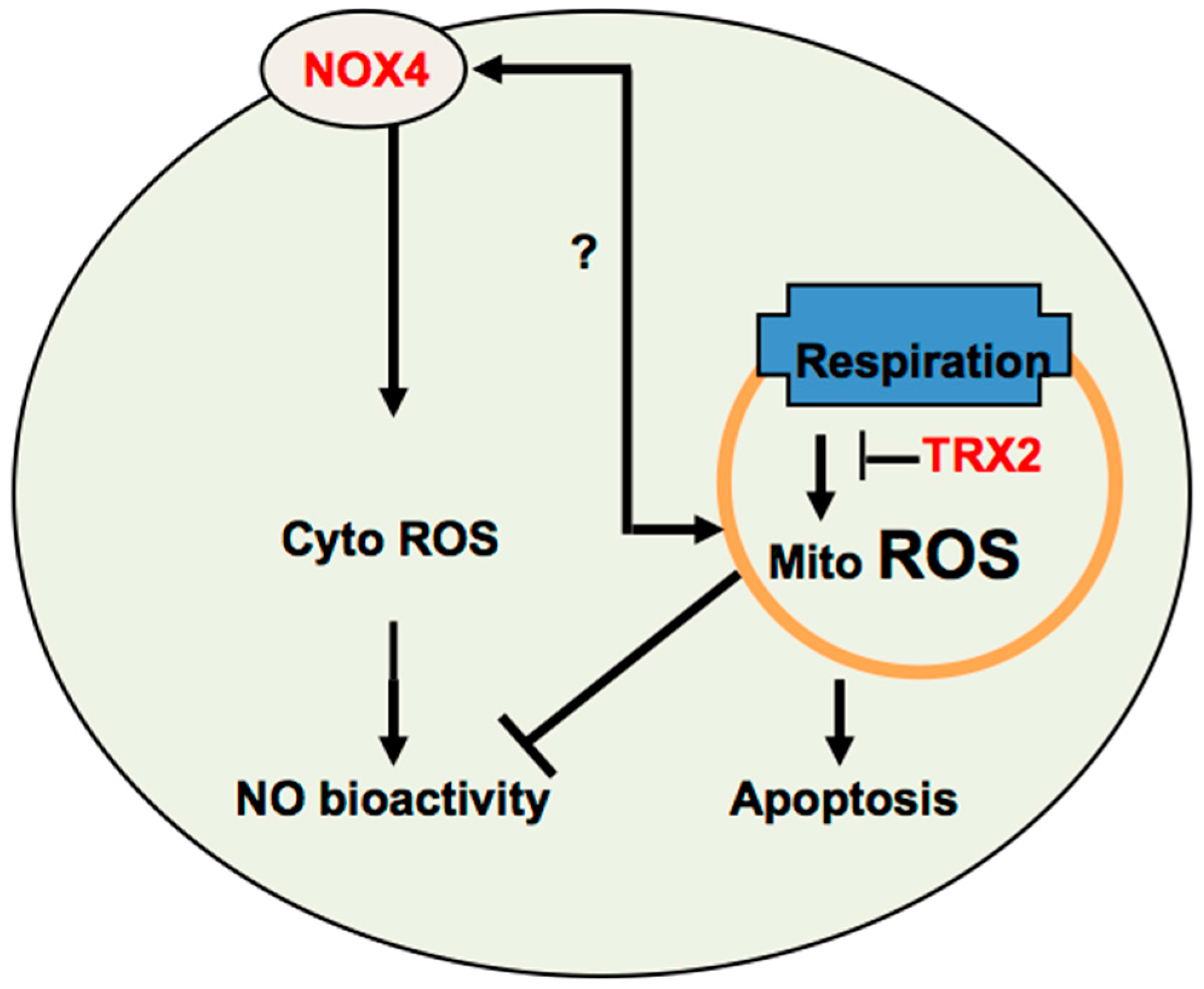
4. The Potential Cross-Talk between NOX4 and TRX2 in Angiogenesis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Levonen, A.L.; Brookes, P.S.; Ceaser, E.; Shiva, S.; Barone, M.C.; Darley-Usmar, V. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic. Biol. Med. 2002, 33, 1465–1474. [Google Scholar] [CrossRef]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via pten oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J. From form to function: The role of NOX4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of NOX4 and NOX2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox. Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vas. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and localization of Nox2 and Nox4 in primary human endothelial cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Von Lohneysen, K.; Noack, D.; Jesaitis, A.J.; Dinauer, M.C.; Knaus, U.G. Mutational analysis reveals distinct features of the Nox4-p22phox complex. J. Biol. Chem. 2008, 283, 35273–35282. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Chan, E.C.; Liu, G.S.; Jiang, F.; Dusting, G.J. Transforming growth factor-beta1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J. Cell. Mol. Med. 2014, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lan, T.; Hou, J.; Li, J.; Fang, R.; Yang, Z.; Zhang, M.; Liu, J.; Liu, B. Nox4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget 2014, 5, 4392–4405. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; Lopez-Luque, J.; Fernando, J.; Sanchez, A.; Fernandez, M.; Navarro, E.; Fabregat, I. The NADPH oxidase Nox4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.X.; Liang, L.; Wang, L.; Han, J.T.; Zhu, X.X.; Han, H.; Hu, D.H.; Zhang, P. Inhibition of notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell. Immunol. 2014, 287, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; Kawut, S.M.; Krowka, M.J.; Brown, R.S.; Trotter, J.F.; Shah, V.; Peter, I.; Tighiouart, H.; Mitra, N.; Handorf, E.; et al. Genetic risk factors for hepatopulmonary syndrome in patients with advanced liver disease. Gastroenterology 2010, 139, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F. NADPH Oxidase 4 Promotes Endothelial Angiogenesis Through Endothelial Nitric Oxide Synthase Activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit Nox4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH oxidase subunit Nox4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell. 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hong, Z.; Zeng, C.; Yu, Q.; Wang, H. NADPH oxidase 4 promotes cardiac microvascular angiogenesis after hypoxia/reoxygenation in vitro. Free Radic. Biol. Med. 2014, 69, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Yu, M.O.; Park, K.J.; Chi, S.G.; Park, D.H.; Chung, Y.G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.O.; Park, K.J.; Park, D.H.; Chung, Y.G.; Chi, S.G.; Kang, S.H. Reactive oxygen species production has a critical role in hypoxia-induced Stat3 activation and angiogenesis in human glioblastoma. J. Neurooncol. 2015, 125, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell. Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Bland, J.M.; Kleinhenz, D.J.; Mitchell, P.O.; Walp, E.R.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Murphy, T.C.; Nanes, M.S.; Hart, C.M. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2010, 299, L559–L566. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Madeddu, P. Angiogenesis gene therapy to rescue ischaemic tissues: Achievements and future directions. Br. J. Pharmacol. 2001, 133, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, B.A.; Cartland, S.P.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.; Thai, T.; Yeung, A.W.; Thomas, S.R.; et al. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Promotes Angiogenesis and Ischemia-Induced Neovascularization Via NADPH Oxidase 4 (Nox4) and Nitric Oxide-Dependent Mechanisms. J. Am. Heart. Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Srisook, K.; Kim, C.; Cha, Y.N. Molecular mechanisms involved in enhancing HO-1 expression: De-repression by heme and activation by Nrf2, the “one-two” punch. Antioxid. Redox. Signal. 2005, 7, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Sudhahar, V.; Kim, S.J.; Chen, G.F.; McKinney, R.D.; Kojda, G.; Fukai, T.; Ushio-Fukai, M. Critical role of endothelial hydrogen peroxide in post-ischemic neovascularization. PLoS ONE 2013, 8, e57618. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase Nox4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I.; Gliki, G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc. Res. 2001, 49, 568–581. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jiang, T.; Lu, D.; Luo, Y.; Zheng, C.; Feng, J.; Yang, D.; Chen, C.; Yan, X. NADPH oxidase 4 mediates reactive oxygen species induction of CD146 dimerization in VEGF signal transduction. Free Radic. Biol. Med. 2010, 49, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Helfinger, V.; Henke, N.; Harenkamp, S.; Walter, M.; Epah, J.; Penski, C.; Mittelbronn, M.; Schroder, K. The NADPH Oxidase Nox4 mediates tumour angiogenesis. Acta. Physiol. 2016, 216, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Carnevale, E.; Corallini, F.; Rizzardi, C.; Zacchigna, S.; Melato, M.; Zauli, G. Evidence for a proangiogenic activity of TNF-related apoptosis-inducing ligand. Neoplasia 2004, 6, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Prado-Lourenco, L.; Khachigian, L.M.; Bennett, M.R.; Di Bartolo, B.A.; Kavurma, M.M. Trail promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFKappaB-dependent manner. Circ. Res. 2010, 106, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Huecksteadt, T.P.; Norman, K.; Sanders, K.; Murphy, T.M.; Chitano, P.; Wilson, K.; Hoidal, J.R.; Kennedy, T.P. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. AJP Lung Cell. Mol. Physiol. 2007, 292, L1543–L1555. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Wong, H.; Shah, M.H.; Dusting, G.J.; Jiang, F.; Peshavariya, H.M. Smad-independent pathway involved in transforming growth factor beta1-induced Nox4 expression and proliferation of endothelial cells. Naunyn-Schmiedeberg Arch. Pharmacol. 2015, 388, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Hock, T.D.; Logsdon, N.; Zhou, Y.; Thannickal, V.J. A far-upstream AP-1/Smad binding box regulates human NOX4 promoter activation by transforming growth factor-beta. Gene 2014, 540, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiao, J.; Kuroda, J.; Ago, T.; Sadoshima, J.; Cohen, R.A.; Tong, X. Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim. Biophys. Acta 2014, 1842, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Dusting, G.J.; Peshavariya, H.M. Trichostatin A, a histone deacetylase inhibitor suppresses NADPH Oxidase 4-Derived Redox Signalling and Angiogenesis. J. Cell. Mol. Med. 2016, 20, 1932–1944. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Liu, G.S.; Chang, C.W.; Jiang, F.; Chan, E.C.; Dusting, G.J. Prostacyclin signaling boosts NADPH oxidase 4 in the endothelium promoting cytoprotection and angiogenesis. Antioxid. Redox Signal. 2014, 20, 2710–2725. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Mei, A.; Liu, J.; Kang, X.; Shi, X.; Qian, R.; Chen, S. NADPH oxidase 4 mediates insulin-stimulated HIF-1alpha and VEGF expression, and angiogenesis in vitro. PLoS ONE 2012, 7, e48393. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Kruse, C.; Zhang, M.; Schroder, K. Nox4 supports proper capillary growth in exercise and retina neo-vascularization. J. Physiol. 2015, 593, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Brewer, A.C.; Schroder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Naruo, A.; Okada, M.; Hayabe, Y.; Yamawaki, H. Brain-derived neurotrophic factor promotes angiogenic tube formation through generation of oxidative stress in human vascular endothelial cells. Acta Physiol. 2014, 211, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Wang, S.; Klco, J.M.; Kaelin, W.G.; Livingston, D.M. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat. Med. 2000, 6, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Giaccia, A.J.; Denko, N.C. The hypoxic tumor microenvironment and gene expression. Semin. Radiat. Oncol. 2004, 14, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.L.; Turner, R.M.; Chang, G.; Joshi, D.; Zhan, Y.; Chen, L.; Maranchie, J.K. NADPH Oxidase NOX4 Supports Renal Tumorigenesis by Promoting the Expression and Nuclear Accumulation of HIF2. Cancer Res. 2014, 74, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, E83. [Google Scholar] [CrossRef] [PubMed]
- Maranchie, J.K.; Zhan, Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, N.; Yin, T.; Huang, L.; Liu, S.; Liu, D.; Xie, C.; Zhang, M. Lentivirus-Mediated Nox4 shRNA Invasion and Angiogenesis and Enhances Radiosensitivity in Human Glioblastoma. Oxid. Med. Cell. Longev. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Ahn, Y.H.; Moon, B.I.; Kim, H.S. Exogenous C2 Ceramide Suppresses Matrix Metalloproteinase Gene Expression by Inhibiting ROS Production and MAPK Signaling Pathways in PMA-Stimulated Human Astroglioma Cells. Int. J. Mol. Sci. 2016, 17, 477. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Oshima, M.; Oshima, H.; Takaku, K.; Maruyama, T.; Yodoi, J.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol 1996, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B. J. Biol. Chem. 1964, 239, 3436–3444. [Google Scholar] [PubMed]
- Moore, E.C. A thioredoxin—Thioredoxin reductase system from rat tumor. Biochem. Biophys. Res. Commun. 1967, 29, 264–268. [Google Scholar] [CrossRef]
- Clarke, F.M.; Orozco, C.; Perkins, A.V.; Cock, I.; Tonissen, K.F.; Robins, A.J.; Wells, J.R. Identification of molecules involved in the “early pregnancy factor” phenomenon. J. Reprod. Fertil. 1991, 93, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, G.; Enmark, E.; Miranda-Vizuete, A.; Gustafsson, J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997, 272, 2936–2941. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Liu, H.; Choe, E.; Yoshioka, J.; Shalev, A.; Bloch, K.D.; Lee, R.T. Nitric oxide-dependent suppression of thioredoxin-interacting protein expression enhances thioredoxin activity. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Turturro, F.; Friday, E.; Welbourne, T. Hyperglycemia regulates thioredoxin-ROS activity through induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer 2007, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, J.; Imahashi, K.; Gabel, S.A.; Chutkow, W.A.; Burds, A.A.; Gannon, J.; Schulze, P.C.; MacGillivray, C.; London, R.E.; Murphy, E.; et al. Targeted deletion of thioredoxin-interacting protein regulates cardiac dysfunction in response to pressure overload. Circ. Res. 2007, 101, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Damdimopoulos, A.E.; Miranda-Vizuete, A.; Pelto-Huikko, M.; Gustafsson, J.A.; Spyrou, G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 2002, 277, 33249–33257. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Zhang, H.; Jones, D.P. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. 2006, 91, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Min, W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ. Res. 2002, 90, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, H.J.; Huang, Q.; Lu, L.; Min, W. Novel action and mechanism of auranofin in inhibition of vascular endothelial growth factor receptor-3-dependent lymphangiogenesis. Anti-Cancer Agent Med. Chem. 2014, 14, 946–954. [Google Scholar] [CrossRef]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; He, Y.; Zhang, H.; Yu, L.; Wan, T.; Xu, Z.; Jones, D.; Chen, H.; Min, W. Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; He, X.; Liu, W.; Lu, M.; Hsieh, J.T.; Min, W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J. Clin. Investig. 2003, 111, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, Y.; Dai, S.; Xu, Z.; Luo, Y.; Wan, T.; Luo, D.; Jones, D.; Tang, S.; Chen, H.; et al. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J. Clin. Investig. 2008, 118, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Tonissen, K.F.; Di Trapani, G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Diccianni, M.B.; Tanaka, T.; Gribi, R.; Yu, A.L.; Pullen, J.D.; Camitta, B.M.; Yu, J. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001, 61, 7333–7338. [Google Scholar] [PubMed]
- Berggren, M.; Gallegos, A.; Gasdaska, J.R.; Gasdaska, P.Y.; Warneke, J.; Powis, G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996, 16, 3459–3466. [Google Scholar] [PubMed]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Gallegos, A.; Gasdaska, J.R.; Taylor, C.W.; Paine-Murrieta, G.D.; Goodman, D.; Gasdaska, P.Y.; Berggren, M.; Briehl, M.M.; Powis, G. Transfection with human thioredoxin increases cell proliferation and a dominant-negative mutant thioredoxin reverses the transformed phenotype of human breast cancer cells. Cancer Res. 1996, 56, 5765–5770. [Google Scholar] [PubMed]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar] [CrossRef] [PubMed]
- Hellfritsch, J.; Kirsch, J.; Schneider, M.; Fluege, T.; Wortmann, M.; Frijhoff, J.; Dagnell, M.; Fey, T.; Esposito, I.; Kolle, P.; et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox. Signal. 2015, 22, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2015. [Google Scholar] [CrossRef] [PubMed]
- Perry, B.N.; Govindarajan, B.; Bhandarkar, S.S.; Knaus, U.G.; Valo, M.; Sturk, C.; Carrillo, C.O.; Sohn, A.; Cerimele, F.; Dumont, D.; et al. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J. Investig. Dermatol. 2006, 126, 2316–2322. [Google Scholar] [CrossRef] [PubMed]
- Lapidoth, M.; Ben-Amitai, D.; Bhandarkar, S.; Fried, L.; Arbiser, J.L. Efficacy of topical application of eosin for ulcerated hemangiomas. J. Am. Acad. Dermatol. 2009, 60, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Y.; Fried, L.E.; Du, Y.; Montano, S.J.; Sohn, A.; Lefkove, B.; Holmgren, L.; Arbiser, J.L.; Holmgren, A.; et al. Disruption of the mitochondrial thioredoxin system as a cell death mechanism of cationic triphenylmethanes. Free Radic. Biol. Med. 2011, 50, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, M.; Praggastis, A.; Li, Y.; Zhou, H.J.; He, Y.; Ghazvinian, R.; Cincotta, D.J.; Rice, K.P.; Min, W. Carbamoylating activity associated with the activation of the antitumor agent laromustine inhibits angiogenesis by inducing ASK1-dependent endothelial cell death. PLoS ONE 2014, 9, e103224. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L.; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, H1600–H1608. [Google Scholar] [PubMed]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.; et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Wade, B.E.; Bijli, K.M.; Kang, B.Y.; Williams, C.R.; Ma, J.; Go, Y.M.; Hart, C.M.; Sutliff, R.L. Hypoxia inhibits expression and function of mitochondrial thioredoxin 2 to promote pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L599–L608. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial Reactive Oxygen Species Generation and Apoptosis Stress Kinase-1 Activity to Maintain Cardiac Function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Li, L.; Zhou, H.J.; Min, W. The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants 2017, 6, 42. https://doi.org/10.3390/antiox6020042
Chen C, Li L, Zhou HJ, Min W. The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants. 2017; 6(2):42. https://doi.org/10.3390/antiox6020042
Chicago/Turabian StyleChen, Chaofei, Li Li, Huanjiao Jenny Zhou, and Wang Min. 2017. "The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk" Antioxidants 6, no. 2: 42. https://doi.org/10.3390/antiox6020042
APA StyleChen, C., Li, L., Zhou, H. J., & Min, W. (2017). The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants, 6(2), 42. https://doi.org/10.3390/antiox6020042