Abstract
The incidence of pancreatic cancer is increasing as the population ages but treatment advancements continue to lag far behind. The majority of pancreatic cancer patients have a K-ras oncogene mutation causing a shift in the redox state of the cell, favoring malignant proliferation. This mutation is believed to lead to nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation and superoxide overproduction, generating tumorigenic behavior. Superoxide dismutases (SODs) have been studied for their ability to manage the oxidative state of the cell by dismuting superoxide and inhibiting signals for pancreatic cancer growth. In particular, manganese superoxide dismutase has clearly shown importance in cell cycle regulation and has been found to be abnormally low in pancreatic cancer cells as well as the surrounding stromal tissue. Likewise, extracellular superoxide dismutase expression seems to favor suppression of pancreatic cancer growth. With an increased understanding of the redox behavior of pancreatic cancer and key regulators, new treatments are being developed with specific targets in mind. This review summarizes what is known about superoxide dismutases in pancreatic cancer and the most current treatment strategies to be advanced from this knowledge.
1. Introduction
Adenocarcinoma of the pancreas is the fourth leading cause of cancer-related death in the United States [1]. Therapeutic responsiveness of pancreatic cancer to surgery, chemotherapy, and radiation therapy is poor, resulting in a dismal 5-year survival of less than 3% [2]. Mutation of the K-ras oncogene is a common event in the early development of pancreatic cancer occurring in 95% of pancreatic cancers, with a resulting overall increase in production of reactive oxygen species (ROS) [3]. Superoxide dismutases (SODs) modulate the oxidative status of the cell by dismutation of two molecules of O2●− into hydrogen peroxide (H2O2) and molecular oxygen (O2) [4]. SODs have been shown to inhibit the in vitro and in vivo growth of pancreatic cancer through various intracellular signaling pathways [5,6]. Loss of MnSOD function leads to intracellular signaling supporting pancreatic acinar malignant cellular programming, while loss of extracellular superoxide dismutase (EcSOD) leads to an influx of regulatory pathways supporting pancreatic tumor microenvironments [7,8]. Therefore, we propose that strategies to scavenge mitochondrial and non-mitochondrial-generated O2●− may prove beneficial in the treatment of pancreatic cancer. This review will focus on the role of SOD-induced inhibition of tumor growth and propagation, and its potential as a targeted pancreatic cancer therapy.
2. Pancreatic Adenocarcinoma
53,670 patients are projected to be diagnosed with pancreatic adenocarcinoma in 2017, making it the tenth most common cancer among both men and women; an incidence of 14.0 and 10.9 per 100,000, respectively [1,2]. Despite the relatively modest incidence, it is the fourth deadliest cancer amongst both men and women in the U.S. [1,2]. Over 43,000 patients are expected to succumb to the disease in 2017, with a death rate of 12.6 men and 9.6 women per 100,000. This high lethality is likely due to a combination of late stage at diagnosis, at which point 29% of patients will already have regional lymph node spread and 52% will have metastatic disease, and poor survival at all stages [1,2]. The relative lack of progress in therapy for pancreatic adenocarcinoma compared to other common cancers has led to opportunities for innovation and the development of new treatment modalities [9].
3. Superoxide and Superoxide Dismutase
Superoxide (O2●−) is a byproduct of respiration and acts as a signaling molecule through one-electron oxidations (via HO2•) or reductions (via O2●2−), while hydrogen peroxide (H2O2), the byproduct of superoxide dismutation via SOD, participates in two-electron signaling circuits [10,11,12]. The aberrant passage of electrons to ~0.1–0.5% of oxygen molecules during respiration results in mitochondrial O2●− generation [13]. In addition to the electron transport chain, other sources of O2●− include NADPH oxidases (NOX) and xanthine oxidase [14]. The primary roles of NOX enzymes are to produce reactive oxygen species such as O2●− [15]. For example, the immune system generates abundant O2●− levels via NOX enzymes as an oxygen-dependent defense mechanism within phagocytes [12]. The NOX family consists of several homologs, which have the ability to deliver electrons across cellular membranes to generate these reactive oxygen species. NOX activation results in the regulation of gene expression, cell signaling, and cell differentiation, and is precisely why this family of enzymes has been linked to an equally wide range of pathologic states [15].
SODs are a class of metalloenzymes that scavenge O2●− to prevent cell toxicity and regulate cell signaling. SODs are enzymes that participate in two-step reactions to dismute two molecules of O2●− into one molecule of O2 and one molecule of H2O2, which undergo either subsequent dismutation by the catalase to H2O and O2 or are reduced to water by peroxidases [4]. An important feature of SOD enzymes is that they are highly compartmentalized. In mammalian species, three SOD isotypes have been identified: Copper Zinc SOD (CuZnSOD, SOD1), Manganese SOD (MnSOD, SOD2), and extracellular SOD (EcSOD, SOD3).
CuZnSOD is present in the cytosol, nucleus, peroxisomes, and intermembrane space of the mitochondria of human cells [16]. Following the translation of the CuZnSOD protein (Molecular Weight 32 kDa), the stable homodimer is held together by hydrophobic contact [17]. It becomes activated by copper chaperone for superoxide dismutase (Ccs1) by introducing copper and creating an intramolecular disulfide bond for enzymatic action [18]. Only the copper subunit is enzymatically active; the zinc subunit purely structural. The enzymatically active copper reacts in a ping-pong mechanism where one superoxide molecule is reduced to hydrogen peroxide and a second superoxide is oxidized to molecular oxygen [19]. MnSOD is composed of four identical 22 kDa subunits and is essential for cell survival because it protects the mitochondria from reactive oxygen species in aerobic systems [20]. In mice, heterozygous MnSOD knock-out leads to a 50% decrease in enzyme activity and age-dependent oxidative DNA damage in the nucleus and mitochondria. Complete knock-out leads to severe cardiomyopathy, oxidative mitochondrial damage, and early postnatal death [21]. MnSOD also plays a crucial role in cellular biology beyond mitigating oxidative stress. It acts as a bifurcation point from the one-electron radicals (O2●−) and two-electron radicals (H2O2) which modulate biological stages of differentiation, proliferation, quiescence, apoptosis, and necrosis [22,23]. The delicate balance of O2●− and H2O2, which MnSOD partially regulates, modulates the expression of critical transcription factors such as HIF-1α, AP-1, NF-κB, and p53 [24].
EcSOD is the only isoform of SOD that is expressed extracellularly and is a tetrameric glycoprotein containing four 30 kDa subunits, which utilizes copper and zinc ions in a similar fashion to the previously described soluble, cytosolic CuZnSOD (SOD1) [25,26,27]. EcSOD is cell type specific and is secreted in greatest amount by adipose tissue but also by the pancreas, lung, kidney, and vasculature [27,28]. It regulates the redox state of the extracellular environment and plays a key signaling role in the recruitment of inflammatory leukocytes and the creation of directly toxic molecules such as peroxynitrite [25]. Deficiency in mice results in increased oxygen-dependent toxicity [29]. A fundamental property of EcSOD, imparted by a heparin-binding domain (HBD) not found in other SODs, is its affinity for heparin sulfate proteoglycans located on the cell surface and in the extracellular matrix, allowing for its binding and adherence to the extracellular milieu [30]. Interestingly, the extracellular presence of superoxide dismutase has been observed to have anti-tumorigenic activity connected to intracellular signaling cascades and their downstream products. For example, the overexpression of EcSOD in vitro has been shown to inhibit the growth of melanoma by blunting tumor neovascularization through the down-regulation of vascular endothelial growth factor expression [31].
In general, cancer cells demonstrate an increase in the amount of reactive oxygen species present compared to normal tissues [32]. This is also true in pancreatic cancer as well [33]. Several studies have demonstrated high concentrations of reactive oxygen species are cytotoxic; however, at low concentrations, they regulate several key physiological processes including cell differentiation, apoptosis, and cell proliferation [32,34]. Redox-sensitive signal transduction pathways may control the regulation of these processes [35].
4. SOD, Superoxide and the Pancreas
The importance of functional SOD is demonstrated by its remarkable conservation. Population studies of functional polymorphisms have shown mixed results regarding risk for pancreatic cancer [36]. One small, case-control study demonstrated a two-fold increased risk for pancreatic cancer in patients with an alanine-to-valine polymorphism at codon 16 of the MnSOD gene, which is a part of the mitochondrial targeting sequence [37]. The limited evidence of genotypic variation is in spite of the very predictable pattern of SOD activity evolution with oncogenesis and suggests that these rare polymorphisms have a similar phenotype as the natural genotype, at least with regards to oncogenesis [38]. Regarding the systemic difference in SOD activity, some data identifies no difference in plasma SOD activity in patients with pancreatic cancer, suggesting any differences in activity driving tumorigenesis may be local phenomena [39].
In pancreatic cancer, increased O2●− acts as an antiapoptotic, pro-survival mediator (Figure 1) [40]. This is supported by increased levels of nitrotyrosine, a footprint of the reactive nitrogen species peroxynitrite (formed by the reaction of O2●− with nitric oxide), in pancreatic cancer when compared to normal pancreas [41]. Growth factors were shown to stimulate O2●− generation in pancreatic cancer cells through the activation of NOX, whereas inhibition of O2●− production induced apoptosis. Similar results have been found in K-ras transformed human keratinocytes [42]. The increased levels of O2●− could be reversed efficiently by intracellular SODs. Most interestingly, the results demonstrated that SOD promoted death in K-ras transformed cells, whereas non-K-ras-expressing or wild type K-ras-expressing cancer cells were unaffected [42]. This is important because 95% of pancreatic cancers are K-ras mutants. Mutation of K-ras alone is enough to induce tumorigenicity in previously non-tumorigenic, immortalized pancreas cells [43] Tumorigenesis from K-ras mutation follows a predictable progression from acinar cells to a progenitor ductal phenotype and, finally, to the early pancreatic intra-epithelial neoplasm (PanIN) [44,45,46]. The point mutation of the K-ras oncogene is a common event in the early development of pancreatic cancer leading to an overall increase in production of reactive oxygen species via activation of NOX [3]. Interestingly, fibroblasts transfected with the viral ras oncogene have a resulting increase in O2●− production that has been proposed to act as a second messenger molecule to promote cell proliferation [47]. Oncogenesis mediated by ras also involves the activation of the Rel/nuclear factor (NF)-κB transcription factors. Indeed, constitutive activation of NF-κB p65 (RelA) has been demonstrated in 67% of pancreatic adenocarcinomas, which raises the possibility that it may itself be an oncogene playing a critical role in pancreatic tumorigenesis [48] Based on these observations, it is hypothesized that ras activates the NOX system to produce O2●−, which then leads to the activation of downstream signal transduction pathways, and ultimately cell proliferation.
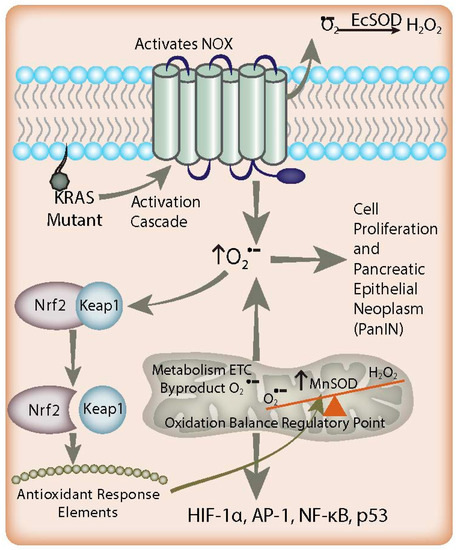
Figure 1.
Redox-based mechanisms in pancreatic cancer. K-ras mutations and NADPH oxidase (NOX) overexpression lead to increased O2●− flux in the early stages of pancreatic adenocarcinoma, resulting in activation of signaling pathways involved in proliferation. EcSOD scavenges the non-mitochondrial generation of O2●−, leading to growth inhibition in pancreatic cancer. In addition, there is an increased flux of mitochondrial O2●− in combination with suppressed MnSOD expression found in pancreatic tumor development, resulting in a redox imbalance leading to cell signaling events contributing to proliferation. Simultaneously, increased O2●− levels favor the dissociation of Nrf2 protein from Keap1. Nrf2 activates antioxidant response elements to increase the expression of MnSOD as a feedback mechanism to counterbalance the increased O2●− levels generated by K-ras mutation via NOX. Overall, cell O2●− levels remain increased and lead to PanIN malignant progression and Nrf2 expression; this paradoxically enables this progression by preventing cells from reaching cytotoxic O2●− levels.
K-ras mutation alone does not appear to decrease the SODs. Rather, upregulation of O2●−-producing NOX-2 and/or NOX-4 is likely an inciting initiating event which increases steady state reactive oxygen species in these cells (Figure 1) [33,44,49]. Furthermore, since K-ras is found in 95% of pancreatic cancers, this hypothesis could also explain the increased susceptibility of pancreatic cancer cells to scavenging of non-mitochondrial-generated O2●− [5,6]. The mechanism by which K-ras is responsible for NOX-2/4 upregulation is still under investigation. Mitochondrial protein isolates from K-ras mutants demonstrate a decrease in the Complex I protein NADH dehydrogenase 1 alpha sub-complex assembly factor 1 (NDUFAF1) [50], with resultant depletion of the NAD+ pool. Recent data show that NOX4 may be able to use NADH as a substrate, which suggests that NOX4 upregulation is a necessary response to NAD+ pool depletion in K-ras mutants. There is also some evidence that inactivation of p16 (CDKN2A; a cyclin dependent kinase) is required for tumorigenesis and further NOX over-activity in vivo [44].
While SOD expression changes do not appear to be the inciting event in development, the subsequent down-regulation of SOD expression in early pancreatic cancer is well documented. In pancreatic cancer cells with low SOD expression, over-expression of all three types of SOD slow tumor growth, suggesting that tight regulation through the SODs is important for tumor survival [6]. The mechanism of slowed growth by SOD in pancreatic cancer is likely multifactorial. Clearly, certain signaling pathways are de-activated by O2●− dismutation. The subsequent increases in H2O2 can be toxic, but smaller amounts can act as another growth signaling molecule, as demonstrated by additive decreased growth when SOD is combined with glutathione peroxidase (GPx) [51]. Thus, pancreatic cancer cells appear to be stimulated by increased levels of O2●− and H2O2, which allows for growth stimulation without cytotoxicity [5,6,33]
One critical function of MnSOD is the redox-based regulation of the cell cycle, an oxidative signaling mechanism that appears to control cell progression through proliferative and quiescent phases. Cellular mitosis is driven by a highly choreographed series of fluctuations in the intracellular oxidative state [22,52,53]. There is an increase in MnSOD levels in the G0/G1 phase that fluctuates inversely with cellular superoxide levels, glycolysis, and oxygen consumption [54]. The “Warburg effect”, described nearly a century ago, is observed in cancer cells whereby mitochondrial damage leads to an increase in glycolysis [55]. There is now abundant data supporting the supposition that cancer cell mitochondrial damage is due to a loss in MnSOD expression leading to aberrant cell proliferation [7,56,57,58,59,60]. Indeed, MnSOD levels have been found to be lower in pancreatic cancer, which correlates with increased cell proliferative capability, an effect that is reversed when the cells are transfected to overexpress MnSOD [7].
The role of CuZnSOD in oncogenesis is poorly defined. Some, but not all, cancers demonstrate decreased amounts of CuZnSOD activity [61,62]. There is increased CuZnSOD activity in the hemolyzed erythrocytes of patients with pancreatic adenocarcinoma, although the clinical significance of this is unclear because, as stated earlier, total SOD activity does not appear to be affected [39,63]. MnSOD, however, appears to follow a very predictable pattern in pancreatic oncogenesis. The transition from an early pancreatic intra-epithelial neoplasm (PanIN) to advanced PanIN correlates strongly with loss of MnSOD expression (Figure 1) [64]. Gong, et al., demonstrated that the loss of Pigment Epithelial Derived Factor (PEDF) during this transition results in decreased NF-κB nuclear translocation and MnSOD promoter binding, resulting in decreased MnSOD transcription [64]. Concomitant is the loss of PEDF regulation of autophagy, which is also increased as early neoplasia matures [64]. Methylation of the MnSOD promoter has also been demonstrated to decrease MnSOD expression in pancreatic cancer cells [65].
In somewhat contradictory form, Pandit, et al., demonstrated increased NF-κB expression in pancreatic cancer via a positive feedback loop with miR301A [66]. While both studies demonstrate decreased MnSOD in pancreatic cancer, the difference may be in comparing PanIN and pancreatic adenocarcinoma where NF-κB seems to have opposite effects. Regardless, many studies have demonstrated increased early aggressiveness of pancreatic cancers expressing low levels of functional MnSOD, which is consistent with the low levels of MnSOD in primary tumors, and the reversal of this phenotype with MnSOD overexpression [7,67,68,69]. Pancreatic cancers also demonstrate lower MnSOD expression relative to normal tissue. Metastases have a trend toward elevated expression of MnSOD relative to primary tumors, but are still lower than the surrounding tissue into which they implant [70]. This adaptation may be necessary to survive epithelial-to-mesenchymal transition and a new tumor microenvironment. Experiments by Wenger, et al., in Syrian golden hamsters with pancreatic adenocarcinoma, induced via N-nitrosobis-2-oxopropylamine, demonstrated that liver metastases also have higher SOD activity than primary tumors, but consistently lower levels than surrounding liver tissue [71]. In contrast, Li et al., have demonstrated that more invasive pancreatic adenocarcinoma cells demonstrate higher levels of SOD− and H2O2− dependent NF-κB and ERK activation, which drives invasiveness [72,73]. The activated ERK pathway appears to activate invasive mediators like matrix metalloprotease 9 (MMP-9) and MMP-2 [73]. Their results may be explained by a few observations, starting with the fact that they were working with human specimens. Cells that survive chemo- or radio-therapy may develop resistance via up-regulation of MnSOD. Another confounding factor may be the heterogeneity of MnSOD expression within tumors. In fact, MnSOD appears to be regulated relative to the cell cycle, as there are specific pathways that will increase MnSOD in quiescent pancreatic cancer cells in a way that is necessary for their survival [74,75]. This makes a heterogeneous tumor environment much less predictable than cell culture, and renders some cancer cells less sensitive to chemo- and radiotherapy.
Variant genotypes of EcSOD have been shown to be a risk factor for pancreatic adenocarcinoma [76]. Decreased EcSOD expression is associated with increased invasiveness secondary to increased O2●− interaction with nitric oxide, while overexpression of EcSOD correlates with decreased HIF-1α and vascular endothelial growth factor (VEGF) expression, and decreased tumor growth [8,77]. These important data may implicate EcSOD as a critical regulator of redox conditions in the tumor microenvironment that facilitates tumor growth.
The pancreatic tumor microenvironment (stroma) consists of fibroblasts, myofibroblasts, pancreatic stellate cells, immune cells, and endothelial cells [78]. Fibroblasts and extracellular matrix components are known to generate O2●− in response to various cytokines to produce an ideal tumor microenvironment for supporting pancreatic cancer growth [79,80]. Cancer-associated fibroblasts (CAFs) have been identified as a key source of interleukin 6 (IL-6), which signals tumor inflammatory and immune responses, tumor proliferation, and tumor angiogenesis [81]. However, normal human fibroblasts (NHFs) have also been shown to exhibit increases in reactive oxygen species and mitochondrial abnormalities when chronically aged [82]. As a result, age-associated metabolic reprogramming may be occurring which could contribute to pancreatic cancer progression [82]. Interestingly, MnSOD activity can reverse increases in O2●− generated by age-related mitochondrial morphology abnormalities. In fact, MnSOD has been shown in vitro to preserve mitochondrial morphology and protect fibroblasts from age-associated damage and chronological aging that can contribute to unregulated and aberrant proliferation [82].
5. Nrf2 Antioxidant Supports Pancreatic Cancer (PDAC) Development
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor that allows healthy cells to maintain steady states in conditions of oxidative stress to prevent carcinogenesis [83]. In fact, Nrf2 is a master transcription factor thought to be responsible for inducing the expression of numerous antioxidant enzymes [84]. Normally, Nrf2 is bound to the Keap1 protein in the cytosol until oxidative stresses increase within the cell, causing Nrf2 to dissociate from the Keap1 protein [85]. Unbound Nrf2 is then able to translocate to the nucleus where it binds to antioxidant response elements (ARE), transcription factors for key antioxidant enzymes, including MnSOD [85,86]. While Nrf2 function suppresses cancer development in normal cells, abnormal constitutive activation in developing cancer cells can facilitate the protection and propagation of growing tumors [87]. In particular, Nrf2 activation was found to be a key regulator of tumor maintenance in pancreatic cancer [88]. This may seem contradictory to conventional wisdom, which suggests K-ras mutations lead to increased levels of ROS in acinar cells required for tumorigenesis [46]. However, while K-ras mutant cells do lead to downstream generation of ROS, and Nrf2 activity is increased in response [85], there is strong evidence supporting the presence of oxidative damage [45]. To reconcile these observations, it has been proposed that Nrf2 activity helps achieve a cellular redox balance allowing K-ras-induced oxidative stress to facilitate proliferation of PanIN lesions without becoming severe enough to induce senescence or cause toxic DNA damage [45]. Furthermore, Nrf2 activity may be an important source of chemoresistance in pancreatic tumor cells as a constant source of oxidative stress [89,90]. Targeting strategies to lower Nrf2 activity in tumor cells have been proposed that would theoretically eliminate the antioxidant counterbalance to K-ras-induced oxidative stress and remove this potentially important source of chemoresistance. Although there has yet to be full elucidation regarding Nrf2 regulation of any SOD isoform in pancreatic acinar cells, an important link does likely exist.
6. Redox-Based Therapy in Pancreatic Cancer
Treatments aimed at modulating oxidative stress in pancreatic adenocarcinoma have focused on both increasing and decreasing reactive oxygen species [91,92]. Drugs that increase O2●− (Methyl-2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oate [CDDO-Me], dicumarol) have been shown to downregulate important growth regulators in pancreatic cancer including hTERT, p-AKT, p-MTOR, NF-κB, c-myc, SP1, and telomerases, and to induce apoptosis [93,94]. Some of these drugs (capsaicin) have been shown to directly downregulate SOD activity, resulting in increased O2●− levels, leading to mitochondrial damage and apoptosis, while other drugs disrupt the mitochondrial transport chain leading to increased O2●− [95,96]. Drugs that decrease O2●− levels appear to act as either scavengers themselves (Lipoxin) or as activations of pathways involved in scavenging (Octreotide, Vitamins A, C, and E) [67,73]. In one experimental animal model, vitamin supplementation decreased hepatic metastatic disease spread with concomitant increased SOD activity in the hepatic metastatic cells, as well as in normal surrounding hepatocytes [97]. This suggests that the oxidative state of potential metastatic sites is also an important factor.
6.1. Pharmacologic Ascorbate
Pharmacologic ascorbate (high dose intravenous Vitamin C) has demonstrated an ability to generate selective cytotoxicity in pancreatic cancer cells when combined with radiation and chemotherapy by acting as a prodrug to increase the flux of H2O2 [98]. Recent studies have further elaborated on the mechanism supporting its selective cytotoxicity in cancer. Abnormal cancer cell mitochondrial metabolism leads to increased levels of O2●− and H2O2, generating increased labile iron through disruptions in iron homeostasis [99]. Oxidation of pharmacologic ascorbate produces extra H2O2 and reacts with the excess pool of labile iron via Fenton chemistry to generate (OH) capable of producing oxidative DNA damage [100]. This hypothesis was supported by producing MnSOD knockout variants in A549 NSCLC cells, which are ordinarily relatively resistant to pharmacologic ascorbate. The MnSOD knockout cells were found to produce increases in O2●− levels leading to increased labile iron stores and enhanced cytotoxic sensitivity to pharmacologic ascorbate.
6.2. Manganoporphyrins
Manganoporphyrins (MnP) are a class of molecules originally developed as SOD mimetics with a central Mn(III) chelated in a porphyrin ring. This molecule behaves as an SOD mimetic (O2●− dismutation) through a two-step cycling between Mn(III) and Mn(II). First, Mn(III) is reduced in the presence of O2●− to Mn(II) and O2. Second, Mn(II) is oxidized by O2●− to Mn(III) and H2O2 [100]. However, in the presence of pharmacologic ascorbate, MnPs act as superoxide reductases, not as dismutases. Mn(III) is reduced by pharmacologic ascorbate to Mn(II), which reacts with O2 to form O2●−, and subsequently H2O2 and O2 [100]. The reason for this altered chemistry is that the porphyrin complex affects the half-cell reduction potential of the Mn (III), a metal favoring reduction by pharmacologic ascorbate [100,101,102]. As a result, MnPs easily redox cycle with pharmacologic ascorbate to enhance oxidation and increase the flux of H2O2 [103]. These results were reproduced in vivo, demonstrating decreased tumor volumes when MnP was combined with pharmacologic ascorbate and gemcitabine in pancreatic cancer mouse xenografts [104]. These experiments also determined decreases in tumor growth to be H2O2 mediated [104]. A Phase I clinical trial was completed, demonstrating the safety and tolerability of pharmacologic ascorbate in combination with gemcitabine for advanced pancreatic cancer [105]. Clinical trials are also underway combining pharmacologic ascorbate with gemcitabine and radiation for advanced local disease. However, the use of MnPs for pancreatic cancer have yet to make it to clinical study. Despite of the original design of MnPs to behave as SOD mimetics, the mechanisms linked to cytotoxicity in pancreatic cancer were not proven to be strictly related to its ability to dismute O2●−, but rather by its ability to enhance ascorbate oxidation [103,104]. More data is needed regarding the safety of MnPs in humans before combination regimens with pharmacologic ascorbate can be explored, but this treatment strategy does show promise.
6.3. Induction of ROS
Drug therapies that work through the induction of reactive oxygen species (2-methoxyestradiol, radiation) increase the expression of MnSOD and contribute to their radio-chemotherapy resistance [106,107]. The increase in MnSOD is likely highly regulated and multi-factorial, but the radiation-induced activation of NF-κB, Nrf-2, and SIRT3, leading to increased MnSOD activity, has been described [108,109]. However, this upregulation may include more than a signal transduction. Cells treated with gemcitabine release exosomes that have relatively high levels of MnSOD and catalase mRNA transcripts. When other cells are pre-treated with these exosomes, they demonstrate increased resistance to gemcitabine, and decreased levels of reactive oxygen species, suggesting lateral transfer as a potential mechanism of resistance to chemotherapy [110]. K-ras mutant pancreatic cancer cells also exhibit intense macropinocytosis, which aids in the uptake of these exosomes, making this pathway a target of new therapies [111].
7. Conclusions
Alteration of the oxidative state of cells is tightly intertwined with genetic mutations and protein expression changes in the development of pancreatic adenocarcinoma. K-ras mutations and NOX overexpression lead to increased O2●− flux early in pancreatic adenocarcinoma. These events lead to activation of the signaling pathways involved in proliferation. EcSOD or pharmacologically scavenging non-mitochondrial sources of O2●− may represent new opportunities for therapeutic intervention in pancreatic cancer. Increased O2●− flux combined with the suppressed MnSOD expression found in pancreatic tumor development also leaves cells vulnerable to therapies designed to take advantage of the imbalance in antioxidants and pro-oxidants. Treatments designed to overwhelm these vulnerable cells with an oxidative burst can activate pro-apoptotic pathways resulting in cell death. The resistance developed in surviving cells appears to be established by upregulation of the previously suppressed SODs.
Acknowledgments
Supported by NIH grants CA184051 and CA148062.
Author Contributions
Justin G. Wilkes and Matthew S. Alexander contributed equally to this manuscript with respect to literature review, organization of the article, and composition of the.
Conflicts of Interest
The submitted paper or any similar paper has not been published in any other scientific journal. There are no financial or other interests with regard to the submitted manuscript that might be construed as a conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M.; et al. Annual report to the nation on the status of cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Nazee, J.; Joseph, J.C. The role of antioxidant enzymes in the growth of pancreatic carcinoma. Curr. Cancer Ther. Rev. 2007, 3, 61–65. [Google Scholar]
- Weydert, C.; Roling, B.; Liu, J.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol. Cancer Ther. 2003, 2, 361–369. [Google Scholar] [PubMed]
- Teoh, M.L.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Modulation of reactive oxygen species in pancreatic cancer. Clin. Cancer Res. 2007, 13, 7441–7450. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.J.; Weydert, C.; Hinkhouse, M.M.; Ritchie, J.; Domann, F.E.; Spitz, D.; Oberley, L.W. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003, 63, 1297–1303. [Google Scholar] [PubMed]
- Sibenaller, Z.A.; Welsh, J.L.; Du, C.; Witmer, J.R.; Schrock, H.E.; Du, J.; Buettner, G.R.; Goswami, P.C.; Cieslak, J.A.; Cullen, J.J. Extracellular superoxide dismutase (EcSOD) suppresses hypoxia-inducible factor-1α (HIF-1α) in pancreatic cancer. Free Radic. Biol. Med. 2014, 69, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.E.; Armstrong, S.A.; Coussens, L.M.; Cruz-Correa, M.R.; DeBerardinis, R.J.; Doroshow, J.H.; Foti, M.; Hwu, P.; Kensler, T.W.; Morrow, M.; et al. Aacr cancer progress report 2016. Clin. Cancer Res. 2016, 22, S1–S137. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Oberley, L.W.; Leuthauser, S.W. The effect of iron on the distribution of superoxide and hydroxyl radicals as seen by spin trapping and on the superoxide dismutase assay. Photochem. Photobiol. 1978, 693–695. [Google Scholar] [CrossRef]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide ion: Generation and chemical implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Pantoliano, M.W.; McDonnell, P.J.; Burger, A.R.; Lippard, S.J. pH-dependent migration of copper(II) to the vacant zinc-binding site of zinc-free bovine erythrocyte superoxide dismutase. Proc. Natl. Acad. Sci. USA 1979, 76, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.J.; Balbirnie, M.M.; Ogihara, N.L.; Nersissian, A.M.; Weiss, M.S.; Valentine, J.S.; Eisenberg, D. A structure-based mechanism for copper-zinc superoxide dismutase. Biochemistry 1999, 38, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Fetherolf, M.M.; Boyd, S.D.; Taylor, A.B.; Kim, H.J.; Wohlschlegel, J.A.; Blackburn, N.J.; Hart, P.J.; Winge, D.R.; Winkler, D.D. Copper-zinc superoxide dismutase is activated through a sulfenic acid intermediate at a copper-ion entry site. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell differentiation, aging and cancer: The possible roles of superoxide and superoxide dismutases. Med. Hypotheses 1980, 6, 249–268. [Google Scholar] [CrossRef]
- Li, S.; Yan, T.; Yang, J.Q.; Oberley, T.D.; Oberley, L.W. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000, 60, 3927–3939. [Google Scholar] [PubMed]
- Marklund, S.L.; Holme, E.; Hellner, L. Superoxide dismutase in extracellular fluids. Clin. Chim. Acta 1982, 126, 41–51. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [PubMed]
- Marklund, S.L. Extracellular superoxide dismutase in human tissues and human cell lines. J. Clin. Invest. 1984, 74, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.M.; Jonsson, J.; Edlund, T.; Marklund, S.L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA 1995, 92, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, J.; Carlsson, L.; Marklund, S.L.; Edlund, T. The heparin-binding domain of extracellular superoxide dismutase C and formation of variants with reduced heparin affinity. J. Biol. Chem. 1992, 267, 18205–18209. [Google Scholar] [PubMed]
- Wheeler, M.D.; Smutney, O.M.; Samulski, R.J. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of B16 melanomas in mice. Mol. Cancer Res. 2003, 1, 871–881. [Google Scholar] [PubMed]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Du, J.; Nelson, E.S.; Simons, A.L.; Olney, K.E.; Moser, J.C.; Schrock, H.E.; Wagner, B.A.; Buettner, G.R.; Smith, B.J.; Teoh, M.L.; et al. Regulation of pancreatic cancer growth by superoxide. Mol. Carcinog. 2013, 52, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Balin, A.K. Oxidative influence on development and differentiation: An overview of a free radical theory of development. Free Radic. Biol. Med. 1989, 6, 631–661. [Google Scholar] [CrossRef]
- Hockenbery, D.M.; Oltvai, Z.N.; Yin, X.M.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993, 75, 241–251. [Google Scholar] [CrossRef]
- Tang, H.; Dong, X.; Day, R.S.; Hassan, M.M.; Li, D. Antioxidant genes, diabetes and dietary antioxidants in association with risk of pancreatic cancer. Carcinogenesis 2010, 31, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Wheatley-Price, P.; Asomaning, K.; Reid, A.; Zhai, R.; Su, L.; Zhou, W.; Zhu, A.; Ryan, D.P.; Christiani, D.C.; Liu, G. Myeloperoxidase and superoxide dismutase polymorphisms are associated with an increased risk of developing pancreatic adenocarcinoma. Cancer 2008, 112, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Marsakova, L.; Vrana, D.; Holcatova, I.; Ryska, M.; Smerhovsky, Z.; Slamova, A.; Schejbalova, M.; Soucek, P. Superoxide dismutase and nicotinamide adenine dinucleotide phosphate: Quinone oxidoreductase polymorphisms and pancreatic cancer risk. Pancreas 2011, 40, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kikuchi, S.; Yagyu, K.; Ishibashi, T.; Kurosawa, M.; Ito, Y.; Watanabe, Y.; Inaba, Y.; Tajima, K.; Nakachi, K.; et al. Serum soluble fas levels and superoxide dismutase activity and the risk of death from pancreatic cancer: A nested case-control study within the Japanese collaborative cohort study. Asian Pac. J. Cancer Prev. 2009, 10 Suppl, 81–85. [Google Scholar] [PubMed]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Greendorfer, J.S.; Vickers, S.M.; Thompson, J.A. Tyrosine nitration of c-SRC tyrosine kinase in human pancreatic ductal adenocarcinoma. Arch. Biochem. Biophys. 2000, 377, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Q.; Li, S.; Domann, F.E.; Buettner, G.R.; Oberley, L.W. Superoxide generation in v-Ha-ras–transduced human keratinocyte hacat cells. Mol. Carcinog. 1999, 26, 180–188. [Google Scholar] [CrossRef]
- Qian, J.; Niu, J.; Li, M.; Chiao, P.J.; Tsao, M.S. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005, 65, 5045–5053. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Ying, H.; Tian, T.; Ling, J.; Fu, J.; Lu, Y.; Wu, M.; Yang, L.; Achreja, A.; Chen, G.; et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 2017, 8, 14437. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Klöppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Lüttges, J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science (New York, N.Y.) 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-κB RelAl transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar] [PubMed]
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014, 345, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Song, M.; Zeng, Z.-L.; Zhu, C.-F.; Lu, W.-H.; Yang, J.; Ma, M.-Z.; Huang, A.M.; Hu, Y.; Huang, P. Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfunction by a proteomic screening approach. Oncotarget 2015, 6, 3947–3962. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Redox regulation of pancreatic cancer cell growth: Role of glutathione peroxidase in the suppression of the malignant phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar] [PubMed]
- Sarsour, E.H.; Kalen, A.L.; Xiao, Z.; Veenstra, T.D.; Chaudhuri, L.; Venkataraman, S.; Reigan, P.; Buettner, G.R.; Goswami, P.C. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012, 72, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den stoffwechsel der tumoren. Biochem. Zeitschrift 1924, 319–344. [Google Scholar]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W. Anticancer therapy by overexpression of superoxide dismutase. Antioxid. Redox Signal. 2001, 3, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, M.; Tamae, D.; Hou, D.X.; Wang, T.; Lebon, T.; Spitz, D.R.; Li, J.J. Response of cyclin B1 to ionizing radiation: Regulation by NF-κB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res. 2004, 24, 2657–2663. [Google Scholar] [PubMed]
- Venkataraman, S.; Jiang, X.; Weydert, C.; Zhang, Y.; Zhang, H.J.; Goswami, P.C.; Ritchie, J.M.; Oberley, L.W.; Buettner, G.R. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005, 24, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Weydert, C.J.; Waugh, T.A.; Ritchie, J.M.; Iyer, K.S.; Smith, J.L.; Li, L.; Spitz, D.R.; Oberley, L.W. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic. Biol. Med. 2006, 41, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Loven, D.P.; Oberley, L.W.; Rousseau, F.M.; Stevens, R.H. Superoxide dismutase activity in 1,2-dimethylhydrazine-induced rat colon adenocarcinoma. J. Natl. Cancer Inst. 1980, 65, 377–381. [Google Scholar] [PubMed]
- Kodydkova, J.; Vavrova, L.; Stankova, B.; Macasek, J.; Krechler, T.; Zak, A. Antioxidant status and oxidative stress markers in pancreatic cancer and chronic pancreatitis. Pancreas 2013, 42, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Belinsky, G.; Sagheer, U.; Zhang, X.; Grippo, P.J.; Chung, C. Pigment epithelium-derived factor (PEDF) blocks Wnt3a protein-induced autophagy in pancreatic intraepithelial neoplasms. J. Biol. Chem. 2016, 291, 22074–22085. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br. J. Cancer 2007, 97, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Pandit, H.; Zhang, W.; Li, Y.; Agle, S.; Li, X.; Li, S.P.; Cui, G.; Li, Y.; Martin, R.C. Manganese superoxide dismutase (MnSOD) expression is negatively associated with microRNA-301a in human Pancreatic Ductal Adenocarcinoma (PDAC). Cancer Gene Ther. 2015, 22, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Kilian, M.; Gregor, J.I.; Heukamp, I.; Helmecke, K.; Hanel, M.; Wassersleben, B.; Walz, M.K.; Schimke, I.; Kristiansen, G.; Wenger, F.A. Impact of Octreotide and SOM-230 on liver metastasis and hepatic lipidperoxidation in ductal pancreatic adenocarcinoma in Syrian Hamster. Clin. Exp. Metastasis 2009, 26, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Smith, B.J.; Oberley, L.W. Enzymatic activity is necessary for the tumor-suppressive effects of MnSOD. Antioxid. Redox Signal. 2006, 8, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Ough, M.; Lewis, A.; Zhang, Y.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic. Res. 2004, 38, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Du, J.; Liu, J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Metastatic progression of pancreatic cancer: Changes in antioxidant enzymes and cell growth. Clin. Exp. Metastasis 2005, 22, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, I.; Gregor, J.I.; Kilian, M.; Kiewert, C.; Jacobi, C.A.; Schimke, I.; Walz, M.K.; Guski, H.; Wenger, F.A. Influence of different dietary fat intake on liver metastasis and hepatic lipid peroxidation in BOP-induced pancreatic cancer in syrian hamsters. Pancreatology 2006, 6, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Z.; Ma, J.; Li, X.; Xu, Q.; Duan, W.; Chen, X.; Lv, Y.; Zhou, S.; Wu, E.; et al. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget 2015, 6, 31119–31133. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Li, J.; Chen, X.; Chen, K.; Li, W.; Li, X.; Zhang, L.; Duan, W.; Lei, J.; Xu, Q.; et al. Lipoxin A4 attenuates cell invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer. Oxid. Med. Cell. Longev. 2016, 2016, 6815727. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Ewton, D.Z.; Hu, J.; Vilenchik, M.; Deng, X.; Luk, K.C.; Polonskaia, A.; Hoffman, A.F.; Zipf, K.; Boylan, J.F.; Friedman, E.A. Inactivation of Mirk/Dyrk1B kinase targets quiescent pancreatic cancer cells. Mol. Cancer Ther. 2011, 10, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Qin, Y.M.; Guo, L.K. Correlations between polymorphisms of extracellular superoxide dismutase, aldehyde dehydrogenase-2 genes, as well as drinking behavior and pancreatic cancer. Chin. Med. Sci. J. 2014, 29, 162–166. [Google Scholar] [CrossRef]
- O’Leary, B.R.; Fath, M.A.; Bellizzi, A.M.; Hrabe, J.E.; Button, A.M.; Allen, B.G.; Case, A.J.; Altekruse, S.; Wagner, B.A.; Buettner, G.R.; et al. Loss of SOD3 (EcSOD) expression promotes an aggressive phenotype in human pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2015, 21, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem. J. 1989, 263, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Goswami, M.; Kalen, A.L.; Goswami, P.C. Mnsod activity protects mitochondrial morphology of quiescent fibroblasts from age associated abnormalities. Mitochondrion 2010, 10, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol Toxicol 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.M.; Kansanen, E.; Polonen, P.; Heinaniemi, M.; Levonen, A.L. Role of the keap1-Nrf2 pathway in cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. Nrf2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kang, H.J.; Kwon, S.Y.; Kim, H.J.; Kwon, K.Y.; Cho, C.H.; Lee, J.-M.; Kallakury, B.V.S.; Bae, I. Nrf2 regulates drug resistance in pancreatic cancer cells. Pancreas 2010, 39, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid. Med. Cell. Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Liu, Y.; Varma, N.R.S.; Arbab, A.S.; Gautam, S.C. Inhibition of telomerase activity by oleanane triterpenoid CDDO-Me in pancreatic cancer cells is ROS-dependent. Molecules 2013, 18, 3250–3265. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Liu, Y.B.; Gautam, S.C. Inhibition of cell proliferation and induction of apoptosis by CDDO-Me in pancreatic cancer cells is ROS-dependent. J. Exp. Ther. Oncol. 2012, 10, 51–64. [Google Scholar] [PubMed]
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PloS ONE 2011, 6, e20151. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Daniels, D.H.; Asbury, C.; Venkataraman, S.; Liu, J.; Spitz, D.R.; Oberley, L.W.; Cullen, J.J. Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. J. Biol. Chem. 2006, 281, 37416–37426. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, I.; Kilian, M.; Gregor, J.I.; Neumann, A.; Jacobi, C.A.; Guski, H.; Schimke, I.; Walz, M.K.; Wenger, F.A. Effects of the antioxidative vitamins A, C and E on liver metastasis and intrametastatic lipid peroxidation in BOP-induced pancreatic cancer in syrian hamsters. Pancreatology 2005, 5, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O●2− and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017, 31, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F.S.; Fridovich, I. The scavenging of superoxide radical by manganous complexes: In vitro. Arch. Biochem. Biophys. 1982, 214, 452–463. [Google Scholar] [CrossRef]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Rawal, M.; Schroeder, S.R.; Wagner, B.A.; Cushing, C.M.; Welsh, J.L.; Button, A.M.; Du, J.; Sibenaller, Z.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013, 73, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Cieslak, J.A.; Strother, R.K.; Rawal, M.; Du, J.; Doskey, C.M.; Schroeder, S.R.; Button, A.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free radic. Biol. Med. 2015, 83, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J.; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, Y. Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol. Cancer Res. R 2012, 10, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J.; Goswami, P.C. Mitochondria-targeted antioxidant enzyme activity regulates radioresistance in human pancreatic cancer cells. Cancer Biol. Ther. 2008, 7, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fan, M.; Candas, D.; Qin, L.; Zhang, X.; Eldridge, A.; Zou, J.X.; Zhang, T.; Juma, S.; Jin, C.; et al. CDK1-mediated SIRT3 activation enhances mitochondrial function and tumor radioresistance. Mol. Cancer Ther. 2015, 14, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Shang, B.-Y.; Sheng, W.-J.; Zhang, S.-H.; Li, Y.; Miao, Q.-F.; Zhen, Y.-S. A recombinantly tailored β-defensin that displays intensive macropinocytosis-mediated uptake exerting potent efficacy against K-ras mutant pancreatic cancer. Oncotarget 2016, 7, 58418–58434. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).