Reactive Oxygen and Nitrogen Species Regulate Key Metabolic, Anabolic, and Catabolic Pathways in Skeletal Muscle


 ,
, 
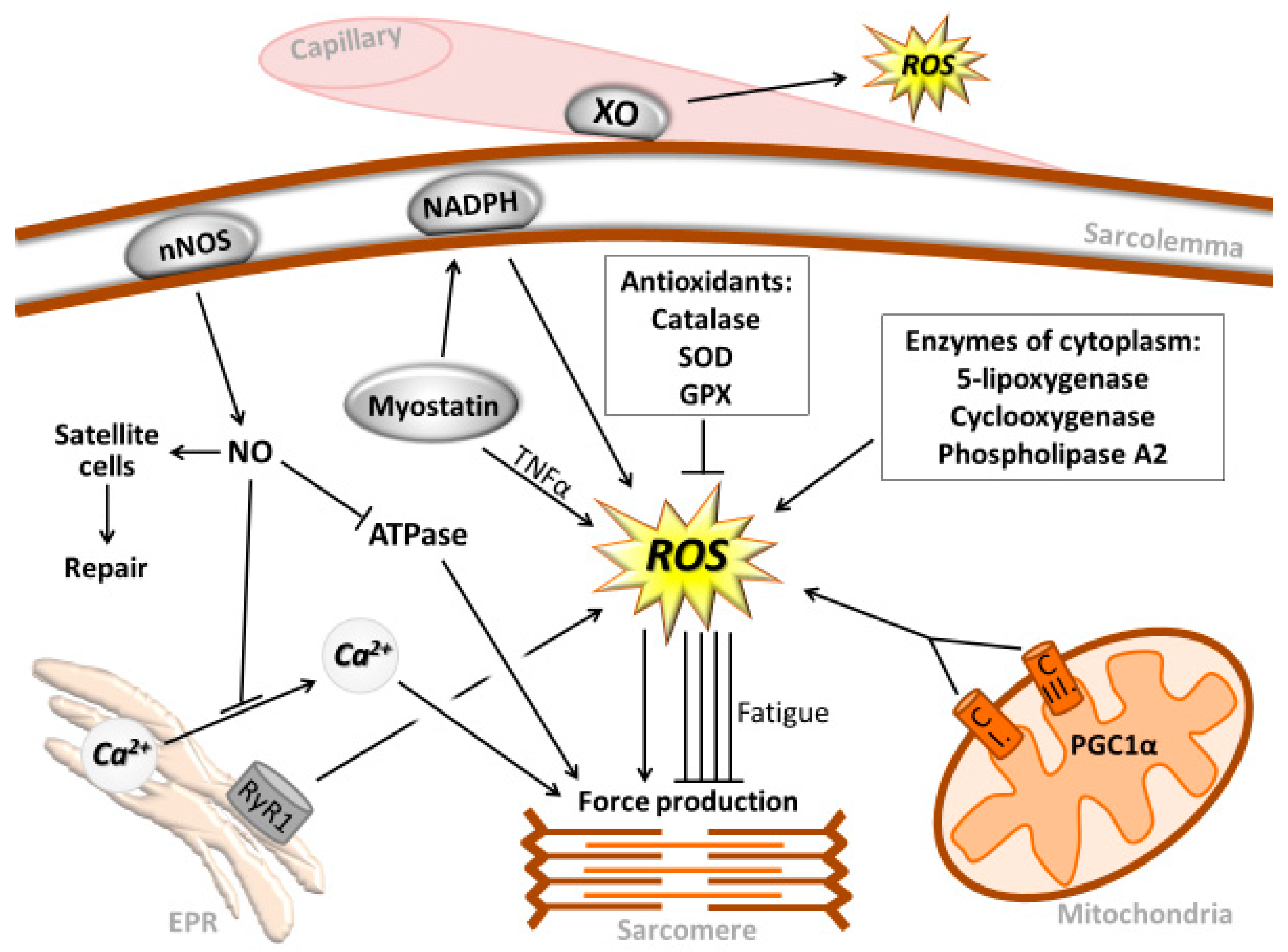
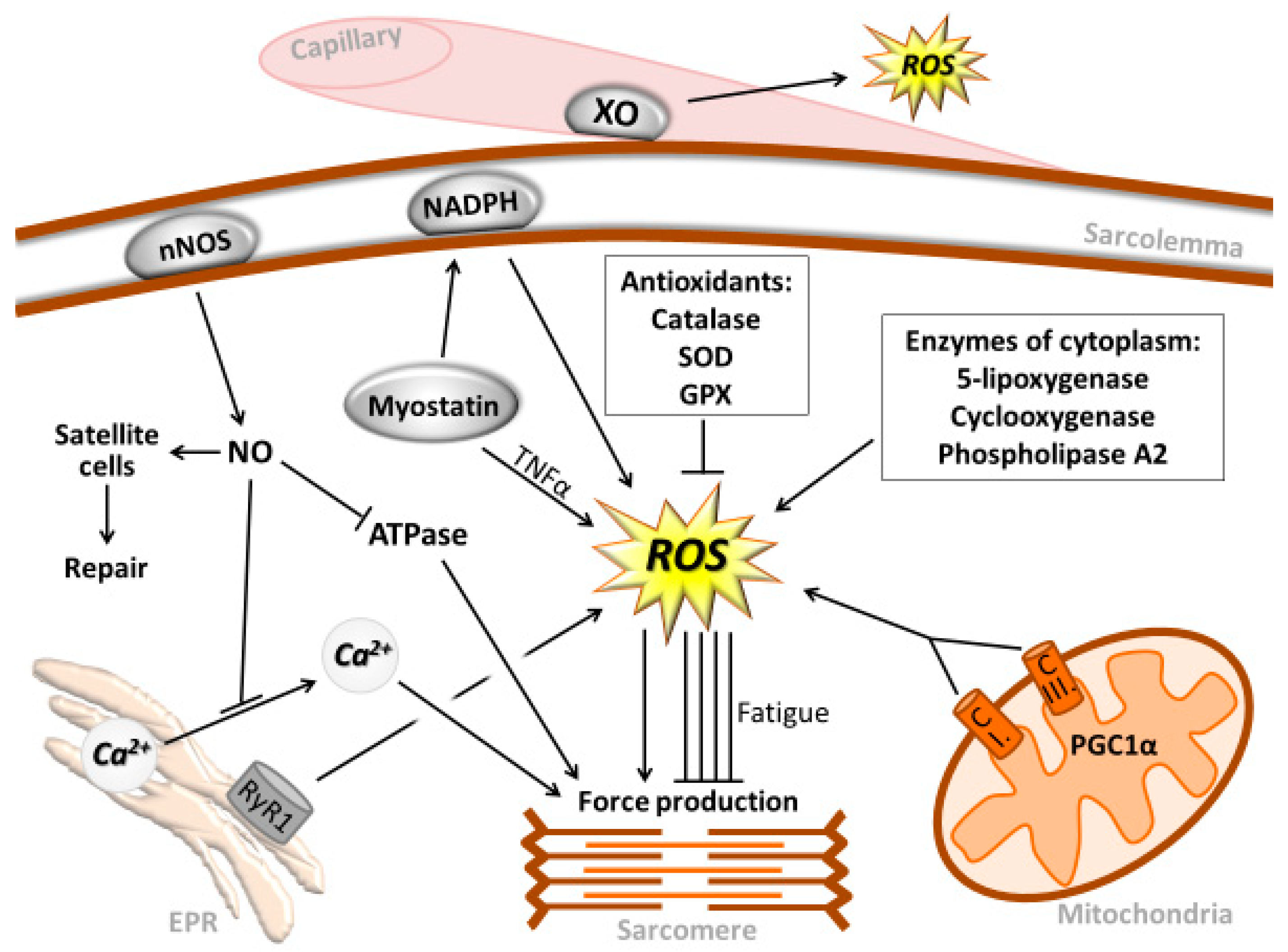{kind=link}
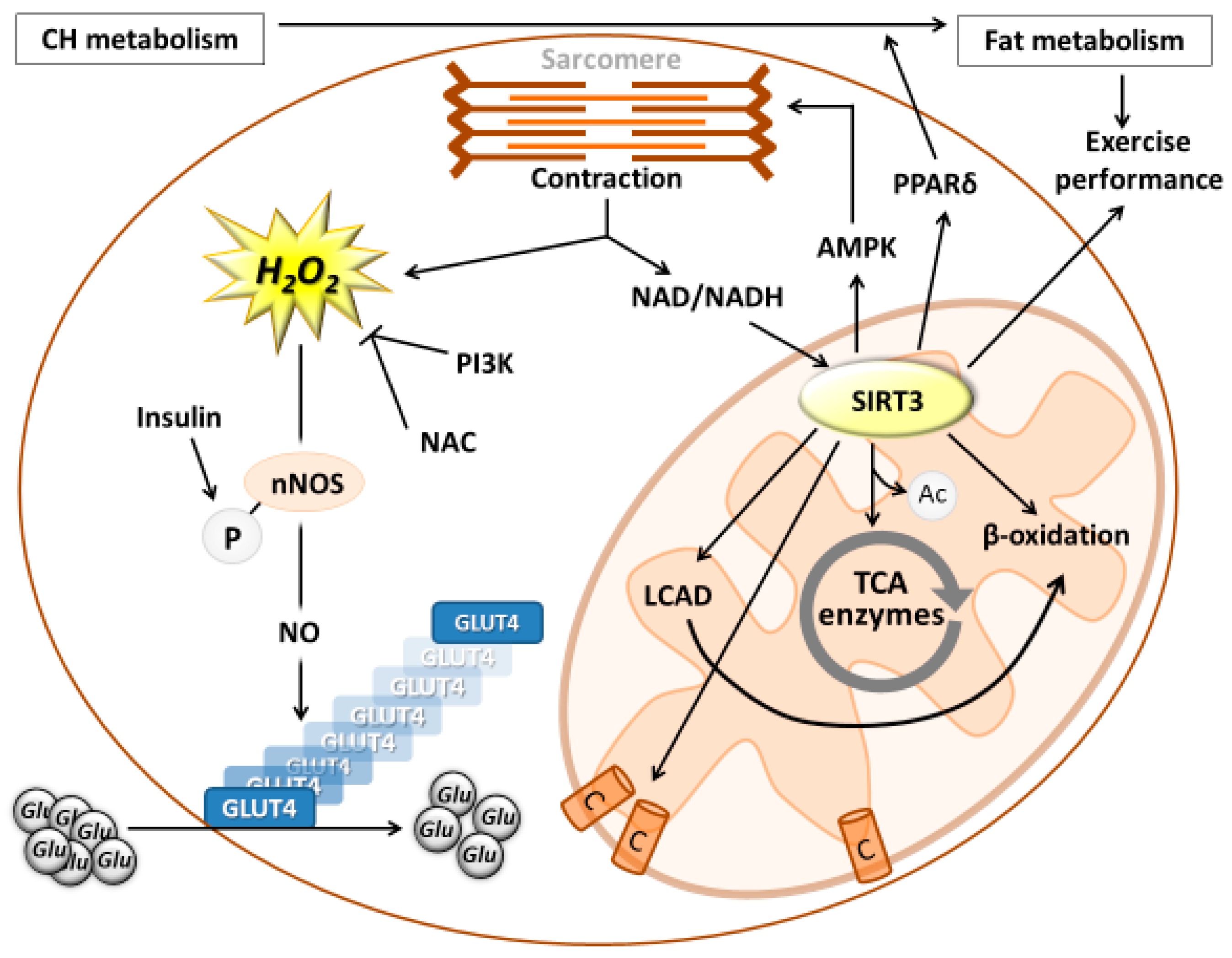
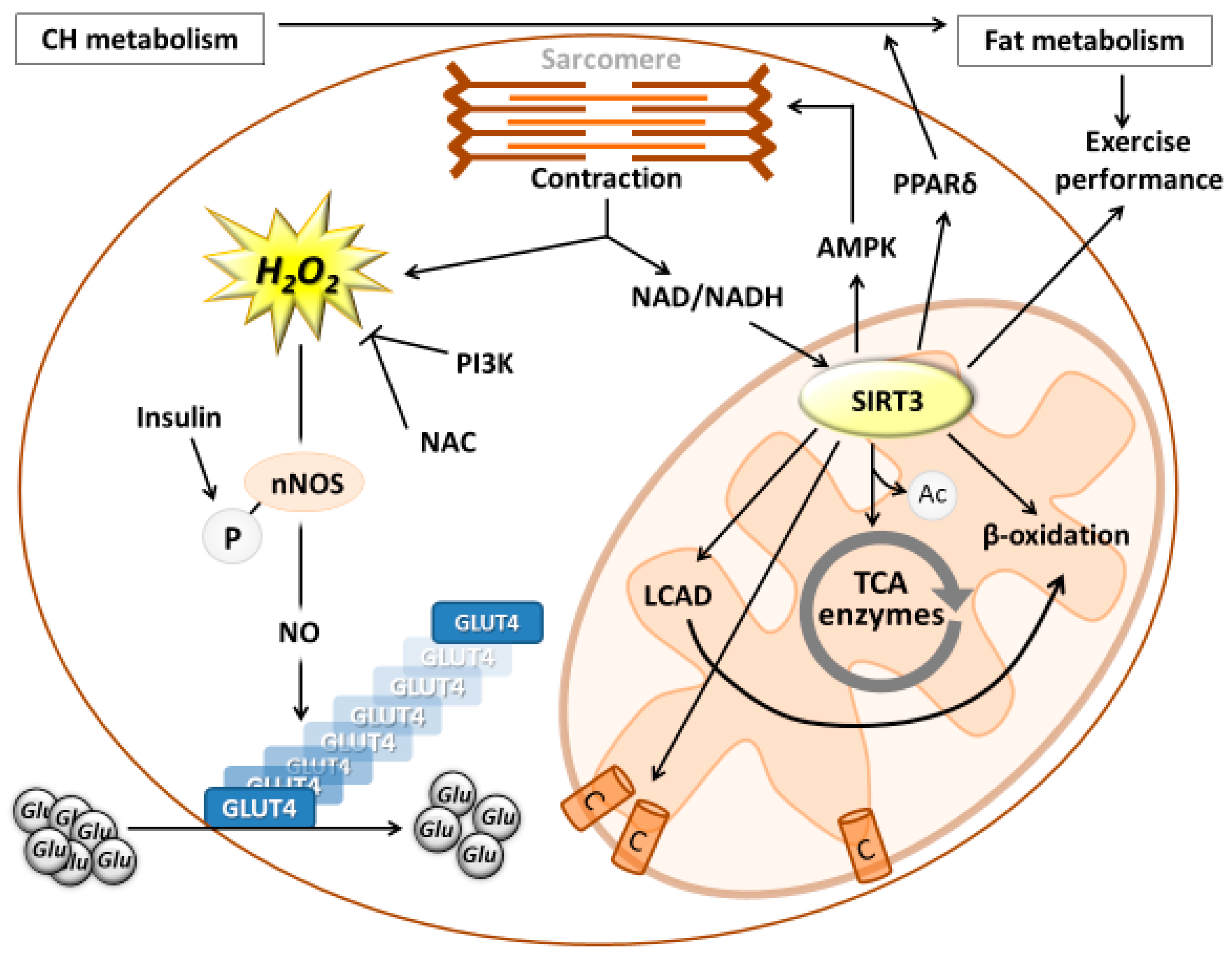{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Muscle Contraction and Reactive Oxygen and Nitrogen Species
3. RONS-Associated Oxidative Damage and Repair
4. The Role of ROS in Exercise-Induced Metabolism
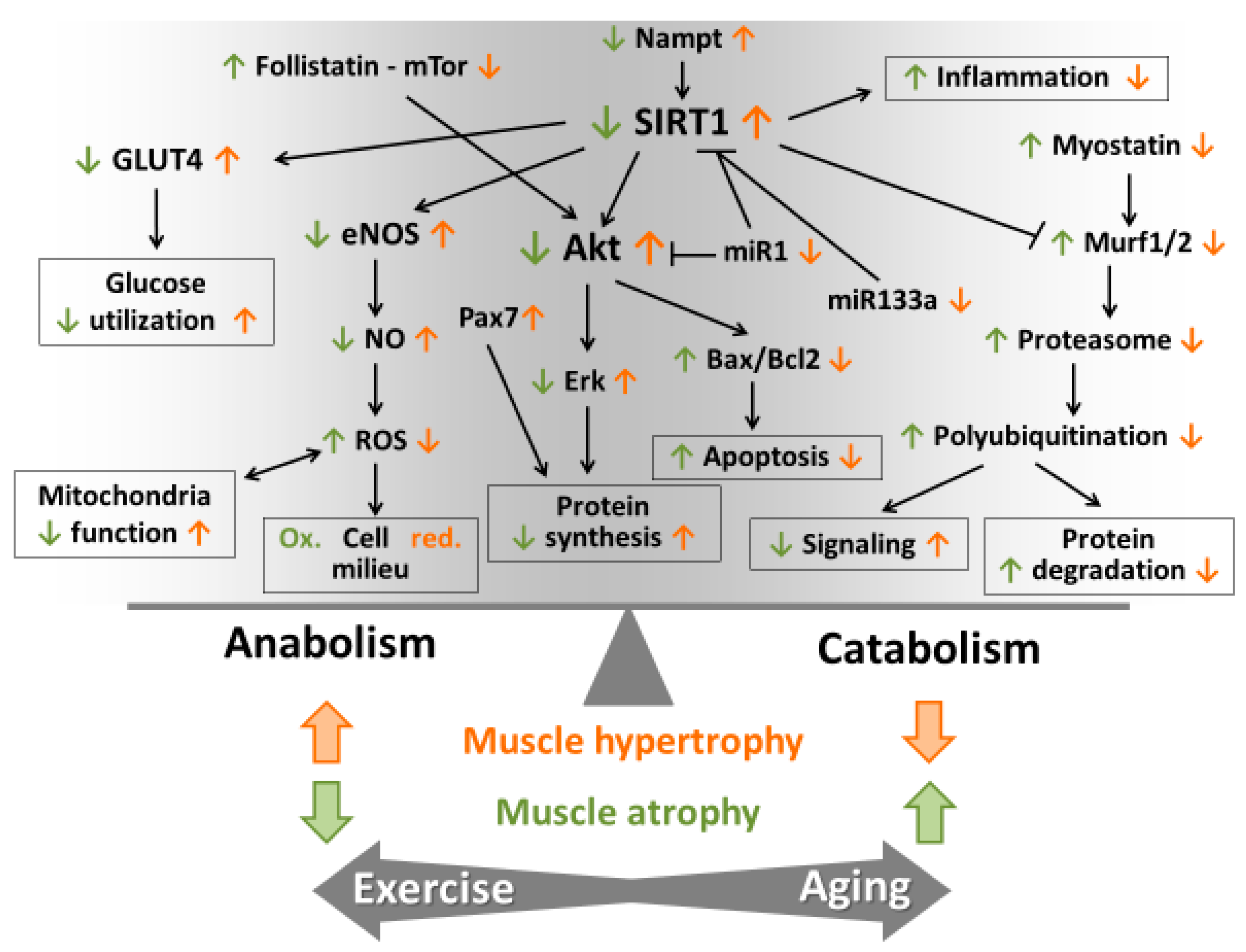
5. Role of ROS in Muscle Hypertrophy and Atrophy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef]
- Reid, M.B.; Khawli, F.A.; Moody, M.R. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. (1985) 1993, 75, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. (1985) 1992, 73, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.B.; Shoji, T.; Moody, M.R.; Entman, M.L. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol. (1985) 1992, 73, 1805–1809. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Naito, H.; Kaneko, T.; Chung, H.Y.; Radak, Z. Hormetic effects of regular exercise in aging: Correlation with oxidative stress. Appl. Physiol. Nutr. Metab. 2007, 32, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Exercise and hormesis: Oxidative stress-related adaptation for successful aging. Biogerontology 2005, 6, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Chung, H.Y.; Koltai, E.; Taylor, A.W.; Goto, S. Exercise, oxidative stress and hormesis. Ageing Res. Rev. 2008, 7, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Saborido, A.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; Megias, A. Stanozolol treatment decreases the mitochondrial ros generation and oxidative stress induced by acute exercise in rat skeletal muscle. J. Appl. Physiol. 2011, 110, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Vasilaki, A.; Mansouri, A.; Remmen, H.; van der Meulen, J.H.; Larkin, L.; Richardson, A.G.; McArdle, A.; Faulkner, J.A.; Jackson, M.J. Free radical generation by skeletal muscle of adult and old mice: Effect of contractile activity. Aging Cell 2006, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Shabalina, I.G.; Mattsson, C.M.; Bakkman, L.; Fernstrom, M.; Rozhdestvenskaya, Z.; Enqvist, J.K.; Nedergaard, J.; Ekblom, B.; Tonkonogi, M. Ultraendurance exercise increases the production of reactive oxygen species in isolated mitochondria from human skeletal muscle. J. Appl. Physiol. 2010, 108, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J. 1973, 134, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; Klimcakova, E.; St-Pierre, J. Impact of pgc-1alpha on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radic. Biol. Med. 2011, 51, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Ortenblad, N.; Young, J.F.; Oksbjerg, N.; Nielsen, J.H.; Lambert, I.H. Reactive oxygen species are important mediators of taurine release from skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2003, 284, C1362–C1373. [Google Scholar] [CrossRef] [PubMed]
- Bejma, J.; Ji, L.L. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J. Appl. Physiol. 1999, 87, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Nelson, W.B.; Hudson, M.B. Exercise-induced oxidative stress in humans: Cause and consequences. Free Radic. Biol. Med. 2011, 51, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J. Control of reactive oxygen species production in contracting skeletal muscle. Antioxid. Redox. Signal. 2011, 15, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide dismutase derivative reduces oxidative damage in skeletal muscle of rats during exhaustive exercise. J. Appl. Physiol. 1995, 79, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zweier, J.L. Substrate control of free radical generation from xanthine oxidase in the postischemic heart. J. Biol. Chem. 1995, 270, 18797–18803. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide dismutase derivative prevents oxidative damage in liver and kidney of rats induced by exhausting exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1996, 72, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Vina, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Nethery, D.; Stofan, D.; Callahan, L.; DiMarco, A.; Supinski, G. Formation of reactive oxygen species by the contracting diaphragm is PLA2 dependent. J. Appl. Physiol (1985) 1999, 87, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy—What causes the increased membrane permeability in skeletal muscle? Int. J. Biochem Cell Biol. 2011, 43, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Azevedo, L.; Jahne, S.; Hoffmann, C.; Stalder, D.; Heller, M.; Pries, A.R.; Zakrzewicz, A.; Baum, O. Up-regulation of the peroxiredoxin-6 related metabolism of reactive oxygen species in skeletal muscle of mice lacking neuronal nitric oxide synthase. J. Physiol. 2009, 587, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Goto, S.; Koltai, E. Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol. Aspects Med. 2011, 32, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Sathiakumar, D.; Venkatesh, R.; Salerno, M.S.; McFarlane, C.D.; Kambadur, R.; Sharma, M. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through nf-kappab. Aging Cell 2011, 10, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Syu, G.D.; Chen, H.I.; Jen, C.J. Severe exercise and exercise training exert opposite effects on human neutrophil apoptosis via altering the redox status. PLoS ONE 2011, 6, e24385. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nakaji, S.; Yamada, M.; Liu, Q.; Kurakake, S.; Okamura, N.; Kumae, T.; Umeda, T.; Sugawara, K. Impact of a competitive marathon race on systemic cytokine and neutrophil responses. Med. Sci. Sports Exerc. 2003, 35, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Totsuka, M.; Nakaji, S.; Yamada, M.; Kudoh, S.; Liu, Q.; Sugawara, K.; Yamaya, K.; Sato, K. Endurance exercise causes interaction among stress hormones, cytokines, neutrophil dynamics, and muscle damage. J. Appl. Physiol. (1985) 1999, 87, 1360–1367. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Mizokami, T.; Niihara, H.; Yada, K.; Suzuki, K. Neutrophil depletion attenuates muscle injury after exhaustive exercise. Med. Sci. Sports Exerc. 2016, 48, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Liu, C.Y.; Merola, A.J.; Berliner, L.J.; Clanton, T.L. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am. J. Physiol. Cell Physiol. 2000, 279, C1058–C1066. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favero, T.G.; Zable, A.C.; Abramson, J.J. Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1995, 270, 25557–25563. [Google Scholar] [CrossRef] [PubMed]
- Scherer, N.M.; Deamer, D.W. Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-atpase. Arch. Biochem. Biophys. 1986, 246, 589–601. [Google Scholar] [CrossRef]
- Matsuo, M. Oxygen dependency of life-span in the nematode. Comp. Biochem. Physiol. Comp. Physiol. 1993, 105, 653–658. [Google Scholar] [CrossRef]
- Zhu, X.; Heunks, L.M.; Ennen, L.; Machiels, H.A.; Van Der Heijden, H.F.; Dekhuijzen, P.N. Nitric oxide modulates neuromuscular transmission during hypoxia in rat diaphragm. Muscle Nerve 2006, 33, 104–112. [Google Scholar] [CrossRef] [PubMed]
- King-Vanvlack, C.E.; Curtis, S.E.; Mewburn, J.D.; Cain, S.M.; Chapler, C.K. Role of endothelial factors in active hyperemic responses in contracting canine muscle. J. Appl. Physiol. (1985) 1995, 79, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Viner, R.I.; Ferrington, D.A.; Williams, T.D.; Bigelow, D.J.; Schoneich, C. Protein modification during biological aging: Selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-atpase in skeletal muscle. Biochem. J. 1999, 340 Pt 3, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, L.G.; Minarovic, I.; Zahradnikova, A. Inhibition of the skeletal muscle ryanodine receptor calcium release channel by nitric oxide. FEBS Lett. 1996, 380, 49–52. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Pucsok, J.; Mecseki, S.; Csont, T.; Ferdinandy, P. Muscle soreness-induced reduction in force generation is accompanied by increased nitric oxide content and DNA damage in human skeletal muscle. Free Radic. Biol. Med. 1999, 26, 1059–1063. [Google Scholar] [CrossRef]
- Wolin, M.S.; Hintze, T.H.; Shen, W.; Mohazzab, H.K.; Xie, Y.W. Involvement of reactive oxygen and nitrogen species in signalling mechanisms that control tissue respiration in muscle. Biochem. Soc. Trans. 1997, 25, 934–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, T.; Kvietys, P.R. NFκB and AP-1 differentially contribute to the induction of mn-sod and enos during the development of oxidant tolerance. FASEB J. 2005, 19, 1908–1910. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Apor, P.; Pucsok, J.; Berkes, I.; Ogonovszky, H.; Pavlik, G.; Nakamoto, H.; Goto, S. Marathon running alters the DNA base excision repair in human skeletal muscle. Life Sci. 2003, 72, 1627–1633. [Google Scholar] [CrossRef]
- Radak, Z.; Naito, H.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Takahashi, R.; Cardozo-Pelaez, F.; Goto, S. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 2002, 445, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Kumagai, S.; Nakamoto, H.; Goto, S. 8-Oxoguanosine and uracil repair of nuclear and mitochondrial DNA in red and white skeletal muscle of exercise-trained old rats. J. Appl. Physiol. (1985) 2007, 102, 1696–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, Z.; Atalay, M.; Jakus, J.; Boldogh, I.; Davies, K.; Goto, S. Exercise improves import of 8-oxoguanine DNA glycosylase into the mitochondrial matrix of skeletal muscle and enhances the relative activity. Free Radic. Biol. Med. 2009, 46, 238–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, Z.; Takahashi, R.; Kumiyama, A.; Nakamoto, H.; Ohno, H.; Ookawara, T.; Goto, S. Effect of aging and late onset dietary restriction on antioxidant enzymes and proteasome activities, and protein carbonylation of rat skeletal muscle and tendon. Exp. Gerontol. 2002, 37, 1423–1430. [Google Scholar] [CrossRef]
- Clavel, S.; Coldefy, A.S.; Kurkdjian, E.; Salles, J.; Margaritis, I.; Derijard, B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis anterior muscle. Mech. Ageing Dev. 2006, 127, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Raue, U.; Slivka, D.; Jemiolo, B.; Hollon, C.; Trappe, S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1407–1412. [Google Scholar] [CrossRef] [PubMed]
- Husom, A.D.; Peters, E.A.; Kolling, E.A.; Fugere, N.A.; Thompson, L.V.; Ferrington, D.A. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 2004, 421, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Ohno, H.; Sasvari, M.; Nyakas, C.; Goto, S. The effect of exercise training on oxidative damage of lipids, proteins, and DNA in rat skeletal muscle: Evidence for beneficial outcomes. Free Radic. Biol. Med. 1999, 27, 69–74. [Google Scholar] [CrossRef]
- Reid, M.B. Response of the ubiquitin-proteasome pathway to changes in muscle activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R1423–R1431. [Google Scholar] [CrossRef] [PubMed]
- Sultan, K.R.; Dittrich, B.T.; Leisner, E.; Paul, N.; Pette, D. Fiber type-specific expression of major proteolytic systems in fast- to slow-transforming rabbit muscle. Am. J. Physiol. Cell Physiol. 2001, 280, C239–C247. [Google Scholar] [CrossRef] [PubMed]
- Kozlovsky, N.; Rudich, A.; Potashnik, R.; Bashan, N. Reactive oxygen species activate glucose transport in l6 myotubes. Free Radic. Biol. Med. 1997, 23, 859–869. [Google Scholar] [CrossRef]
- Sandstrom, M.E.; Zhang, S.J.; Bruton, J.; Silva, J.P.; Reid, M.B.; Westerblad, H.; Katz, A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol. 2006, 575, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative stress stimulates skeletal muscle glucose uptake through a phosphatidylinositol 3-kinase-dependent pathway. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E889–E897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellogg, D.L., 3rd; McCammon, K.M.; Hinchee-Rodriguez, K.S.; Adamo, M.L.; Roman, L.J. Neuronal nitric oxide synthase mediates insulin- and oxidative stress-induced glucose uptake in skeletal muscle myotubes. Free Radic. Biol. Med. 2017, 110, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Heng, B.; He, W.; Shi, L.; Lai, C.; Xiao, L.; Ren, H.; Mo, S.; Su, Z. Chronic reactive oxygen species exposure inhibits glucose uptake and causes insulin resistance in C2C12 myotubes. Biochem. Biophys. Res. Commun. 2016, 478, 798–803. [Google Scholar] [CrossRef] [PubMed]
- White, A.T.; Schenk, S. NAD+/NADH and skeletal muscle mitochondrial adaptations to exercise. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E308–E321. [Google Scholar] [CrossRef] [PubMed]
- Edington, D.W.; McCafferty, W.B. Mitochondrial size distribution analysis in the soleus muscle of trained and aged rats. Experientia 1973, 29, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radak, Z.; Bori, Z.; Koltai, E.; Fatouros, I.G.; Jamurtas, A.Z.; Douroudos, I.I.; Terzis, G.; Nikolaidis, M.G.; Chatzinikolaou, A.; Sovatzidis, A.; et al. Age-dependent changes in 8-oxoguanine-DNA glycosylase activity are modulated by adaptive responses to physical exercise in human skeletal muscle. Free Radic. Biol. Med. 2011, 51, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas-Ortiz, K.; Perez-Vazquez, V.; Figueroa, A.; Diaz, F.J.; Montano-Ascencio, P.G.; Macias-Cervantes, M.H. Aerobic training but no resistance training increases SIRT3 in skeletal muscle of sedentary obese male adolescents. Eur. J. Sport Sci. 2018, 18, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Palacios, O.M.; Carmona, J.J.; Michan, S.; Chen, K.Y.; Manabe, Y.; Ward, J.L., 3rd; Goodyear, L.J.; Tong, Q. Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1α in skeletal muscle. Aging (Albany N. Y.) 2009, 1, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Chen, K.; Abdel Khalek, W.; Ward, J.L., 3rd; Yang, H.; Chabi, B.; Wrutniak-Cabello, C.; Tong, Q. Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3. PLoS ONE 2014, 9, e85636. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARδ promotes running endurance by preserving glucose. Cell Metab. 2017, 25, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R. Interventions based on the possibility that oxidative stress contributes to sarcopenia. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50A, 157–161. [Google Scholar]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Dubouchaud, H.; Mosoni, L.; Chardigny, J.M.; Oudot, A.; Fontaine, E.; Vergely, C.; Keriel, C.; Rochette, L.; Leverve, X.; et al. Abnormalities of mitochondrial functioning can partly explain the metabolic disorders encountered in sarcopenic gastrocnemius. Aging Cell 2007, 6, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R.; et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degens, H. The role of systemic inflammation in age-related muscle weakness and wasting. Scand. J. Med. Sci. Sports 2010, 20, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Exercise at old age: Does it increase or alleviate oxidative stress? Ann. N. Y. Acad. Sci. 2001, 928, 236–247. [Google Scholar] [CrossRef] [PubMed]
- McArdle, A.; Jackson, M.J. The role of attenuated redox and heat shock protein responses in the age-related decline in skeletal muscle mass and function. Essays Biochem. 2017, 61, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Halade, G.V.; El Jamali, A.; Fernandes, G. Conjugated linoleic acid (CLA) prevents age-associated skeletal muscle loss. Biochem. Biophys. Res. Commun. 2009, 383, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavers, K.M.; Beavers, D.P.; Serra, M.C.; Bowden, R.G.; Wilson, R.L. Low relative skeletal muscle mass indicative of sarcopenia is associated with elevations in serum uric acid levels: Findings from nhanes III. J. Nutr. Health Aging 2009, 13, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, A.P.; McCormick, R.; Nye, G.A.; McArdle, A. Mechanisms of skeletal muscle ageing; avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2014, 16, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tian, Z.; Kadomatsu, T.; Xie, P.; Miyata, K.; Sugizaki, T.; Endo, M.; Zhu, S.; Fan, H.; Horiguchi, H.; et al. Age-dependent increase in angiopoietin-like protein 2 accelerates skeletal muscle loss in mice. J. Biol. Chem. 2018, 293, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Ziaaldini, M.M.; Koltai, E.; Csende, Z.; Goto, S.; Boldogh, I.; Taylor, A.W.; Radak, Z. Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp. Gerontol. 2015, 67, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Siu, P.M.; Alway, S.E. Aging alters the reduction of pro-apoptotic signaling in response to loading-induced hypertrophy. Exp. Gerontol. 2006, 41, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Koltai, E.; Bori, Z.; Chabert, C.; Dubouchaud, H.; Naito, H.; Machida, S.; Davies, K.J.; Murlasits, Z.; Fry, A.C.; Boldogh, I.; et al. SIRT1 may play a crucial role in overload-induced hypertrophy of skeletal muscle. J. Physiol. 2017, 595, 3361–3376. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemes, R.; Koltai, E.; Taylor, A.W.; Suzuki, K.; Gyori, F.; Radak, Z. Reactive Oxygen and Nitrogen Species Regulate Key Metabolic, Anabolic, and Catabolic Pathways in Skeletal Muscle. Antioxidants 2018, 7, 85. https://doi.org/10.3390/antiox7070085
Nemes R, Koltai E, Taylor AW, Suzuki K, Gyori F, Radak Z. Reactive Oxygen and Nitrogen Species Regulate Key Metabolic, Anabolic, and Catabolic Pathways in Skeletal Muscle. Antioxidants. 2018; 7(7):85. https://doi.org/10.3390/antiox7070085
Chicago/Turabian StyleNemes, Roland, Erika Koltai, Albert W. Taylor, Katsuhiko Suzuki, Ferenc Gyori, and Zsolt Radak. 2018. "Reactive Oxygen and Nitrogen Species Regulate Key Metabolic, Anabolic, and Catabolic Pathways in Skeletal Muscle" Antioxidants 7, no. 7: 85. https://doi.org/10.3390/antiox7070085
APA StyleNemes, R., Koltai, E., Taylor, A. W., Suzuki, K., Gyori, F., & Radak, Z. (2018). Reactive Oxygen and Nitrogen Species Regulate Key Metabolic, Anabolic, and Catabolic Pathways in Skeletal Muscle. Antioxidants, 7(7), 85. https://doi.org/10.3390/antiox7070085